

Abstract
DNA methylation is an epigenetic mechanism for gene silencing engaged by DNA methyltransferase (Dnmt)-catalyzed methyl group transfer to cytosine residues in gene-regulatory regions. It is unknown whether aberrant DNA methylation can cause neurodegeneration. We tested the hypothesis that Dnmts can mediate neuronal cell death. Enforced expression of Dnmt3a induced degeneration of cultured NSC34 cells. During apoptosis of NSC34 cells induced by camptothecin, levels of Dnmt1 and Dnmt3a increased fivefold and twofold, respectively, and 5-methylcytosine accumulated in nuclei. Truncation mutation of the Dnmt3a catalytic domain and Dnmt3a RNAi blocked apoptosis of cultured neurons. Inhibition of Dnmt catalytic activity with RG108 and procainamide protected cultured neurons from excessive DNA methylation and apoptosis. In vivo, Dnmt1 and Dnmt3a are expressed differentially during mouse brain and spinal cord maturation and in adulthood when Dnmt3a is abundant in synapses and mitochondria. Dnmt1 and Dnmt3a are expressed in motor neurons of adult mouse spinal cord, and, during their apoptosis induced by sciatic nerve avulsion, nuclear and cytoplasmic 5-methylcytosine immunoreactivity, Dnmt3a protein levels and Dnmt enzyme activity increased preapoptotically. Inhibition of Dnmts with RG108 blocked completely the increase in 5-methycytosine and the apoptosis of motor neurons in mice. In human amyotrophic lateral sclerosis (ALS), motor neurons showed changes in Dnmt1, Dnmt3a, and 5-methylcytosine similar to experimental models. Thus, motor neurons can engage epigenetic mechanisms to drive apoptosis, involving Dnmt upregulation and increased DNA methylation. These cellular mechanisms could be relevant to human ALS pathobiology and disease treatment.
Introduction
DNA methylation is an epigenetic process whereby a DNA methyltransferase (Dnmt) catalyzes the covalent transfer of a methyl group from S-adenosyl-methionine to the 5′ carbon position of a substrate cytosine residue (Cheng, 1995; Pradhan et al., 1999; Bestor, 2000; Szyf, 2005). This reaction is part of a complex mechanism for gene regulation. DNA methylation changes protein–DNA interactions that lead to alterations in chromatin structure and rate of transcription (Jones and Takai, 2001). Generally, when DNA methylation is involved in gene silencing, cytosine methylation occurs in cytosine–phosphate–guanine (CpG) dinucleotides within the 5′ regulatory regions of genes (Bird, 2002; Brenner and Fuks, 2006). CpG-rich areas are referred to as CpG islands (Bird, 2002; Brenner and Fuks, 2006) and have been defined as a region of 200 bp where guanine-cytosines make up >50% of the region and CpGs are >60% of the observed/expected ratio within that region (Gardiner-Garden and Frommer, 1987). There are ∼3 × 107 residues of 5-methylcytosine in the mammalian genome, and 40% of genes contain CpG islands in their promoters (Bestor, 2000; Fatemi et al., 2005). CpG-rich areas in promoters are one of the initiation sites where transcription factors bind to the DNA allowing transcription of the corresponding gene. When these regulatory regions are methylated, transcription factors are unable to bind DNA, thus inhibiting gene transcription (Kimura and Shiota, 2003). The transcriptional repression involves the methylated cytosines operating in a stable multiprotein complex of corepressors to modify chromatin structure and inhibit transcription (Razin, 1998; Curradi et al., 2002).
Several isoforms of Dnmts have been cloned. Dnmt1 is the most abundant isoform in proliferating cells, displays a preference for hemimethylated substrates, and is targeted to replication forks, acting to maintain methylation patterns during cell replication (Robertson, 2001). Dnmt2 transfers methyl groups to RNA, not DNA (Schaefer and Lyko, 2010). Dnmt3a and Dnmt3b function as de novo methyltransferases because they methylate hemimethylated DNA and also completely unmethylated DNA (Okano et al., 1999; Xie et al., 1999). Dnmt3L functions as an essential regulatory cofactor (Jia et al., 2007). Dnmts are essential for proper nervous system development (Martinowich et al., 2003) and are present in adult brain and in neurons (Feng et al., 2005, 2010). Aberrant methylation of DNA has been implicated in neurological disorders, including Rett syndrome (Kondo et al., 2000); mutations in the Dnmt3b gene cause immunodeficiency centromeric region instability and facial anomalies syndrome (Okano et al., 1999). The functions of Dnmts in postmitotic neurons are not understood. Recent studies have shown that Dnmts function in synaptic plasticity (Levenson et al., 2006; Miller and Sweatt, 2007) and behavioral plasticity (LaPlant et al., 2001). Reduced levels of Dnmt1 in heterozygous null mice confer protection against stroke (Endres et al., 2000). However, it is not known whether epigenetic mechanisms through DNA methylation and Dnmts have any direct role in neurodegeneration. Here, we examined epigenetically driven neuronal cell death signaling and found evidence for Dnmt- and cytosine methylation-mediated motor neuron apoptosis.
Materials and Methods
Generation of green fluorescent protein fusion protein constructs.
To generate Dnmt1– and Dnmt3a–green fluorescent protein (GFP) fusion protein constructs, the mouse Dnmt1 and Dnmt3a mRNA (GenBank accession numbers Dnmt1-NM_010066 and Dnmt3a-NM_007872) sequences were PCR amplified from commercial mammalian expression vectors for Dnmt1 (InvivoGen) and Dnmt3a (Invitrogen). PCR oligonucleotides were modified by eliminating the stop codons in both cDNAs, and MluI and HpaI restriction sites (New England Biolabs) were added to the 5′ and 3′ends, respectively. The oligonucleotides were designed (Dnmt1 sequence 1, CGCCACGCGTCAGCATGCCAGCGAACAGCTCC; sequence 2, AGAAGTTAACGTCCTTGGTAGCAGCCTCCTC; Dnmt3a sequence 1, CGCCACGCGTCCACCATGCCCTCCAGCGGCCC; sequence 2, CGACGTTAACCACACAAGCAAAATATTCCTTCA), annealed, and subcloned into pORF9 vector (InvivoGen), which had previously been digested with MluI and HpaI (Integrated DNA Technologies). The GFP sequence was PCR amplified from Vitality-GFP vector (Stratagene), and HpaI and SwaI restriction sites were added to the 5′ and 3′ ends, respectively. Oligonucleotides were designed (sequence 1, CGCCGTTAACATGGTGAGCAAGCAGATCCTG; sequence 2, AGAAATTTAAATTTACACCCACTCGTGCAGGCT), annealed, and subcloned into the existing Dnmt-pORF9 plasmid.
Mutant Dnmt3a-GFP.
To express a mutant form of Dnmt3a without methytransferase activity, we used restriction enzyme digestion for targeted deletion of the catalytic domain. Aat II (New England Biolabs) was used to digest the full construct plasmid. The digestion by Aat II deleted the gene fragment encoding the protein amino acid sequence 630 through 903 of Dnmt3a. This deletion eliminates enzyme catalytic activity because of the incapacity to bind the regulatory Dnmt3L (Jia et al., 2007). The plasmid was run on a 1% agarose gel to confirm the correct size after the deletion and the product was ligated using a T4 DNA ligase (Invitrogen).
Cell lines, cell culture, and transfection.
Human embryonic kidney 293 (HEK293) cells (Invitrogen) were grown at 37°C in 95% air–5% CO2 in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Mouse brain C8-D1A astrocytes (CRL-2467; American Type Culture Collection) were grown at 37°C in 95% air-5% CO2 in DMEM supplemented with 10% FBS, 2 mm l-glutamine, and antibiotics. Mouse brain EOC13.31 microglia (CRL-2468; American Type Culture Collection) were grown at 37°C in 95% air–5% CO2 in DMEM supplemented with 4 mm l-glutamine, 1.5 g/l sodium bicarbonate, 4.5 g/l sucrose, 10% FBS, 20% mouse bone marrow-producing colony-stimulating factor-1 conditioned medium, and antibiotics. The NSC34 cell line (Cashman et al., 1992) was obtained from Cellutions Biosystems. The cells were maintained and subcultured every 3 d in DMEM with 10% fetal bovine serum. The NSC34 cell differentiation culture medium consisted of high-glucose DMEM supplemented with 10% heat-inactivated FBS (Invitrogen), 2 mm l-glutamine, 0.1 mm nonessential amino acids, and antibiotics at 37°C in 95% air–5% CO2.
Transient transfections with Dnmt expression plasmids were performed using Lipofectamine 2000 (Invitrogen), and cells were harvested 48 h later. Cell cultures were lysed in TNE buffer (10 mm Tris-HCl, pH7.4, 5 mm EDTA, and 150 mm NaCl) supplemented with protease inhibitors (Roche). NSC34 cells were also cotransfected with Dnmt-GFP fusion protein expression constructs and mitochondrial-targeted red fluorescent protein (Su9-RFP plasmid; kindly provided by Drs. H. Sesaki and R. Jensen, Johns Hopkins Medical Institutions, Baltimore, MD).
NSC34 cell cultures were differentiated over 12–20 d. The sustained culturing and differentiation was done to determine whether NSC34 cells could achieve large, polygonal motor neuron-like somatodendritic morphologies. NSC34 cells at 5 d in vitro (DIV5) were compared with NSC34 cells cultured for 14–20 d (DIV14–20). To assess morphological differentiation in living cells, transfection with Vitality-GFP vector (Stratagene) was done. Differentiation was also specified by immunophenotyping for neurofilament, microtubule-associated protein 2 (MAP2), and synaptophysin (Chang and Martin, 2011). For immunocytochemistry, DIV5 and DIV14–20 NSC34 cultures grown on chamber slides were fixed for 20 min in 4% paraformaldehyde in 4% sucrose–PBS. Cells were immunostained with a mouse monoclonal antibody to neurofilament-H (SMI32; Sternberger Monoclonals), a mouse monoclonal antibody to MAP2 (Roche), and a rabbit polyclonal antibody to synaptophysin (Dako). Alexa-488- and Alexa-594-conjugated secondary antibodies were used. Cells were imaged with a Meta confocal microscope (Carl Zeiss Microimaging), and pictures were captured using ZEN 2007 Light Edition software (Carl Zeiss Microimaging). The nuclear sizes of NSC34 cells, stained with Hoechst 33258, were quantified using ImageJ software (NIH).
Caspase-3 activity assay.
NSC34 neurons were disrupted in cell lysis buffer and microcentrifuged (10,000 × g), and supernatants were evaluated for the activity of caspases that recognize the Asp-Glu-Val-Asp (DEVD) sequence using a colorimetric assay kit based on the detection of p-nitroaniline after cleavage from DEVD (Millipore Corporation). Absorbance was read spectrophotometrically at 405 nm. Human recombinant active caspase-3 (Millipore Corporation) was used as a positive control. Treatment of samples with caspase-3 inhibitor (Ac-DEVD-CHO) was the negative control.
siRNA knockdown of Dnmt1 and Dnmt3a in cell culture.
NSC34 cells were transiently transfected using Dharmacon siGENOME SMART Pool predesigned siRNAs (Thermo Fisher Scientific). Transfections using siRNAs were designed to target independently Dnmt1 and Dnmt3a. siRNA targeting β-amyloid cleaving enzyme-1 (BACE1) was used as an independent target negative control. A total of 5 nmol per well of the siGENOME was used with 4 μl of DharmaFECT-4 lipid reagent. The NSC34 cells were transfected for 4 h before removing the transfection medium and replacing it with cell culture medium. The cells were maintained for an additional 48 h before harvesting of total cell lysates for Western blotting.
Neuronal cell culture model of apoptosis using camptothecin.
To induce apoptosis in NSC34 cells, they were treated with the DNA topoisomerase-I inhibitor camptothecin (CPT) (Sigma). CPT induces DNA single-strand breaks and apoptosis robustly in mouse cortical neurons as described previously (Martin et al., 2009a). Because our previous experiments have shown that the molecular regulation of DNA damage-induced apoptosis can be different in cortical neurons at different stages of differentiation and synaptic maturation, we studied Dnmt-related changes during CPT-induced apoptosis in NSC34 cells at different states of in vitro maturation at DIV5 and DIV20. CPT was dissolved in distilled water supplemented with sodium hydroxide as described previously (Lesuisse and Martin, 2002) to give a final stock concentration of 10 mm. NSC34 cells were treated by replacing the culture medium with fresh medium containing 10 μm CPT or vehicle at DIV14–20. Cell lysates were harvested from vehicle and CPT-treated NSC34 cells exposed for 4, 8, 16, and 24 h.
Dnmt enzyme activity assay.
Total Dnmt enzymatic activity (maintenance and de novo) was measured in NSC34 cells and spinal cord extracts using a biochemical assay (Epigentek). In this ELISA-like assay, Dnmts transfer a methyl group to a cytosine-rich substrate DNA that is then detected by 5-methycytosine (5-mC) antibody. The enzymatic activity was determined by colorimetric measurement at 450 nm. Validation tests for this assay included pharmacologic inhibition of Dnmts with N-phthalyl-l-tryptophan (RG108) and procainamide, as well as genetic knockdown of Dnmts with siRNA.
Dnmt inhibition in CPT-treated NSC34 cells.
To determine whether Dnmt inhibition exerts neuroprotective consequences, established small molecule inhibitors of Dnmts were used. The inhibitors were RG108 and procainamide hydrochloride. Both drugs are cell permeable and direct inhibitors of Dnmts (Brueckner et al., 2005; Lee et al., 2005). RG108 (Calbiochem) was solubilized in DMSO at a stock concentration of 10 mm. Procainamide (Sigma) was dissolved in distilled DNase/RNase-free H20 at a stock concentration of 1 m. NSC34 cells were treated with high-glucose DMEM containing either 20 μm RG108 or 500 μm procainamide for 2.5 h. The medium containing RG108 and procainamide was removed and replaced with culture medium (50% DMEM, 50% optimem) containing 10 μm CPT for 24 h. The cells were fixed and stained with Hoechst 33258 to assess apoptotic cell number or they were used to prepare cell lysates for Western blotting.
After Dnmt knockdown by siRNAs was validated by Western blotting, Dnmt RNAi was assessed in a neuroprotection experimental design. NSC34 cells were transfected with Dnmt3a siRNA 24 h before treatment with 10 μm CPT. NSC34 cell lysates were harvested for Western blotting 24 h later.
Dnmt protein expression in developing and adult mouse CNS.
The temporal profiles of Dnmt1 and Dnmt3a protein expression were determined in immature and mature mouse brain and spinal cord. Wild-type C57BL/6J mice were used. Brains and spinal cords were harvested from mouse pups at postnatal day 0 (P0) (day of birth), P5, P10, P15, P20, P25, P30, and from 6- to 8-week-old adults. The mice were deeply anesthetized by hypothermia or overdosed with sodium pentobarbital and decapitated, and the brain and spinal cord were removed quickly and placed in ice-cold HBSS. Whole brains and spinal cords were used to prepare nuclear protein, soluble protein, and mitochondrial-enriched membrane protein fractions by subcellular fractionation using a confirmed protocol (Martin et al., 2003) and further validation by Western blotting for tissue fraction-specific proteins. For each time point, two to four mice were harvested. Both genders were used. With adult mouse brain and spinal cord, a Nycodenz gradient procedure also was used to isolate pure mitochondria for Western blot detection of Dnmt1 and Dnmt3a. The tissue was placed in homogenization buffer at a 1:5 (w/v) ratio, sonicated, and centrifuged at 1300 × g for 10 min. The pellet was resuspended in 0.5 volume of homogenization buffer and centrifuged at 1300 × g for 10 min. The supernatant was centrifuged at 17,000 × g for 15 min in an ultracentrifuge with a swinging bucket rotor. The supernatant was then removed, and the pellet was resuspended in 25% Nycodenz. The resuspended pellet was layered onto 2 ml of 30% Nycodenz, and 2 ml of 23% Nycodenz was layered on top of the 25% Nycodenz. The Nycodenz gradient was centrifuged at 52,000 × g for 1.5 h in an ultracentrifuge, and the pure mitochondria fraction was removed from between the 25 and 30% layer interface. The subcellular fractionation was confirmed by Western blotting with monoclonal antibodies against nuclear and mitochondrial proteins. The nuclear compartment was tracked by the presence of NeuN, and the mitochondrial compartment was tracked by the presence of complex V.
Mouse model of motor neuron apoptosis.
A unilateral sciatic nerve avulsion (SNA) served as the model for inducing apoptosis in motor neurons in vivo to determine whether Dnmts function in this cell death. The validation and reproducibility of this motor neuron degeneration model in mouse has been described previously (Martin and Liu, 2002; Martin et al., 2005). SNA lesions were done on adult wild-type male C57BL/6J mice and on B6.Cg-Tg (Hlxb9-gfp)1Tmj/j transgenic mice expressing EGFP driven by the mouse Hlxb9 (Hb9) promoter (Wichterle et al., 2002) originally obtained from The Jackson Laboratory. Adult Hb9-eGFP mice have subsets of spinal motor neurons highly expressing EGFP (Chang and Martin, 2011; Martin, 2011). SNA mice were used for biochemical and histological experiments. A cohort of wild-type mice with SNA lesions was killed at 1, 2, 3, 4, 5, or 6 d later (n = 4 mice per time) by anesthesia overdose and decapitation for harvesting fresh spinal cord samples. Samples of lumbar spinal cord ventral horn columns (ipsilateral and contralateral) were collected by careful microdissection under a microsurgical stereomicroscope as described previously (Martin et al., 2005). Samples from the different mice were pooled and fractionated into nuclear-enriched, mitochondrial-enriched, and soluble compartments as described previously (Martin et al., 2003). A cohort of Hb9-eGFP mice with SNA lesions was killed at 1, 2, 3, 4, 5, 6, 7, 10, and 14 d later (n = 4 mice per time) by anesthesia overdose and perfusion fixation with 4% paraformaldehyde for immunohistochemical studies of Dnmts and 5-methylcytosine in lumbar spinal cord. A cohort of wild-type mice was implanted with intracerebroventricular (ICV) cannulas and received the SNA. These mice were ICV treated with 5 or 10 μg of RG108 (in 2 μl of sterile saline) or with vehicle on day 0 (day of lesion) and on every other postlesion day over 3 weeks and were killed at postlesion day 21. Another group of RG108- or saline-treated mice was killed at 3, 4, and 7 d after lesion. The mice were killed by anesthesia overdose and perfusion fixation in 4% paraformaldehyde for spinal cord histology, immunohistochemistry, and neuroprotection analysis. The institutional Animal Care and Use Committee approved the animal protocols.
Western blotting.
The protein concentrations of cell lysates or brain and spinal cord extracts and subcellular fractions were determined using the Bio-Rad protein assay (Pierce Chemical). Proteins from cultured cell lysates and tissue extracts of brain and spinal cord fractions were subjected to SDS-PAGE (4–12%) and transferred to nitrocellulose membrane by electroelution as described previously (Martin, 1999). The reliability of sample loading and electroblotting in each blot was evaluated by staining nitrocellulose membranes with Ponceau S before immunoblotting. If transfer was not uniform, blots were discarded and gels were run again. Blots were blocked with 2.5% nonfat dry milk with 0.1% Tween 20 in 50 mm Tris-buffered saline, pH 7.4, and incubated overnight at 4°C with an antibody. The primary antibodies used were mouse monoclonal anti-Dnmt1 (clone 60B1220.1; Alexis Laboratories), rabbit polyclonal anti-Dnmt1 (1:500; Bethyl Laboratories), mouse monoclonal anti-Dnmt3A (clone 64B1446; Alexis Laboratories), rabbit polyclonal anti-Dnmt3a (1:1000; Abgent), mouse monoclonal anti-phospho-p53 (1:10,000; Cell Signaling Technology or R&D Systems), rabbit polyclonal anti-cleaved caspase-3, (1:500; Cell Signaling Technology), mouse monoclonal anti-NeuN (1:2000; Millipore Corporation), mouse monoclonal anti-complex V (1:10,000; Invitrogen), mouse monoclonal anti-MAP2 (Roche Diagnostics), or mouse monoclonal anti-α-actin (1:10,000; Millipore Corporation). The antibodies were used at concentrations for visualizing protein immunoreactivity within the linear range (Lok and Martin, 2002). After the primary antibody incubation, blots were washed and incubated with horseradish peroxidase-conjugated secondary antibody (0.2 μg/ml), developed with enhanced chemiluminescence (Pierce Chemical), and exposed to x-ray film or imaged using a ChemiDoc XRS Gel documentation system. Western blots were analyzed quantitatively by densitometry using Quantity One analysis software (Bio-Rad). Western blots were done in triplicate or greater, and the fold change was calculated by dividing the optical density of each time point by the loading control optical density. Protein loading controls were blots probed for α-actin, MAP2, or nitrocellulose membranes stained with Ponceau S. In some instances, the reprobing failed to give a reliable signal and the Ponceau S-stained membrane was used for normalization. In developmental, subcellular fraction, and lesion experiments, standard housekeeping proteins are unsuitable for protein loading because of developmental changes in cytoskeletal proteins, normal differences in cellular compartmentalization, and injury-induced changes in cytoskeletal proteins, respectively; thus, Ponceau S was used for normalization. Other studies have recognized some of the limitations of housekeeping proteins for references in Western blotting (Ferguson et al., 2005; Dittmer and Dittmer, 2006; Liu and Xu, 2006).
Immunohistochemistry.
After perfusion fixation, Hb9-eGFP and wild-type mouse spinal cords were removed from the vertebral column by dorsal laminectomy and cryoprotected in phosphate-buffered 20% glycerol. Dnmt1, Dnmt3a, and 5-methylcytosine were localized in lumbar spinal cord by a peroxidase–anti-peroxidase detection method using diaminobenzidine (DAB) as chromogen. The primary antibodies used (determined as appropriate for use in immunohistochemistry when Western blotting showed detection of monospecific bands or by nucleic acid digestion) were mouse monoclonal anti-Dnmt1 (1:1000, Alexis Laboratories), rabbit polyclonal anti-Dnmt3a (1:500; Abgent), or mouse monoclonal anti-5-methylcytosine (clone 162 33 D3, 1:1000; Calbiochem). After counterstaining with cresyl violet, the sections were viewed by bright-field microscopy and imaged using a Dxm 1200 digital camera (Nikon) and ACT-1 software.
Hb9-eGFP and wild-type mouse spinal cord sections were also used to detect immunofluorescence of Dnmt1 and Dnmt3a specifically in motor neurons using Alexa-594-conjugated secondary antibody (Invitrogen). Immunofluorescence was also used for colocalization of Dnmt3a with the mitochondrial marker cyclophilin D (Martin et al., 2009b), Dnmt3a with the synaptic marker synaptophysin (Calhoun et al., 1996), and 5-methylcytosine with the mitochondrial marker superoxide dismutase-2 (SOD2) (Martin et al., 2007; Chang and Martin, 2009) using combinations of primary antibodies generated in different species. AlexaFluor-488- and AlexaFluor-594-conjugated secondary antibodies (Invitrogen) were used for detection. After counterstaining with DAPI and coverslipping, the sections were viewed using a LSM 510 confocal microscope (Carl Zeiss Microimaging).
Motor neuron measurements.
Mouse lumbar spinal cord enlargements were dissected under a surgical microscope, and L4, L5, L6, and S1 segments were cryoprotected in 20% glycerol–PBS and frozen under pulverized dry ice. Transverse serial symmetrical sections (40 μm) through the spinal cord were cut using a sliding microtome and stored individually in 96-well plates. Sections were selected with a random start and systematically sampled (every ninth section) to generate a subsample of sections from each spinal cord that were mounted on glass slides and stained with cresyl violet for neuronal counting. Neuronal counts in the ipsilateral and contralateral ventral horns were made at 1000× magnification using the stereological optical dissector method as described previously (Calhoun et al., 1996; Martin et al., 1999). Motor neurons without apoptotic structural changes were counted using strict morphological criteria. These criteria included a round, open, pale nucleus (not condensed and darkly stained); globular Nissl staining of the cytoplasm; and a diameter of ∼30–45 μm. With these criteria, astrocytes, oligodendrocytes, and microglia were excluded from the counts. Nissl-stained sections of lumbar spinal cord sections were used to determine motor neuron cell body volume. Motor neurons were observed at 1000× magnification, and with a calibrated ocular micrometer, the major axis diameter (including the largest proximal dendrite) and the minor axis diameter were measured for 25–50 different motor neurons in each mouse. Cell body volume was calculated from the equation V = (π/6)ab2, where a is the diameter of the major axis and b is the diameter of the minor axis (Wong and Martin, 2010). Neuronal counts and volumes in the contralateral ventral horn always served as controls for the ipsilateral ventral horn. Neuronal counts and volumes were used to determine group means and variances, and comparisons among groups were analyzed using a one-way ANOVA and a Newman–Keuls post hoc test.
Human CNS tissues.
Postmortem human brain and spinal cord samples were derived from the Human Brain Resource Center at the Department of Pathology, Johns Hopkins University School of Medicine. Control (n = 12) and sporadic amyotrophic lateral sclerosis (ALS) cases (n = 12) were matched for age and postmortem delay (Martin, 1999, 2000; Shaikh and Martin, 2002). Snap-frozen samples of motor cortex and spinal cord were used for Western blotting for Dnmt1 and Dnmt3a after subcellular fractionation as described previously (Martin, 2000). Nuclear and mitochondrial enrichment of human CNS tissue fractions was confirmed by detection of NeuN and CyPD, respectively. Paraffin-embedded CNS samples were used for immunohistochemical localization of Dnmt1, Dnmt3a, and 5-methylcytosine using DAB as the chromogen and counterstaining with cresyl violet.
Results
Dnmt protein expression in neural cell lines
We screened several commercial antibodies to Dnmt isoforms for reliability in detecting monospecifically Dnmt1 and Dnmt3a in neural and non-neural cell lines (Fig. 1). For positive controls, mouse Dnmt1 and Dnmt3a genes were overexpressed in different cell lines by transient transfection, and cell lysates were used for Western blotting; also, purified E. coli-derived recombinant human Dnmt1 and Dnm3a (R&D Systems) were loaded onto gels. Negative control experiments were probing blots with a primary antibody preadsorbed against recombinant Dnmt1 and Dnmt3a protein and with a secondary antibody alone. Figure 1A shows the relative endogenous levels of putative Dnmt1 and Dnmt3a in cultured HEK293 cells, NSC34 cells, astrocytes, and microglia. The immunoreactive band identified as Dnmt1 had a molecular mass of ∼190 kDa, consistent with its predicted size (Inano et al., 2000). The immunoreactive band identified as Dnmt3a had a molecular mass of ∼100 kDa, consistent with its predicted size (Feng et al., 2005). Endogenous levels of Dnmt1 were much higher in HEK293 cells compared with neural cell lines (Fig. 1A), whereas the endogenous levels of Dnmt3a were similar among the different cell lines analyzed.
Figure 1.

Dnmt protein expression and enzyme activity in non-neural and neural cell lines. A, Immunoblot showing endogenous expression of Dnmt1 and Dnmt3a in cultured HEK293 cells and mouse spinal cord neuron cells (NSC34), mouse brain astrocytes, and mouse brain microglia. B, Negative control antibody preadsorption assay. NSC34 cells were harvested and lysed, and the protein lysates were used for the preadsorption test or nonpreadsorbed controls. Antibody to Dnmt1 or Dnmt3a diluted to 1:1000 (w/v) was incubated with Dnmt1 or Dnmt3a recombinant protein (1:10 and 1:100, ratios of antibody to recombinant protein). After incubation to form the antibody/protein complex, the samples were centrifuged, and the supernatant was collected and used to probe Western blot (20 μg of protein). Ponceau S staining was used to verify protein loading in each lane. C, Cultured HEK and mouse neural cells were transfected with Dnmt1 or Dnmt3a vectors to enforce expression or were mock transfected as a control. The transfected cells were harvested and lysed, and proteins were used for Western blotting. Ponceau S-stained membrane shows protein loading. D, E, RNAi knockdown of Dnmts. NSC34 cells were transiently transfected using siRNAs targeting independently Dnmt1 and Dnmt3a. The cultured cells were harvested 48 h later. siRNA-Dnmt cell lysates (20 μg of total protein) were used for immunoblotting for Dnmt1 and Dnmt3a. The graph displays the Dnmt1 and Dnmt3a immunoreactivity levels as percentage of control optical density (OD; mean ± SD) after siRNA-Dnmt1 and siRNA-Dnmt3a in NSC34 cells (asterisks indicate significant difference from control; see Results). Independent experiments were done in triplicate. F, Knockdown of Dnmt enzyme activity with RNAi. NSC34 whole-cell lysates after Dnmt1- and Dnmt3a-siRNA construct transfection were used to determine Dnmt enzyme activity biochemically. The total enzyme activities are represented as percentage of activity (mean ± SD) compared with control mock-transfected NSC34 cells (asterisks indicate significant difference, p < 0.01). Independent experiments were done twice.
The proteins assumed to be Dnmt1 and Dnmt3a were verified as such by adsorption of a primary antibody with human recombinant Dnmt1 and Dnmt3a (Fig. 1B), by transfection of cells with Dnmt1 and Dnmt3a expression plasmids (Fig. 1C), and by RNAi knockdown (Fig. 1D). Enforced expression of Dnmt1 and Dnmt3a above endogenous (mock-transfected) levels was demonstrated in HEK293 cells, NSC34 cells, astrocytes, and microglia (Fig. 1C). Transfection of siRNAs designed to target Dnmt1 and Dnmt3a independently confirmed antibody specificity by robustly reducing the proteins levels of Dnmt1 and Dnmt3a (Fig. 1D,E), but the knockdown was not completely isoform specific. The Western blot analysis for siRNA-Dnmt1 showed a significant (p = 0.005) decrease in Dnmt1 immunoreactivity (Fig. 1D,E). Dnmt3a immunoreactivity was also significantly (p = 0.04) decreased by siRNA-Dnmt1 (Fig. 1D,E). Western blot analysis of siRNA-Dnmt3a cells showed a highly significant (p = 0.0002) decrease in Dnmt3a immunoreactivity in NSC34 cells and also a significant (p = 0.001) decrease in Dnmt1 immunoreactivity (Fig. 1D,E). RNAi knockdown of Dnmt1 and Dnmt3A was also successful in causing a significant reduction in biochemical enzyme activity for Dnmt (Fig. 1F). The siRNA-Dnmt1 had a 28% decrease in activity in the NSC34 cells compared with control cells, and the siRNA-Dnmt3a had a 26% decrease in activity (Fig. 1F). These results demonstrate our capacity to accurately assess Dnmts at protein and enzyme activity levels.
Overexpression of Dnmt3a induces neurodegeneration
In the course of cell transfection experiments for antibody validation, we observed serendipitously that enforced expression of Dnmt3a, but not Dnmt1, had cytopathic effects. We then engineered mammalian expression constructs for Dnmt1- and Dnmt3a-GFP fusion proteins. NSC34 cells were transfected with the Dnmt-GFP constructs or a GFP expression plasmid as a control. Control cells expressing GFP showed a diffuse expression pattern throughout the entire cell (Fig. 2A). In Dnmt1-GFP transfectants, Dnmt1 was localized entirely to the cell nucleus and seen as discrete small foci (Fig. 2B). NSC34 cells transfected with Dnmt3a-GFP construct exhibited a markedly different pattern of localization from those transfected with Dnmt1. Dnmt3a-GFP was primarily cytoplasmic and was seen as discrete punctate structures (Fig. 2C). Moreover, the overexpression of Dnmt3a-GFP caused the neurons to degenerate (Fig. 2C). This degeneration was characterized by nuclear condensation as well as cellular fragmentation and blebbing that occurred over 48–72 h. Dnmt3a overexpression caused a 2.1-fold decrease in nuclear size, but enforced expression of Dnmt1 did not cause nuclear shrinkage or cell fragmentation (Fig. 2D). To confirm that the overexpressed Dnmts were catalytically active forms of enzyme, we measured biochemically total Dnmt activity in transfected NSC34 cell lysates. Total Dnmt enzyme activity was increased 2- and 1.6-fold in the Dnmt1- and Dnmt3a-transfected cells, respectively, compared with the mock-transfected controls (Fig. 2E). Because the neurodegeneration induced by enforced expression of Dnmt3a had some nuclear morphology features reminiscent of apoptosis, we assayed for caspase-3. NSC34 cells overexpressing Dnmt3a for 24 h, but not Dnmt1, showed significance increases in caspase-3 enzyme activity (Fig. 2F) and formation of the active small subunit as seen by Western blotting (Fig. 2G).
Figure 2.
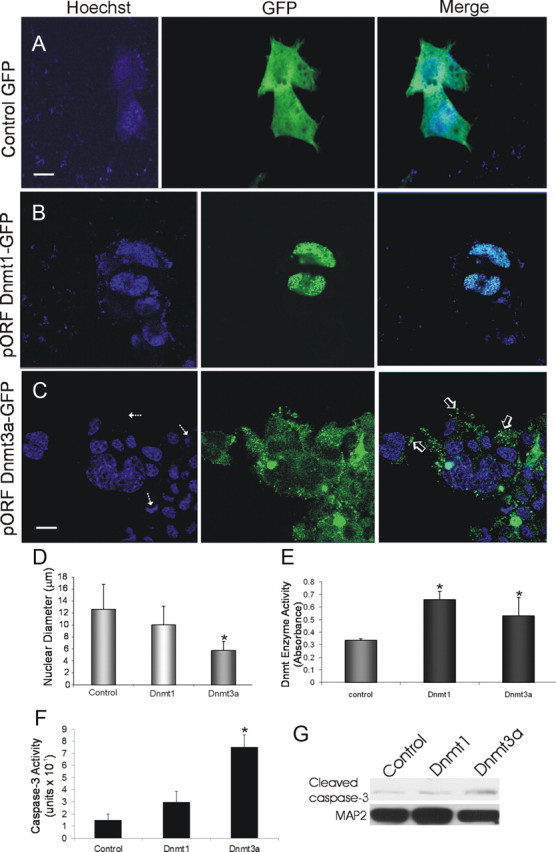
Transient transfection of NSC34 cells with Dnmt-GFP fusion protein expression constructs and induction of neurodegeneration. Cells were transfected, and 48 h later fixed and stained with the nuclear dye Hoechst 33258 (blue). A, Control transfection with GFP expression plasmid (2 μg) shows diffuse green fluorescence throughout NSC34 cells. Scale bar: A, B, 5 μm. B, Cells transfected with plasmid pORF9 Dnmt1-GFP (2 μg) show that Dnmt1 is localized selectively in the nucleus with a striking punctate labeling pattern. C, Cells transfected with plasmid pORF9 Dnmt3a-GFP (2 μg) exhibit nuclear and marked punctate cytoplasmic localizations (open arrows) of Dnmt3a and also show nuclear condensation and fragmentation (hatched arrows). Scale bar, 7 μm. D, Graph showing measurements of nuclear diameters (mean ± SD; asterisk indicates significant difference, p < 0.05) in transfected cells (n = 20 cells per group). E, Total Dnmt enzyme activity (mean ± SD of absorbance) in NSC34 cell lysates after Dnmt1 and Dnmt3a transfection. Asterisks indicate significant difference (p < 0.05) compared with control. F, Caspase-3 enzyme activity in NSC34 cells overexpressing Dmnt1 and Dnmt3a at 24 h after plasmid or mock transfection. Asterisk indicates significant difference (p < 0.05) compared with control. G, Western blot determination of cleaved capsase-3 formation in NSC34 cells overexpressing Dmnt1 and Dnmt3a at 24 h after plasmid or mock transfection. MAP2 immunoreactivity was used as a protein loading control.
Mutant catalytically inactive Dnmt3a is impotent at inducing cell degeneration
We engineered an expression construct containing the Dnmt3a gene with a deletion in the DNA sequence encompassing >93% of the catalytic domain of the encoded protein (Fig. 3A) to determine whether the cell degeneration is related specifically to the methytransferase activity of Dnmt3a. NSC34 cells transfected with the Dnmt3a-GFP deletion plasmid (Dnmt3aΔ-GFP) survived, differentiated neuron-like cells, and remained viable for beyond 16 d after transfection without evidence of nuclear condensation or cell fragmentation (Fig. 3B,C). Moreover, NSC34 cells transfected with mutant Dnmt3a did not show evidence for caspase-3 activation compared with cells transfected with active Dnmt3a, which showed robust accumulation of active caspase-3 subunit at 24 h after transfection (Fig. 3F,G).
Figure 3.
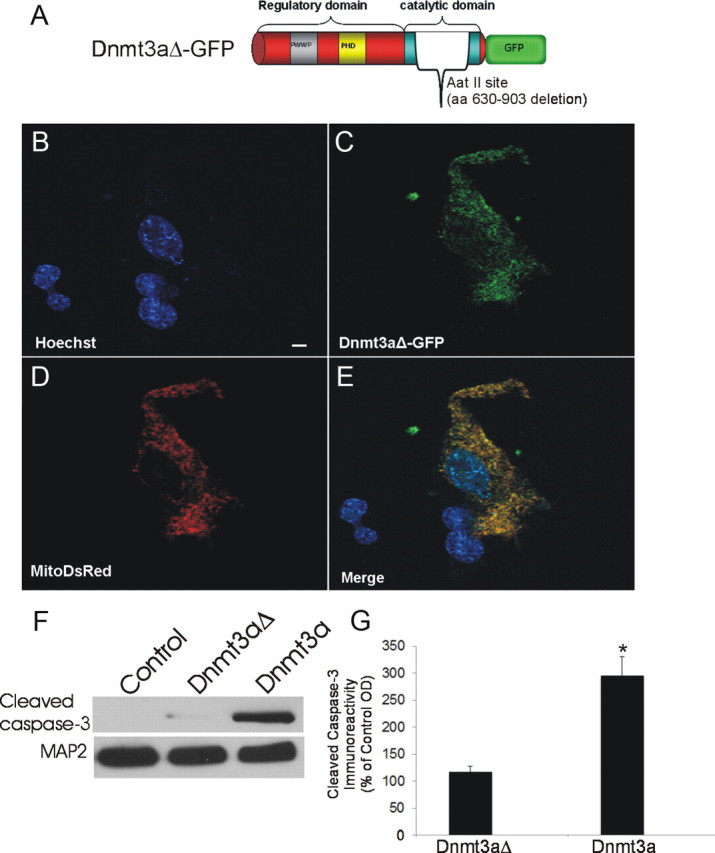
Mutant Dnmt3a-GFP localizes to mitochondria. A, Schematic diagram showing design of mutated Dnmt3a gene construct with a deleted catalytic domain. B–E, Cultured NSC34 cells were cotransfected with Dnmt3aΔ-GFP (C) and mitochondrial-targeted DsRed (D) plasmids and cultured for 48 h to 16 d. Fixed NSC-34 cells were stained with Hoechst 33258 (B). Dnmt3aΔ-GFP was localized to the nucleus and cytoplasm (C). E, Cytoplasmic Dnmt3aΔ-GFP showed near-complete localization to mitochondria (yellow). Scale bar (in B), 7 μm. F, NSC34 cells transfected with catalytic domain mutant Dnmt3a did not show accumulation of cleaved caspase-3 by Western blotting as did cells transfected with wild-type Dnmt3a. MAP2 immunoreactivity was used as a protein loading control. G, Western blot quantification of cleaved caspase-3 immunoreactivity in lysates of NSC34 cells transfected with catalytic domain mutant Dnmt3a (Dnmt3aΔ) and wild-type Dnmt3a compared with mock-transfected control cells. Values (percentage of control) are mean ± SD of three experiments. Asterisk indicates significant difference (p < 0.01) compared with control and Dnmt3aΔ cells.
Dnmt3a localizes to mitochondria in NSC34 cells
The survival of neurons overexpressing Dnmt3aΔ-GFP allowed for determination of the subcellular localization of Dnmt3a without the confounding effects of rapid degeneration. Dnmt3aΔ-GFP was observed within the nucleus where it was seen as small foci distributed within the nuclear matrix (Fig. 3B,C). Dnmt3aΔ-GFP was prominent in the cytoplasm where it was seen as punctuate structures resembling mitochondria in the cell body and processes (Fig. 3C). We used to transiently cotransfect NSC34 cells our Dnmt3a-GFP deletion plasmid and a plasmid expressing DsRed monomer fluorescent protein with a mitochondrial localization (SU9) sequence to determine whether Dnmt3a localizes to mitochondria. Cytoplasmic Dnmt3a showed considerable localization to mitochondria in NSC34 cells (Fig. 3C–E).
Motor neuron-like cell culture model of apoptosis
Most reports using NSC34 cells show scant or no evidence for their differentiation into cells resembling neurons (Tovar-y-Romo et al., 2009), although NSC34 cells are frequently referred to in the literature as a motor neuron-like cell model (Cashman et al., 1992); therefore, we evaluated the ability of NSC34 cells to differentiate into motor neuron-like cells. We compared NSC34 cells cultured for 5 d (DIV5) and cells cultured for 14 and 20 d (DIV14–20). During this differentiation process, there was an underlying cell-survival selection process as witnessed by an ∼50% reduction cell number (Fig. 4A). Morphological differentiation was determined by transfection with the GFP expression plasmid to visualize the entire cell and by immunostaining fixed NSC34 cells with dendritic and synaptic markers. DIV5 NSC34 cells were predominantly immature, and undifferentiated cells were seen as small (<10 μm) in diameter and mostly round or bipolar in shape (Fig. 4A). In contrast, many DIV20 NSC34 cells were large (>20 μm in diameter) and multipolar (Fig. 4A) as shown by GFP expression and SMI-32 monoclonal antibody (Fig. 4A), which recognizes a nonphosphorylated epitope of the neurofilament protein SMI-32 within neurons and is used to identify motor neurons in culture (Chang and Martin, 2011). Differentiated SMI-32-positive NSC34 cells had broad and long dendrites, with some spanning >50 μm (Fig. 4A), and were choline acetyltransferase positive (data not shown). Differentiated NSC34 cells also possessed elaborate dendritic arbors seen by MAP2 immunostaining that were decorated by numerous presynaptic terminals detected by synaptophysin immunostaining (Fig. 4B). Synaptophysin-positive presynaptic boutons were not detected in DIV5 NSC34 cell cultures (data not shown).
Figure 4.
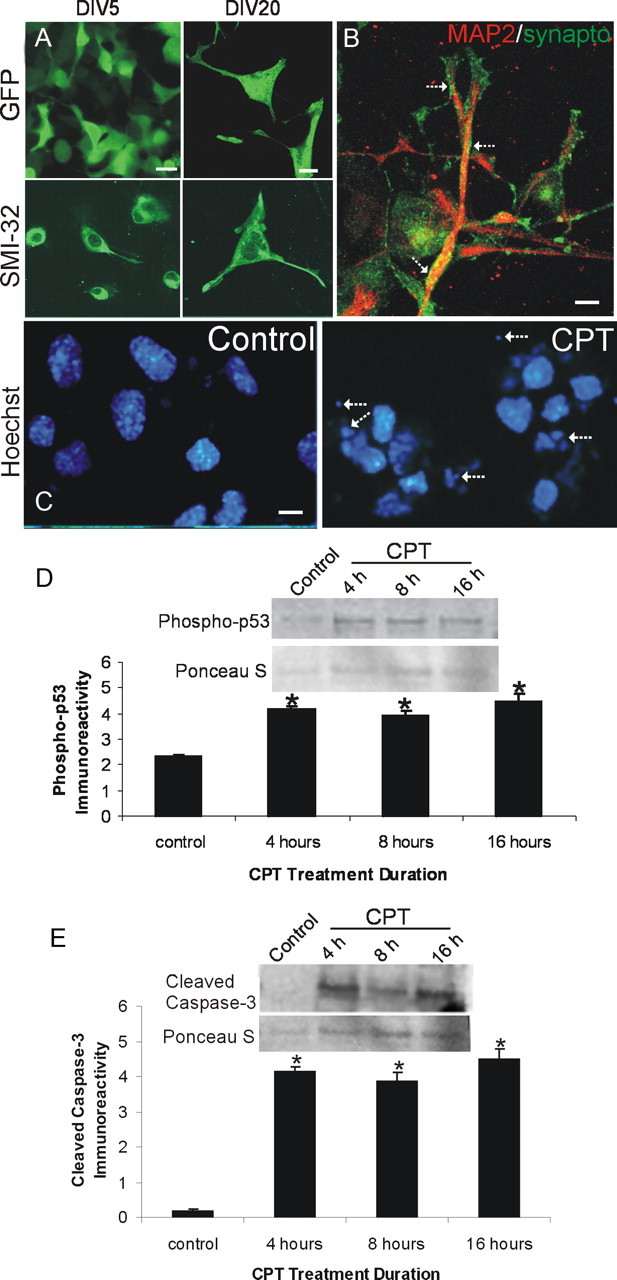
NSC34 neuron morphological characterization and camptothecin-induced apoptosis. A, Morphological characterization of the differentiation of cultured NSC34 cells. NSC34 cells were cultured for 5 d (DIV5) and differentiated over 20 d (DIV20). Living cells at DIV5 and DIV20 were transfected with GFP expression plasmid or were fixed and used for immunofluorescence for neurofilament protein with SMI-32 antibody. Immature DIV5 NSC34 cells are small and bipolar as seen by GFP expression and neurofilament staining. In contrast, differentiated DIV20 NSC34 cells have features of motor neurons with large multipolar cell bodies and broad dendrites. Over a 20 d culture, the original plating density is reduced ∼50%. Scale bar, 10 μm. B, Differentiated NSC34 neurons form elaborate dendrites (red, MAP2) contacted by numerous synapses (hatched arrows, green, synaptophysin). Scale bar, 5 μm. C–E, DIV20 NSC34 cells treated with 10 μm CPT, a DNA topoisomerase I inhibitor that induces DNA single-strand breaks (Martin et al., 2009b), undergo robust apoptosis. C, Hoechst 33258 staining shows that nuclei of NSC34 cells treated with CPT undergo apoptosis and show nuclear condensation, shrinkage, and fragmentation (hatched arrows). The same batch of DIV20 cultures treated with vehicle (control) has normal large nuclei. Scale bar, 10 μm. D, E, Whole-cell lysates of DIV20 NSC34 cells treated with 10 μm CPT show increased immunoreactivity for activated phospho-p53 (D) and cleaved-caspase 3 (E) at 4, 8, and 16 h of CPT treatment compared with vehicle-treated control cells. Values in graphs are mean ± SEM of optical density units. Asterisks denote a significant difference from control for phospho-53 (p < 0.01) and cleaved caspase-3 (p < 0.001). Representative Western blots are shown along with Ponceau S-stained membranes to show protein loading. The results were replicated in at least three separate cell culture experiments.
We wanted to study Dnmt expression patterns in a setting where immature, less differentiated, and differentiated NSC34 cells were induced to undergo apoptosis by treatment with the DNA-damaging agent CPT. CPT treatment of cultured mouse primary cortical neurons induces DNA single-strand break accumulation and robust p53- and caspase-dependent apoptosis (Martin et al., 2009a). We determined that CPT treatment of NSC34 cells is a valid cell culture model of spinal cord neuron apoptosis. CPT induced robust apoptosis in NSC34 cells, as shown morphologically with Hoechst 33258-stained nuclei (Fig. 4C) and biochemically by Western blot analysis showing increased levels of phospho-p53 (Fig. 4D) and cleaved-caspase 3 (Fig. 4E).
Dnmt expression in apoptotic NSC34 cells
To identify possible relationships between Dnmts and apoptosis, we measured Dnmt protein expression patterns in NSC34 cells undergoing apoptosis. The cells were evaluated at DIV5 and DIV20, because cell death signaling mechanisms in mouse cortical neurons seem to vary depending on stage of maturation (Martin et al., 2009a). The levels of Dnmt1and Dnmt3a in CPT-treated undifferentiated DIV5 NSC34 cells remained stable during the progression of apoptosis (Fig. 5A,B). In contrast, a significant threefold increase in Dnmt1 levels over control values occurred in differentiated DIV20 NSC34 cells (p < 0.05) at 4 h after CPT treatment (Fig. 5C). Subsequently, Dnmt1 levels were increased by 5.3-, 5.9-, and 5.4-fold at 8, 16, and 24 h, respectively, after CPT treatment (Fig. 5C). Dntm3a levels were significantly increased (p < 0.05) in DIV20 NSC34 cells, with 2.1-, 1.8-, 2.1-, and 2.1-fold increases being observed at 4, 8, 16 and 24 h, respectively, after CPT treatment (Fig. 5D).
Figure 5.

Dnmts can mediate DNA damage-induced apoptosis of differentiated neurons. A, B, Dnmt1 and Dnmt3a protein levels do not change significantly in DIV5 NSC34 cells exposed to 10 μm CPT for 4–24 h. Representative Western blots are shown along with membranes reprobed for actin to show protein loading. Immunoreactivity optical densities for DNMT1 and Dnmt3a were normalized to actin immunoreactivity. Values in graphs are mean ± SEM of optical density units. C, D, Dnmt1 and Dnmt3a protein levels increase in DIV20 NSC34 cells exposed to 10 μm CPT for 4–24 h. Values in graphs are mean ± SEM of optical density units. Asterisks denote a significant difference from control for Dnmt1 (p < 0.01) and Dnmt3a (p < 0.05). Representative Western blots are shown along with membranes reprobed for actin to show protein loading. E, Dnmt inhibitors protect against CPT-induced apoptosis. DIV20 NSC34 cells were treated with either 20 μm RG108 or 0.5 mm procainamide for 2.5 h in optimem medium. The medium with inhibitors was removed, and the cells were treated with 10 μm CPT for 24 h. The cells were fixed and stained with Hoechst 33258. The percentages of apoptotic cells were determined by counting total cell nuclei (n = 1000 cells per condition) and the proportion of apoptotic nuclei. Values in graphs are mean ± SEM. Double asterisks denote a significant difference from control for CPT (p < 0.001). There is a significant difference from CPT-treated cells for RG108 (++p < 0.01) and procainamide (+p < 0.05). F, RNAi for Dnmt3a protects against CPT-induced apoptosis. DIV14 NSC34 cells were transfected with siRNA construct targeting Dnmt3a or BACE1 (as an independent target), and 24 h later, the cells were treated with 10 μm CPT for 24 h. Cells were prepared as in E. Values in graphs are mean ± SEM. ++Significant difference from control for CPT (p < 0.001); +Significant difference from CPT-treated cells for siRNAi cells (p < 0.01). BACE1 knockdown (control siRNA + CPT) did not protect. G, Western blot analysis of phospho-p53Ser15 in NSC34 cells treated with RG108 (20 μm) and Dnmt3a-siRNA and challenged with 10 μm CPT for 24 h. RG108 dramatically blocked the CPT-induced activation of p53. Dnmt3a-siRNA attenuated the activation of p53 in CPT-treated cells.
Dnmt drug inhibitors and RNAi protect against apoptosis and cytosine methylation in cultured neurons
To further assess the role of Dnmts in the direct mechanisms of neuronal apoptosis, we used drug inhibitors. RG108 is an isoform nonspecific DNA methyltransferase inhibitor (Brueckner et al., 2005), whereas procainamide is a Dnmt1-specific inhibitor (Lee et al., 2005). NSC34 cells were cultured and treated with either RG108 or procainamide before apoptosis induction by CPT. Both of the inhibitors decreased significantly the amount of apoptosis in differentiated NSC34 neurons (Fig. 5E). Procainamide protected ∼25% of the cells, whereas RG108 protected ∼60% of the cells (Fig. 5E). RNAi using Dnmt3a-siRNA, which mediates a more robust knockdown of Dnmt protein compared with Dnmt1-siRNA (Fig. 1D), also decreased significantly the amount of NSC34 cell apoptosis induced by CPT (Fig. 5F). siRNA knockdown of an independent target (BACE1) did not affect the levels of CPT-induced apoptosis (Fig. 5F). The protection afforded by Dnmt3a-siRNA was confirmed at a biochemical level by Western blotting revealing that RG108 potently abolished the CPT-induced activation of p53 (Fig. 5G) and caspase-3 cleavage (data no shown). Dnmt3a-siRNA also attenuated p53 phosphorylation in CPT-treated NSC34 cells (Fig. 5G).
We examined the cellular localizations of Dnmt3a and a direct marker of DNA methylation (i.e., 5-methylcytosine) in CPT-treated NSC34 cells. By immunofluorescence, endogenous mouse Dnmt3a in control NSC34 cells is localized in the cytoplasm as a reticulum and discrete puncta (Fig. 6A), consistent with the patterns seen in human Dnmt3a-transfected NSC34 cells (Figs. 2C, 3C), and basal levels appeared low in the nucleus (Fig. 6A). Similarly, 5-methylcytosine immunoreactivity is detectable, but low, in the nucleus of control NSC34 cells (Fig. 6C). In contrast, CPT-treated NSC34 cells exhibited intense cytoplasmic immunoreactivity and definite nuclear staining for endogenous Dnmt3a (Fig. 6B,D) and for 5-methylcytosine (Fig. 6D). 5-Methylcytosine immunoreactivity accumulated diffusely in the nuclear matrix and as prominent masses in the nucleus (Fig. 6D). RG108-treated cells markedly attenuated the immunostaining for Dnmt3a and 5-methylcytosine in CPT-treated NSC34 cells (Fig. 6E).
Figure 6.

Cellular localization of endogenous Dnmt3a and 5-methylcytosine in NSC34 cells treated with CPT. A, Immunofluorescence for Dnmt3a in control DIV20 NSC34 cells shows its localization mostly in the cytoplasm, some of which are seen as discrete puncta, with low detection in the nucleus (asterisks). B, In DIV20 NSC34 cells treated with 10 μm CPT, Dnmt3a immunoreactivity is highly enriched in the cytoplasm and processes and is also present robustly in the nucleus (asterisk). C–E, Immunofluorescent colocalization of Dnmt3a (green) and 5-mC (red) in control (C) and CPT-treated DIV14 NSC34 (D, E) cells. C, Control cells have a primarily cytoplasmic perinuclear localization of Dnmt3a (green) and very low 5-mC (red) limited to a few faint specks in the nuclei, some of which are also faintly yellow because of colocalization with 5-mC. D, In contrast, CPT-treated cells have very high levels of immunoreactivity for both Dnmt3 and 5-mC as witnessed by the intense yellow indicating colocalization. E, RG108 treatment attenuated markedly the immunoreactivities of both Dnmt3a and 5-mC in CPT-treated cells as indicated by the dissipation of the yellow signal. Scale bar (in A), 6 μm.
Endogenous Dnmt expression in developing mouse brain and spinal cord
To have a better understanding of DNA methylation enzymes in the intact CNS, we examined, by immunoblotting, the endogenous expression levels of Dnmt1 and Dnmt3a in the brain and spinal cord of mice. We determined whether Dnmt expression is developmentally regulated.
Western blotting of nuclear-enriched subcellular fractions from brain showed that there were no major changes in Dnmt1 protein levels in the developing mouse brain from the day of birth (P0) to P30 (Fig. 7A). In contrast, Dnmt3a protein levels increased progressively in the developing mouse brain from P0 to P30 (Fig. 7A). There were 2.76- and 3.38-fold increases from P0 to P10 and P0 to P15, respectively; the highest expression was reached at P15 (Fig. 7A).
Figure 7.

Developmental expression, regional distribution, and subcellular localization of Dnmt1 and Dnmt3a in mouse CNS. A, Dnmt1 protein levels are very stable during normal postnatal development of mouse brain, whereas Dnmt3a levels increase. Mouse whole brains at P0 (day of birth) through P30 and into adulthood (8 weeks) were homogenized and used for immunoblotting for Dnmt1 and Dnmt3a. Membranes were stained with Ponceau S to show protein loading. Values in graphs are mean ± SEM of optical density units. Asterisks denote a significant difference from P0 for Dnmt3a (p < 0.05). B, Dnmt1 protein levels decrease during postnatal development of mouse spinal cord, whereas Dnmt3a levels increase. Mouse whole spinal cords at P0 (day of birth) through P20 were homogenized and used for immunoblotting for Dnmt1 and Dnmt3a. Membranes were stained with Ponceau S to show protein loading. Values in graphs are mean ± SEM of optical density units. Asterisks denote a significant reduction (p < 0.05) from P0 for Dnmt1 and a significant increase (p < 0.05) from P0 for Dnmt3a. C, Dnmt1 protein levels are uniform within different regions of adult mouse brain, whereas Dnmt3a levels show regional differences. Adult mouse brains were carefully microdissected into different regions that were homogenized and used for immunoblotting for Dnmt1 and Dnmt3a. Membranes were stained with Ponceau S to show protein loading. Values in graphs are mean ± SEM of optical density units. D, Dnmt1 is found primarily in the nucleus and cytosol, whereas Dnmt3a is found in the nucleus, cytosol, and mitochondria. Adult mouse whole brains and whole spinal cords were homogenized; fractionated into nuclear (Nuc), cytosol (Cyto), and pure mitochondria (Mito) compartments; and used for immunoblotting for Dnmt1 and Dnmt3a. In both brain and spinal cord, the nuclear enrichment was confirmed by NeuN immunoreactivity, and mitochondrial enrichment was confirmed by mitochondrial complex V immunoreactivity. Ponceau S-stained nitrocellulose membranes show protein loading. Values in graphs are mean ± SEM of optical density units.
Nuclear-enriched fractions of spinal cord were studied at P0–P20. Dnmt1 exhibited a twofold decrease from P5 to P10, and this decrease persisted throughout P15 and P20 (Fig. 7B). Interestingly, Dnmt3a levels had an inverse relationship to Dnmt1 with a twofold increase from P0 to P15, before a slight reduction between P15 and P20 (Fig. 7B).
Endogenous Dnmt expression in adult mouse brain
The levels of Dnmts were examined in different regions of the adult mouse brain. Dnmt1 protein levels were similar across many adult brain regions (Fig. 7C). In contrast, levels for Dnmt3a showed brain-regional differences. Dnmt3a protein level was lowest in olfactory bulb and about threefold higher in diencephalon and brainstem (Fig. 7C). Cerebral cortex, hippocampus, striatum, and cerebellum had comparable intermediate levels of Dnmt3a protein (Fig. 7C).
Endogenous Dnmt expression in adult brain and spinal cord subcellular fractions
Because cell culture transfection experiments demonstrated targeting of Dnmt3a to mitochondria (Fig. 3B–E), we examined brain and spinal cord subcellular fractions for Dnmt1 and Dnmt3a expression. Brain and spinal cord from C57/BL6 mice were fractionated in nuclear-enriched (P1), cytosol (S1), and pure mitochondria. The nuclear-enriched fraction of brain and spinal cord contained high levels of NeuN, whereas NeuN was not detected in the mitochondrial fractions (Fig. 7D). The mitochondrial fraction of brain and spinal cord contained high levels of complex V, whereas complex V was not detected in the nuclear fraction (Fig. 7D). Dnmt1 was present in the nuclear and cytosolic fractions in the brain and spinal cord (Fig. 7D); however, it was undetectable in the mitochondrial fraction (Fig. 7D). Dnmt3a was present in not only the nuclear and cytosolic fractions but also in the pure mitochondrial fractions of both the brain and spinal cord (Fig. 7D). Compared with Dnmt1, Dnmt3a levels were 3.62- and 3.66-fold higher in the brain nuclear and cytosolic fractions and 2.14- and 2.5-fold higher in the spinal cord, respectively (Fig. 7D).
Endogenous Dnmts are upregulated during apoptosis of motor neurons in vivo
To examine the potential pro-apoptotic role of Dnmts in adult mouse spinal cord, we used a model of SNA that induces robust apoptosis of spinal motor neurons (Martin and Liu, 2002; Martin et al., 2005). Unilateral lesions were made, and lumbar spinal cord microsamples were assayed for Dnmt protein and enzyme activity. By Western blotting, Dnmt1 levels were unchanged at 3 d after lesion but were elevated in both ipsilateral and contralateral sides at 4 and 5 d after lesion (Fig. 8A). In contrast, Dnmt3a was markedly elevated (fourfold) on the lesioned side at 3 d (Fig. 8A). At 4 d, the ipsilateral Dnmt3a elevation was less prominent. At 5 d, Dnmt3a levels were similar in ipislateral and contralateral sides (Fig. 8A). Biochemical assay for total Dnmt enzyme activity revealed significant increases ipsilaterally at 3, 4, and 5 d (Fig. 8B).
Figure 8.
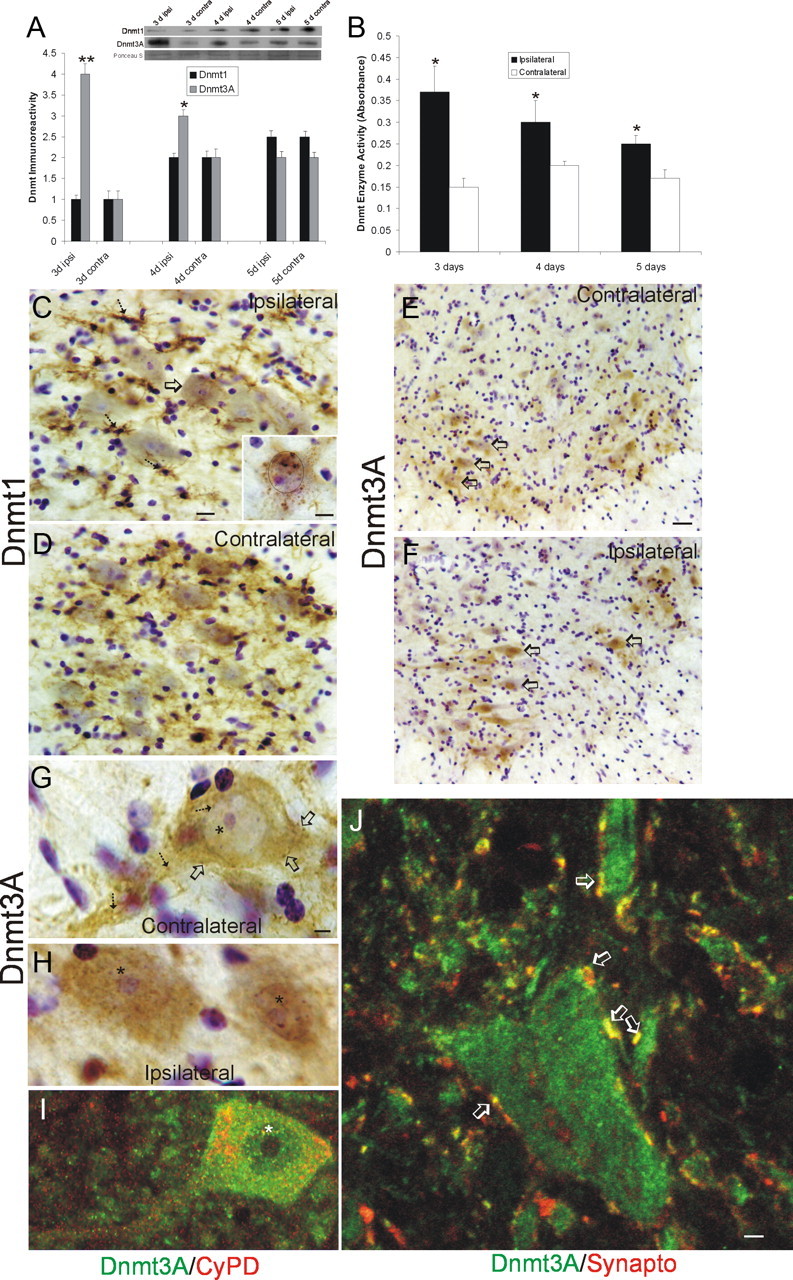
Dnmts are upregulated in adult mouse spinal cord during motor neuron apoptosis. A, Dnmt3a protein levels in spinal cord increase ipsilaterally after SNA. Adult mice received unilateral SNA, and 3, 4, and 5 d later (n = 4 per time), the lumbar spinal cords were microdissected into the nonlesioned contralateral control ventral horn (contra) and the lesioned ipsilateral ventral horn (ipsi). Crude homogenates were used for immunoblotting for Dnmt1 and Dnmt3a. Membranes were stained with Ponceau S to show protein loading. Values in graphs are mean ± SEM of optical density units. Asterisks denote a significant difference from contralateral for Dnmt3a at 3 d (p < 0.001) and 4 d (p < 0.05). See Figure 7B for basal levels of Dnmt1 and Dnmt3a in mature (P20) whole mouse spinal cord. B, Dnmt enzyme activity in spinal cord increases ipsilaterally after SNA. Adult mice received unilateral SNA, and 3, 4, and 5 d later (n = 4 per time), the lumbar spinal cords were microdissected into the nonlesioned contralateral control ventral horns (contra) and the lesioned ipsilateral ventral horns (ipsi). Crude homogenates were used for biochemical assessment of total Dnmt enzyme activity. Values in graphs are mean ± SD. Asterisks denote a significant difference from contralateral at 3 and 4 d (p < 0.01) and at 5 d (p < 0.05). C, D, Immunohistochemical localization of Dnmt1 in mouse lumbar spinal cord at 4 d after SNA. Dnmt1 immunoreactivity was visualized with diaminobenzedine (brown), and sections were counterstained with cresyl violet. C, The ipsilateral ventral horn showed Dnmt1 immunoreactivity within the cytoplasm and nucleus of motor neurons (arrow and inset) and in small cells resembling microglia (hatched arrows). Some motor neurons with chromatin condensation typical of apoptosis (as seen by cresyl violet counterstaining) displayed aggregates of Dnmt1 immunoreactivity in the nucleus and perinuclear region (inset; black circle delineates the nucleus). Scale bars: C (same for D), 20 μm; C, inset, 10 μm. D, In contrast, the contralateral ventral horn showed Dnmt1 immunoreactivity diffusely in the neuropil. E–H, Immunohistochemical localization of Dnmt3a in mouse lumbar spinal cord at 3 d after SNA as seen at low (E, F) and high (G, H) magnifications. Dnmt3a immunoreactivity was visualized with diaminobenzedine (brown), and sections were counterstained with cresyl violet. E, The contralateral control ventral horn showed Dnmt3a immunoreactivity within and around motor neurons (open arrows), but the nuclei of most motor neurons have low immunoreactivity in the nucleus. G, As seen at high resolution, Dnmt3a in normal motor neurons was seen faintly as diffuse immunoreactivity in the nucleus (asterisk) and in the cytoplasm as discrete puncta (hatched arrows). Pericellular bouton-like Dnmt3a immunoreactivity appeared in apposition to motor neurons (open arrows). F, H, In contrast, most ipsilateral motor neurons (F, open arrows) showed prominent Dnmt3a immunoreactivity in the nucleus (H, asterisks). Scale bars: in E (same for F), 20 μm; in G (same for H, I), 5 μm I, Confocal microscopy shows that some cytoplasmic Dnmt3a immunoreactivity in normal motor neurons is localized to mitochondria as demonstrated by its colocalization with the mitochondrial marker cyclophilin D (CyPD). Dnmt3a immunoreactivity is seen as green, and CyPD immunoreactivity is seen as red. Some cytoplasmic Dnmt3a immunoreactivity associates with mitochondria (seen as yellow), but single signals for Dnmt3a and CyPD can also be seen. Dnmt3a immunoreactivity is also seen in the nucleus (asterisk). J, Confocal microscopy shows that some Dnmt3a immunoreactivity is localized to presynaptic terminals as demonstrated by its colocalization with the presynaptic terminal marker synaptophysin. Dnmt3a immunoreactivity is seen as green, and synpatophysin (Synapto) immunoreactivity is seen as red. Perineuronal and peridendritic Dnmt3a is localized to axon terminals (hatched arrows, seen a yellow). Scale bar, 10 μm.
Immunohistochemistry was used to study the cellular localizations of Dnmts in lumbar spinal cord motor neurons after SNA. At 5–14 d after lesion, Dnmt1 immunoreactivity was present ipsilaterally in numerous small ramified cells resembling microglia (Fig. 8C, hatched arrows) and in subsets of motor neurons (Fig. 8C, open arrow), whereas, contralaterally, Dnmt1 was seen diffusely in the neuropil but was generally unapparent in motor neurons (Fig. 8D). Ipsilaterally, Dnmt1 was found enriched in the nucleus and cytoplasm of motor neurons with chromatin condensation suggestive of apoptosis (Fig. 8C, inset). Dnmt3a was present in contralateral motor neurons (Fig. 8E, open arrows), where it was seen as discrete particles in the cytoplasm (Fig. 8G, hatched arrows), faint and particulate nuclear matrix labeling (Fig. 8G, asterisk), and as bouton-like structures near the plasma membrane (Fig. 8G, open arrows). Subsets of ipsilateral motor neurons (Fig. 8F, open arrows) showed a robust presence of Dnmt3a immunoreactivity in the nucleus (Fig. 8H, asterisks). To confirm that the Dnmt changes were occurring in spinal motor neurons rather than other spinal neurons that can be large or in non-neuronal cells, SNA lesions were done on adult transgenic mice expressing EGFP driven by the Hb9 promoter (Wichterle et al., 2002), which have EGFP expressed in subsets of motor neurons at high levels (Chang and Martin, 2011; Martin, 2011). Immunofluorescence for Dnmt1 confirmed the upregulation in the cytoplasm and nucleus of ipsilateral EGFP-positive motor neurons (Fig. 9A) compared with contralateral motor neurons after SNA. Similarly, immunofluorescence for Dnmt3A suggested upregulation of Dnmt3A in the cytoplasm and nucleus of ipsilateral EGFP-positive motor neurons (Fig. 9B, open arrow and hatched arrow) compared to contralateral motor neurons after SNA. Moreover, Dnmt3A immunoreactivity was strongly upregulated in dendrites of ipsilateral motor neurons (Fig. 9B, double arrow).
Figure 9.
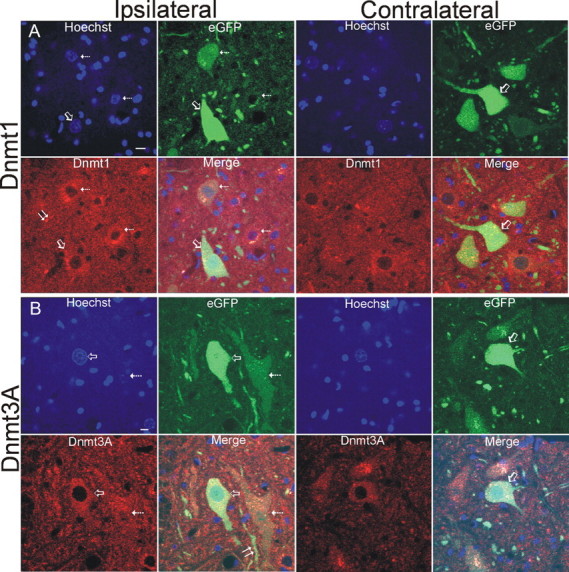
SNA lesions in transgenic mice expressing eGFP selectively in motor neurons. Hb9-eGFP transgenic mice confirm the expression of Dnmt1 (A) and Dnmt3a (B) in adult motor neurons and their upregulation in ipsilateral motor neurons after SNA compared with contralateral nonlesioned motor neurons, and they potentially serve as a reported model for DNA methylation silencing of gene expression during apoptosis. Motor neurons are labeled genetically with eGFP. Dnmt1 or Dnmt3a immunoreactivities are seen as red, and nuclear labeling is blue (Hoechst). Some ipsilateral lumbar motor neurons can be found with a high eGFP signal (A, B, open arrows), whereas in other motor neurons, the eGFP signal is attenuated or nearly abolished (A, B, hatched arrows). Motor neurons with greater Dnmt1 or Dnmt3a signal generally have a lower eGFP signal (hatched arrows). Dnmt1 immunoreactivity was also seen associated with small cells (A, double arrow). Dnmt3a immunoreactivity was also upregulated in dendrites of motor neurons (B, double arrow). Scale bars, 7 μm.
Dnmt3a is localized to spinal cord synapses and mitochondria in vivo
Immunohistochemical assessments revealed that Dnmt3a might be located normally in some axosomatic synapses (Fig. 8G). Immunofluorescence for Dnmt3a and the synaptic marker synpatophysin demonstrated that Dnmt3a is localized to synapses in mouse spinal cord (Fig. 8J). Many neurons were found to be decorated by perineuronal synapses positive for Dnmt3a (Fig. 8J, open arrows). Cell culture-transfection experiments (Fig. 3) and nervous tissue fractionation experiments (Fig. 7D) revealed that Dnmt3a is present in mitochondria. Immunofluorescence for Dnmt3a and the mitochondrial marker cyclophilin D (Martin et al., 2009b) demonstrated that Dnmt3a is localized to mitochondria in mouse spinal cord motor neurons (Fig. 8I).
DNA methylation is increased rapidly in motor neurons during apoptosis in vivo
Changes in Dnmts relate indirectly to DNA methylation. As a readout for DNA methylation directly in motor neurons during apoptosis, 5-mC was detected using an antibody. Antibody-based detection of 5-mC has been used before in studies of DNA methylation (Kang et al., 2006; Zhang et al., 2006). In nonlesioned motor neurons, 5-mC immunoreactivity was observed as numerous small puncta within the cytoplasm and as delicate granules or strands in the nucleus (Fig. 10A). In motor neurons destined to undergo apoptosis, obvious changes in the patterns of 5-mC were seen as early as 2 d after SNA (Fig. 10B), and increases in the levels of immunoreactivity were detected as soon as 3 d after injury (Fig. 10O). The early-occurring changes in the patterns of 5-mC staining in lesioned motor neurons included accumulation of particulate immunoreactivity in the cell body, proximal dendrites, and nucleus (Fig. 10B). To validate that these changes were indeed related to DNA modification, sections were treated with DNase or RNase before immunostaining with 5-mC antibody. DNase treatment completely abolished the 5-MeC immunoreactivity (Fig. 10C), but digestion with RNase did not change the staining pattern (Fig. 10D). Evaluation of motor neurons in SNA mice at 5 d after lesion and thereafter revealed pronounced changes in 5-mC. As motor neurons underwent somatodendritic attrition, the entire nucleus became strongly positive for 5-mC and the cytoplasm showed numerous large 5-mC-positive aggregates (Fig. 10E). Progressive cellular attrition and loss of processes was associated with the presence of 5-mC-positive apoptotic body-like fragments in the neuropil (Fig. 10F). In advanced apoptotic attrition, the nucleus disintegrated into 5-mC-positive fragments (Fig. 10G).
Figure 10.
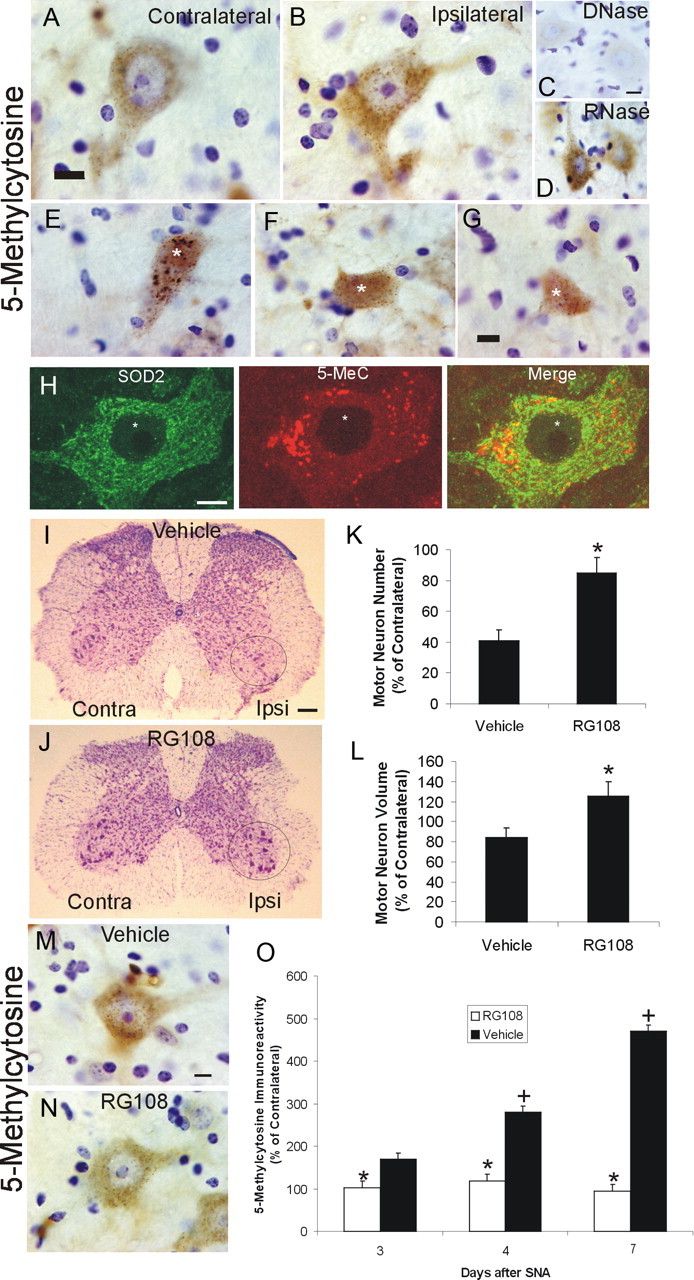
DNA methylation increases in adult mouse motor neurons undergoing apoptosis, and Dnmt inhibition blocks the DNA methylation and the apoptosis. A, B, Immunohistochemical localization of 5-mC in nonlesioned contralateral (A) and SNA-lesioned (B) lumbar spinal cord motor neurons. 5-mC immunoreactivity was visualized with diaminobenzedine (brown), and sections were counterstained with cresyl violet. In control motor neurons (A), 5-mC immunoreactivity was present in the cytoplasm as puncta and in the nucleus as puncta and finely beaded strands. In motor neurons at 4 d after SNA (B), 5-mC immunoreactivity was enriched in the cytoplasm and nucleus. Scale bar (same for B), 7 μm. C, DNase digestion of SNA spinal cord sections abolished completely the 5-mC immunoreactivity in motor neurons. Scale bar (same for D), 10 μm. D, D. RNase digestion of SNA spinal cord sections did not alter the 5-mC immunoreactivity in motor neurons. E–G, Adult motor neurons at different morphological stages of the apoptotic process (Martin et al., 1999; Martin and Liu, 2002) show marked changes in 5-mC immunoreactivity. E, In the chromatolytic stage, the nucleus becomes completely positive for 5-mC immunoreactivity (asterisk) and large aggregates of 5-mC accumulate in the cytoplasm. F, In the somatodendritic attrition stage, characterized by cell-body shrinkage and loss of processes, the nucleus remains strongly positive for 5-mC (asterisk), and 5-mC immunoreactive debris is found in the neuropil (arrow). G, At near end stage, characterized by a residual cell body without processes, the 5-mC immunoreactivity dissipates, and the nucleus is seen as fragments of 5-MeC immunoreactivity (asterisk). H, Confocal microscopy shows that some cytoplasmic 5-mC immunoreactivity in nonlesioned motor neurons is localized to mitochondria, as demonstrated by its colocalization with the mitochondrial marker SOD2. 5-mC immunoreactivity is seen as red, and SOD2 immunoreactivity is seen as green. Much of the cytoplasmic Dnmt3a immunoreactivity associates with mitochondria (seen as yellow), but single signals for 5-mC and SOD2 can also be seen. Scale bar, 10 μm. I, J, Dnmt inhibitor RG108 completely blocks the degeneration of adult motor neurons after SNA. Mice were treated intracerebroventricularly with 10 μg of RG108 (in 2 μl of sterile saline) or with saline (vehicle) on day 0 (day of lesion) and on every other postlesion day over 3 weeks and were killed at 21 d after SNA. I, Vehicle-treated mice show loss of motor neurons in the ventral horn ipsilateral to the lesion. J, RG108-treated mice show no loss of motor neurons after SNA. Circles delineate lateral ventral horn regions. Scale bar, 120 μm. K, Graph showing motor neuron cell counts in SNA mice treated with vehicle (saline) or RG108 and that survived for 21 d. Values (mean ± SD) are represented as percentage of the ipsilateral motor neuron number relative to the contralateral nonlesioned side. The asterisk denotes significant difference (p < 0.01) compared with vehicle. L, Graph showing motor neuron cell volumes in SNA mice treated with vehicle (saline) or RG108 and that survived for 21 d. Values (mean ± SD) are represented as percentage of the remaining ipsilateral motor neuron volumes relative to the motor neuron volumes in the contralateral nonlesioned side. The asterisk denotes significant difference (p < 0.01) compared with vehicle. M, N, Dnmt inhibitor RG108 completely blocks the rise in DNA methylation in adult motor neurons after SNA. Mice were treated intracerebroventricularly with 10 μg of RG108 (in 2 μl of sterile saline) or with saline (vehicle) on day 0 (day of lesion) and days 2 and 4 and were killed at 4 d after SNA. Ipsilateral lumbar motor neurons in vehicle mice displayed an increase in 5-MeC immunoreactivity in the nucleus and cytoplasm (M), and this rise in 5-mC immunoreactivity was blocked by RG108 (N). Scale bar, 7 μm. O, Graph showing motor neuron 5-mC immunoreactivity in SNA mice treated with vehicle (saline) or RG108 and that survived for 3, 4 or 7 d. Quantification was done by single-cell densitometry (Martin et al., 2005). Values (mean ± SD) are the levels of 5-mC immunoreactivity in ipsilateral motor neurons as a percentage of the levels of 5-mC immunoreactivity in contralateral motor neurons. Asterisks denote significant differences at 3, 4, and 7 d (p < 0.05, p < 0.01, and p < 0.001, respectively) compared with vehicle. Plus signs denotes significant differences at 4 and 7 d (p < 0.01, and p < 0.001, respectively) compared with 3 d vehicle.
5-mC immunoreactivity localizes to mitochondria
Because Dnmt3a can localize to mitochondria (Figs. 3, 8I) and because DNA digestion eliminated the cytoplasmic punctate staining for 5-mC (Fig. 10C), we examined whether some of the cytoplasmic structures positive for 5-mC were mitochondria, providing evidence for mitochondrial DNA methylation. An appreciable amount of the cytoplasmic punctate 5-mC immunoreactivity in motor neurons colocalized with the mitochondrial marker SOD2 (Fig. 10H). However, some of the larger aggregates of cytoplasmic 5-mC did not colocalize with SOD2 (Fig. 10H) and have been shown to colocalize with an autophagosome marker (B. Gertz and L. J. Martin, unpublished observations).
Dnmt inhibition potently protects against apoptosis of motoneurons in vivo
Neuronal cell culture experiments showed that Dnmt inhibition and knockdown protect against apoptosis (Fig. 5). To determine whether this finding translates to an in vivo setting, mice with SNA were treated repetitively with intracerebroventricular RG108. Treatment of SNA mice with RG108 nearly completely protected motor neurons against apoptosis (Fig. 10I–K). The effect was sustained through 21 d after injury. In addition, RG108 fostered hypertrophy of injured motor neurons but not contralateral motor neurons, as evidence by the increased cell body volume (Fig. 10L). RG108 also profoundly blocked the postlesion increase in 5-mC immunoreactivity in motor neurons (Fig. 10M–O). Intraventricular RG108 had no overt untoward effects on mice.
Dnmt protein expression and 5-mC are upregulated in human ALS
Because our results in cell culture and mouse models indicate that DNA methylation can participate in the mechanisms of motor neuron degeneration, we examined whether these findings might be relevant to human sporadic ALS. As in wild-type mouse brain (Fig. 7D), Dnmt1 was primarily confined to the nuclear fraction in extracts of control human motor cortex, but barely detectable Dnmt1 was also present in the mitochondrial fraction (Fig. 11A). The nuclear fraction was enriched in NeuN (Fig. 11A). In human ALS motor cortex, Dnmt1 protein levels were increased in the nuclear fraction (Fig. 11B), and Dnmt1 was also found in the mitochondrial fraction, which was also enriched in cyclophilin D (Fig. 11A,B). Dnmt3a in extracts of human control motor was highly enriched in nuclear and mitochondrial fractions, but low in the soluble fraction (Fig. 11A), in contrast to mouse brain where Dnmt3a was also enriched in the soluble fraction (Fig. 7D). In human ALS motor cortex, Dnmt3a protein levels were increased in nuclear, soluble, and mitochondrial fractions (Fig. 11A,B).
Figure 11.
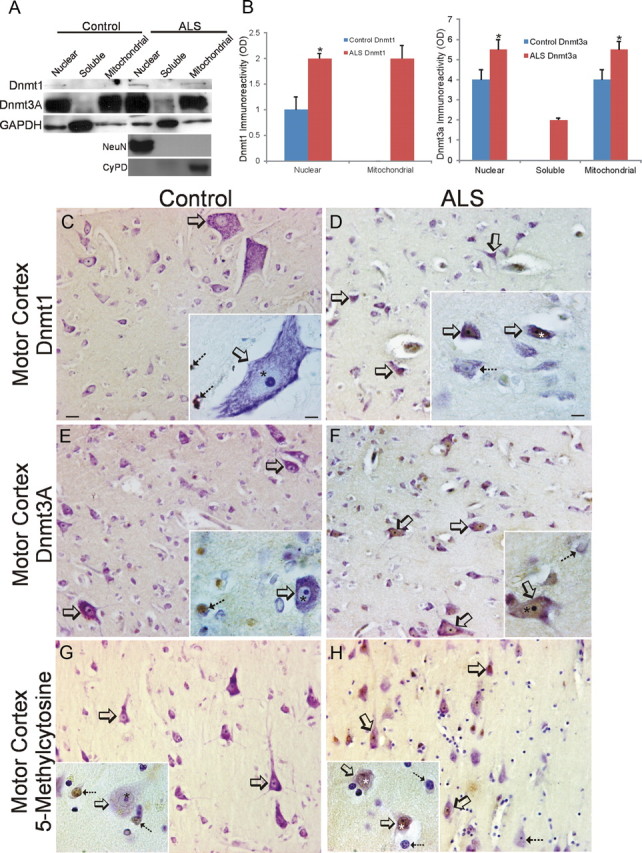
Dnmts and 5-mC are upregulated in human sporadic ALS motor cortex. A, Westerns blots for Dnmt1 and Dnmt3a in human control and sporadic ALS primary motor cortex homogenates fractionated into nuclear-enriched, soluble, and mitochondrial-enriched protein compartments. The blots were reprobed for GAPDH to show protein loading. B, Graphs showing the analysis of Dnmt1 and Dnmt3a Western blots on human control and ALS primary motor cortex tissue fractions. Values represent mean ± SD of optical density (OD) units. Dnmt1 was barely detectable or undetectable in control mitochondrial fractions and was undetectable in control and ALS soluble fractions. Dnmt3a was barely detectable or undetectable in control soluble fractions. Asterisks denote significant differences (p < 0.05) compared with control. C, D, Immunohistochemical localization of 5-mC in human control (C) and ALS (D) motor cortex. Panoramic images of deep layers and higher-magnification images (insets) are shown. C, In control motor cortex, Dnmt1 immunoreactivity is very low to not detectable in neurons, including large Betz cells (open arrows), but is present in small non-neuronal cells (inset, hatched arrows). D, In ALS motor cortex, numerous neurons are distinctly positive for Dnmt1 (open arrows). Dnmt1 immunoreactivity is enriched in the nucleus of some neurons (inset, open arrows and asterisk), but nearby neurons have scant immunoreactivity (inset hatched arrow). Scale bars: (in C, same for D–H), 22 μm; (in A, inset; same for D–H, insets), 10 μm. E, F, Immunohistochemical localization of Dnmt3a in human control (E) and ALS (F) motor cortex. E, In control motor cortex, Dnmt3a immunoreactivity is present in the cytoplasm of neurons (open arrows), including Betz cells, and is present in non-neuronal cells (inset, hatched arrow) and in the neuropil as fine puncta. The nucleus of control neurons has low Dnmt3a immunoreactivity (inset asterisk). F, In ALS motor cortex, numerous neurons, including Betz cells, are distinctly positive for Dnmt3a (open arrows). Dnmt3a immunoreactivity is enriched in the nucleus of some neurons (inset, open arrow and asterisk), but nearby cells are negative (inset, hatched arrow). G, H, Immunohistochemical localization of 5-mC in human control (G) and ALS (H) motor cortex. 5-mC immunoreactivity was visualized with diaminobenzedine (brown), and sections were counterstained with cresyl violet. G, In control motor cortex, most pyramidal neurons (open arrows) have low 5-mC immunoreactivity. 5-mC immunoreactivity was present faintly in pyramidal neurons (inset, open arrow) as fine particles in the nucleus (asterisk) and cytoplasm, but in nearby glial cells (inset, hatched arrows), 5-mC immunoreactivity was enriched in the nucleus. H, In ALS motor cortex, many pyramidal neurons (open arrows) show obvious 5-mC immunoreactivity. 5-mC immunoreactivity was enriched in the nucleus (inset, asterisk) of pyramidal neurons (open arrow), whereas nearby glial cells (inset, hatched arrows) showed scant 5-mC immunoreactivity.
Immunohistochemistry was used to determine the cellular localizations of Dnmt1, Dnmt3a, and 5-mC in control and ALS brain and spinal cord. In control human motor cortex, Dnmt1 was difficult to detect in neurons (Fig. 11C), consistent with the low levels seen by immunoblotting (Fig. 11A), whereas small non-neuronal cells were occasionally positive (Fig. 11C, inset). In ALS motor cortex, Dnmt1-positive neurons were easily identified (Fig. 11D), and Dnmt1 immunoreactivity was found accumulated in the nucleus of cortical neurons (Fig. 11D, inset). Dnmt3A in control motor cortex was seen faintly in the cytoplasm of subsets of neurons (Fig. 11E), in the neuropil (Fig. 11E, inset), and in non-neuronal cells (Fig. 11E, inset). In ALS motor cortex, Dnmt3A-positive neurons were numerous (Fig. 11F). In control human motor cortex, 5-mC immunoreactivity was conspicuously low in pyramidal neurons, but the nuclei of apparent glial cells were stained intensely (Fig. 11G). In contrast, pyramidal neurons in ALS motor cortex displayed strong 5-mC immunoreactivity, whereas nearby glial cells showed scant immunoreactivity (Fig. 11H). In control human spinal motor neurons, low Dnmt1 immunoreactivity was detected in the nucleus (Fig. 12A, inset, asterisk), whereas obvious Dnmt1 accumulations were found peripherally displaced in the cytoplasm (Fig. 12, inset, open arrow). Many remaining attritional spinal motor neurons in ALS cases showed strong immunoreactivity for Dnmt1 in the nucleus and in perinuclear punctuate structures in the cytoplasm (Fig. 12B, hatched arrows). Dnmt3a immunoreactivity in human control motor neurons was localized in the cytoplasm where it appeared as numerous discrete puncta (Fig. 12C) and was also present as delicate beaded strands in the nuclear matrix and decorated the surface of the nucleolus (Fig. 12C, inset, hatched arrow). As in adult mouse spinal cord (Fig. 8J), Dnmt3a immunoreactivity was also seen as bouton-like structures in the neuropil of human spinal cord (Fig. 12C, inset, open arrows). In ALS cases, Dnmt3a immunoreactivity was found enriched in the perinuclear cytoplasm and accumulated in the nucleus of most remaining motor neurons (Fig. 12D). In control spinal cord, 5-mC immunoreactivity was present conspicuously in the nucleus of motor neurons and seen as a reticulum consistent with a chromatin pattern (Fig. 12E). In ALS motor neurons, the entire nucleus was immunoreactive for 5-mC, and many very attritional cells were completely immunoreactive (Fig. 12F, inset).
Figure 12.
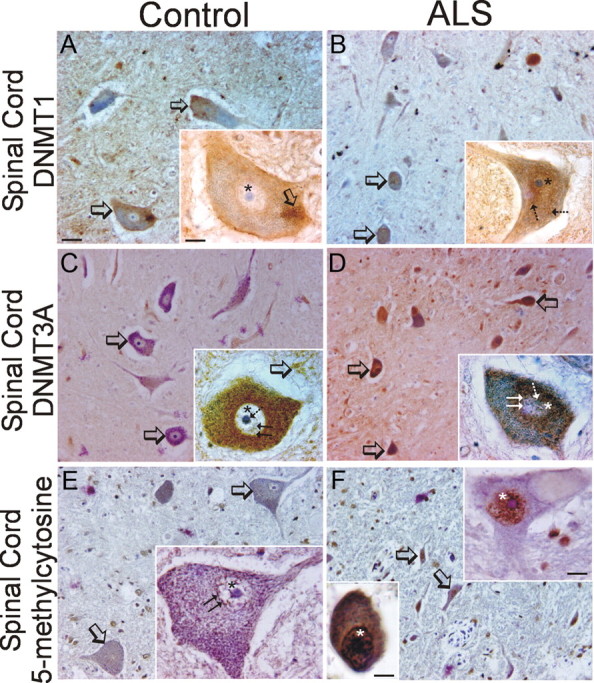
Localizations of Dnmts and 5-mC in human control and sporadic ALS spinal motor neurons. A, B, Immunohistochemical localization of Dnmt1 in human control (A) and ALS (B) spinal motor neurons. Dnmt1 immunoreactivity was visualized with diaminobenzedine (brown), and sections were counterstained with cresyl violet. Panoramic images of several motor neurons in ventral horn and single-cell high-magnification images (insets) are shown. A, In control ventral horn, Dnmt1 immunoreactivity localizes to motor neurons (open arrows), small non-neuronal cells, and the neuropil. In control motor neurons (inset) Dnmt1 immunoreactivity is found in the nucleus (asterisk) as clusters around the nuclear envelop and as fine strands. In the cytoplasm, Dnmt1 immunoreactivity is seen conspicuously as large aggregates peripherally located (open arrow). B, In ALS ventral horn, Dnmt1 immunoreactivity is conspicuous in attritional motor neurons (open arrows). In ALS motor neurons (inset) Dnmt1 is enriched completely throughout the nucleus (asterisk) and in the cytoplasm where it can be seen as puncta (hatched arrows). Scale bars, (in A, same for B–F), 7 μm; (in A, inset; same for B–F, insets), 7 μm. C, D, Immunohistochemical localization of Dnmt3a in human control (C) and ALS (D) spinal motor neurons. Dnmt3a immunoreactivity was visualized with diaminobenzedine (brown) and sections were counterstained with cresyl violet. C, In control ventral horn, Dnmt3a immunoreactivity localizes to motor neurons (open arrows) and small non-neuronal cells. In control motor neurons (inset), Dnmt3a immunoreactivity is present in the cytoplasm, where it is seen as numerous mitochondria-like structures, and is less enriched in the nucleus (asterisk), where it associates with the nucleolus (hatched arrow) and strand-like structures (double arrow). Dnmt3a is also present throughout the neuropil in a bouton-like pattern (open arrows). D, In ALS ventral horn, Dnmt3a immunoreactivity is highly enriched in attritional motor neurons (open arrows) and in small non-neuronal cells. In ALS motor neurons (inset), Dnmt3a immunoreactivity is enriched in the perinuclear cytoplasm and also in the nucleus (asterisk), where it is aggregated around the nucleolus (hatched arrow) and throughout the nuclear matrix (double arrow). E, F, Immunohistochemical localization of 5-mC in human control (E) and ALS (F) spinal motor neurons. 5-mC immunoreactivity was visualized with diaminobenzedine (brown), and sections were counterstained with cresyl violet. E, In control ventral horn, 5-mC immunoreactivity is present in motor neurons (open arrows) and in numerous small non-neuronal cells. In control motor neurons (inset) 5-mC immunoreactivity is found in the nucleus (asterisk) as small aggregates and strands near the nuclear envelope (double arrow) and nucleolus. In the cytoplasm of motor neurons, 5-mC immunoreactivity is seen as round particles. F, In ALS ventral horn, 5-mC immunoreactivity is conspicuous in attritional motor neurons (open arrows). In ALS residual motor neurons, 5-mC is enriched throughout the nucleus in some cells (top right inset, asterisk) and in other cells, 5-mC completely fills the nucleus and cytoplasm (bottom left inset).
Discussion
This study was conducted to test the hypothesis that the mechanisms used to regulate DNA methylation in neurons can be used as mechanisms of neurodegeneration. We used a combination of cell culture and animal experimental designs to show that Dnmt activity can drive neuronal apoptosis. Dnmt3a has a particularly prominent pro-apoptotic function in motor neurons. Other new findings in this work include the identification of Dnmt3a in presynaptic axon terminals and its presence in mitochondria, as well as the presence of 5-mC in mitochondria, suggesting methylation of the mitochondrial genome by Dnmt3a. We also found upregulation of Dnmts and 5-mC in human ALS, suggesting that aberrant regulation of DNA methylation is part of the pathobiology of ALS.
We show for the first time that enforced function of Dnmts can drive neuronal apoptosis. In cultured spinal cord neurons, overexpression of Dnmt3a, but not Dnmt1, induced degeneration; furthermore, robust upregulation of Dnmt1 and Dnmt3a was antecedent to neuronal apoptosis induced by DNA damage. Mutation of the catalytically active site of Dnmt3a rendered it incompetent in inducing neurodegeneration, and inhibition of Dnmts with RG108 and procainamide and Dnmt RNAi protected cultured neurons from apoptosis. The finding that procainamide, a Dnmt1-specific inhibitor (Lee et al., 2005), afforded much less neuroprotection than the more general inhibitor RG108 (Brueckner et al., 2005) supports our conclusion that Dnmt3a has stronger pro-apoptotic actions than Dnmt1 in neurons. The work was extended to an animal model of neurodegeneration where we showed several biochemical and pharmacological end points indicative of a postlesion Dnmt gain of function in mediating neuronal apoptosis.
Only three previous studies have suggested connections between Dnmt and neurodegeneration in brain or spinal cord injury models. Two studies have shown that Dnmt1 haploinsufficiency reduces infarct volume in adult mice (Endres et al., 2000, 2001). However, it is difficult to interpret this neuroprotection vis-à-vis cell death, as stroke models are confounded by a highly heterogeneous, asynchronous neuropathology and coactivation of several different cell death signaling pathways, including apoptosis, necrosis, and autophagy (Martin et al., 1998, 2000; Puyal et al., 2009; Northington et al., 2011). The other study used a bilateral dorsal column transection and sciatic nerve transection paradigm in rat or mouse to show that axon degeneration is paralleled by reduced Dnmt3a levels and genome hypomethylation in whole spinal cord (Iskandar et al., 2010). However, relationships between epigenetic-related changes and neuronal cell death are also unclear in this model.
To study the possible role of Dnmts in neuronal apoptosis specifically within the CNS, divorced from other forms of cell death, we used an animal model featuring homogeneous and synchronous cellular pathology in a discrete CNS region. SNA induces selective retrograde neuronal degeneration in the spinal cord (Martin et al., 1999). The motor neurons die by a morphological process that is unequivocal apoptosis and requires the presence of the bax and p53 genes (Martin and Liu, 2002; Martin et al., 2005). In this model, we showed that motor neuron apoptosis was associated with transient upregulation of Dnmt enzyme activity and Dnmt3a protein, subcellular changes in the localizations of Dnmt1 and Dnmt3a, and increased 5-mC in motor neurons. Importantly, treatment of mice with RG108 completely blocked the injury-induced enhancement of DNA methylation and the apoptosis of motor neurons. Previous studies of epigenetic regulation of neurodegeneration have focused primarily on histone acetylation (Sleiman et al., 2009); our study extends the concept of epigenetic mechanisms in the regulation of neuronal cell death by demonstrating a role for DNA methylation.
Previous studies have noted surprisingly high levels of Dnmt enzyme activity, mRNA, and protein in adult rodent brain (Goto et al., 1994; Brooks et al., 1996; Inano et al., 2000; Feng et al., 2005). It has also been realized that mature neurons highly express Dnmts (Goto et al., 1994; Inano et al., 2000; Feng et al., 2005). Our data are consistent with these studies by showing that Dnmt1 and Dnmt3a are expressed in adult mouse brain and spinal cord, and in adult human cerebral cortex and spinal cord, and that mouse and human motor neurons are enriched in these proteins. We also contribute new findings that Dnmt3a is present in presynaptic axon terminals and in motor neuron mitochondria. The explanation for why Dnmts are present in postmitotic, terminally differentiated neurons is unclear. Brooks et al. (1996) proposed that Dnmts in the adult brain remethylate newly incorporated cytosines from G-T mismatch repair after 5-MeC deamination. Electrophysiological studies using hippocampal slices (Levenson et al., 2006) or dissociated hippocampal cultures (Nelson et al., 2008) and Dnmt inhibitors have revealed that gene expression-mediated synaptic plasticity is regulated by DNA methylation. Recently, mice with conditional knockout of Dnmt1 and Dnmt3a and with hypomethylation in forebrain show deficits in learning and memory distinct from single knock-out mice, suggesting that Dnmt1 and Dnmt3a have redundant functions in neurons (Feng et al., 2010). Our finding that Dnmt3a is located in presynaptic axon terminals is exciting because it identifies a more local presynaptic possibility for Dnmt3a in regulating synaptic function. Moreover, the presence of 5-mC in neuronal mitochondria indicates in vivo methylation of mitochondrial DNA. Methylated cytosine residues have been detected in mouse and human mitochondrial DNA (Shmookler Reis and Goldstein, 1983; Pollack et al., 1984), but it has not been known how this is achieved. The existence of Dnmt3a and 5-mC in mitochondria enhances the idea of DNA methylation involvement in synaptic mechanisms by implicating epigenetic modification of the neuronal mitochondrial genome as a new possible function of Dnmt3a in adult neurons.
We find that another function of Dnmts in the adult nervous system is in neuronal cell death. Dnmt-mediated cell death seems to be another program for cell death. The specific gene targets of apoptosis-related DNA methylation remain to be identified, but it is apparent that motor neurons destined for apoptosis in vivo and in culture engage rapid and marked increases in DNA methylation as shown by the dramatic change in 5-mC levels. Moreover, motor neurons engaged in the apoptotic process show an intensification of nuclear Dnmts. The finding that RG108 completely occludes apoptosis of motor neurons, without shifting the cell death to another form according the cell death continuum concept (Martin, 2010), indicates that DNA methylation is a very upstream agency in the apoptotic process. As part of the genetic program of apoptosis, perhaps gene silencing through DNA methylation is a facet of the chromatin remodeling process preceding DNA cleavage.
It is interesting that nervous tissue mitochondria contain Dnmt. We found Dnmt3a in mitochondrial fractions of mouse and human CNS; in addition, Dnmt1 was found at low levels in human brain mitochondria. Recently, a mitochondrial targeting sequence was identified in Dnmt1, and it has been shown that Dnmt1, but not Dnmt3a, can localize to mitochondria in cultured mouse embryonic fibroblasts and human colon carcinoma cells (Shock et al., 2011). The discrepancy here may be caused by the types of cells/tissues analyzed, because we have found the mitochondrial localization of Dnmt3a is tissue specific (Gertz and Martin, unpublished observations).
The involvement of Dnmts and DNA methylation in neuronal cell death could be relevant to human neurodegenerative disorders. Spinal muscular atrophy (SMA) and ALS are motor neuron diseases. SMA, occurring in 1 in 10,000 live births, is caused by mutations in the Survival of Motor Neuron (Smn) gene (Lefebvre et al., 1995). A centromeric human-specific Smn2 gene is intact in a variable number of copies, but it fails to generate sufficient amounts of full-length SMN; however, the greater the SMN2 copy number, the milder the form of SMA. Interestingly, Smn2 gene silencing by DNA methylation correlates with SMA disease severity (Hauke et al., 2009). In contrast to SMA, ALS is a fatal adult-onset disease, and the majority of ALS cases are sporadic with no known inherited component. Epidemiological studies implicate some environmental and acquired factors in its pathogenesis; however, previous genetic studies have been unable to uncover the genes involved in elucidating the molecular mechanisms of sporadic ALS. DNA methylation in sporadic ALS has been examined previously in two candidate genes, SOD1 and VEGF (Oates and Pamphlett, 2007); in members of the metallothione gene family (Morahan et al., 2007); and in the astroglial EAAT2 gene promoter (Yang et al., 2010). None of these studies found differences in methylation patterns in sporadic ALS cases compared with control cases. Another study using genome-wide analysis of brain DNA methylation in sporadic ALS, accomplished by chromatin immunoprecipitation followed by CpG microarray hybridization, revealed significant hypermethylation of genes involved in calcium dynamics, oxidative stress, and synapses (Morahan et al., 2009). We show in human sporadic ALS that Dnmt1 and Dnmt3a proteins are upregulated in motor cortex and spinal cord motor neurons and that immunoreactivity for 5-mC is increased in motor cortical pyramidal neurons. Moreover, our previous work reveals that motor neurons in human ALS degenerate with biochemical and morphological features resembling a variant form of apoptosis (Martin, 2010). Our study thus supports the possibility that disease mechanisms involving aberrant DNA methylation could be relevant to human ALS pathobiology and therapeutic targeting.
Footnotes
This work was supported by grants from the U.S. Public Health Service, National Institutes of Health, National Institute on Aging (AG016282), and National Institute of Neurological Disorders and Stroke (NS034100, NS065895, and NS052098). We thank Yan Pan for technical assistance and advice.
References
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Brenner C, Fuks F. DNA methyltransferases: facts, clues, mysteries. Curr Top Microbiol Immunol. 2006;301:45–66. doi: 10.1007/3-540-31390-7_3. [DOI] [PubMed] [Google Scholar]
- Brooks P, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65:6305–6311. doi: 10.1158/0008-5472.CAN-04-2957. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR. Comparative evaluation of synaptophysin-based methods for quantification of synapses. J Neurocytol. 1996;25:821–828. doi: 10.1007/BF02284844. [DOI] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Chang Q, Martin LJ. Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a confocal quantitative analysis. Am J Pathol. 2009;174:574–585. doi: 10.2353/ajpath.2009.080557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Martin LJ. Glycine receptor channels in spinal motoneurons are abnormal in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2011;31:2815–2827. doi: 10.1523/JNEUROSCI.2475-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. Structure and function of DNA methyltransferases. Annu Rev Biophys Biomol Struct. 1995;24:293–318. doi: 10.1146/annurev.bb.24.060195.001453. [DOI] [PubMed] [Google Scholar]
- Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol Cell Biol. 2002;22:3157–3173. doi: 10.1128/MCB.22.9.3157-3173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer A, Dittmer J. β-Actin is not a reliable loading control in Western blot analysis. Electrophoresis. 2006;27:2844–2845. doi: 10.1002/elps.200500785. [DOI] [PubMed] [Google Scholar]
- Endres M, Meisel A, Biniskiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U. DNA methytransferase contributes to delayed ischemia brain injury. J Neurosci. 2000;20:3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Fan G, Meisel A, Dirnagl U, Jaenisch R. Effects of cerebral ischemia in mice lacking DNA methyltransferase I in post-mitotic neurons. NeuroReport. 2001;12:3763–3766. doi: 10.1097/00001756-200112040-00032. [DOI] [PubMed] [Google Scholar]
- Fatemi M, Pao MM, Jeong S, Gal-Yam EN, Egger G, Weisenberger DJ, Jones PA. Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005;33:e176. doi: 10.1093/nar/gni180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Chang H, Li E, Fan G. Dynamic expression of de novo DNA methytransferase DNMT3A and DNMT3B in the central nervous system. J Neurosci Res. 2005;79:734–746. doi: 10.1002/jnr.20404. [DOI] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson RE, Carroll HP, Harris A, Maher ER, Selby PJ, Banks RE. Housekeeping proteins: a preliminary study illustrating some limitations as useful references in protein expression studies. Proteomics. 2005;5:566–571. doi: 10.1002/pmic.200400941. [DOI] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Goto K, Numata M, Komura JI, Ono T, Bestor TH, Kondo H. Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation. 1994;56:39–44. doi: 10.1046/j.1432-0436.1994.56120039.x. [DOI] [PubMed] [Google Scholar]
- Hauke J, Riessland M, Lunke S, Eyüpoglu IY, Blümcke I, El-Osta A, Wirth B, Hahnen E. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet. 2009;18:304–317. doi: 10.1093/hmg/ddn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inano K, Suetake I, Ueda T, Miyake Y, Nakamura M, Okada M, Tajima S. Maintenance-type DNA methyltransferase is highly expressed in post-mitotic neurons and localized in the cytoplasmic compartment. J Biochem. 2000;128:315–321. doi: 10.1093/oxfordjournals.jbchem.a022755. [DOI] [PubMed] [Google Scholar]
- Iskandar BJ, Rizk E, Meier B, Hariharan N, Bottiglieri T, Finnell RH, Jarrard DF, Banerjee RV, Skene JH, Nelson A, Patel N, Gherasim C, Simon K, Cook TD, Hogan KJ. Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J Clin Invest. 2010;120:1603–1616. doi: 10.1172/JCI40000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–251. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- Kang JS, Wanibuchi H, Morimura K, Totsuka Y, Yoshimura I, Fukushima S. Existence of a no effect level for MeIQx hepatocarcinogenicity on a background of thioacetaminde-induced liver damage in rats. Cancer Sci. 2006;97:453–458. doi: 10.1111/j.1349-7006.2006.00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. 2003;278:4806–4812. doi: 10.1074/jbc.M209923200. [DOI] [PubMed] [Google Scholar]
- Kondo T, Bobek MP, Kuick R, Lamb B, Zhu X, Narayan A, Bourc'his D, Viegas-Péquignot E, Ehrlich M, Hanash SM. Whole-genome methylation scan in ICF syndrome: hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum Mol Genet. 2000;9:597–604. doi: 10.1093/hmg/9.4.597. [DOI] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, 3rd, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iñiguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolaños CA, Kabbaj M, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2001;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem. 2005;280:40749–40756. doi: 10.1074/jbc.M505593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frezal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lesuisse C, Martin LJ. Immature and mature cortical neurons engage different apoptotic mechanisms involving caspase-3 and the mitogen-activated protein kinase pathway. J Cereb Blood Flow Metab. 2002;22:935–950. doi: 10.1097/00004647-200208000-00005. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:5763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Liu N-K, Xu X-M. β-Tubulin is a more suitable internal control that β-actin in western blot analysis of spinal cord tissues after traumatic injury. J Neurotrauma. 2006;23:1794–1801. doi: 10.1089/neu.2006.23.1794. [DOI] [PubMed] [Google Scholar]
- Lok J, Martin LJ. Rapid subcellular redistribution of Bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. J Neurotrauma. 2002;19:815–828. doi: 10.1089/08977150260190410. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- Martin LJ. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis. 2000;7:613–622. doi: 10.1006/nbdi.2000.0314. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals. 2010;3:839–915. doi: 10.3390/ph3040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. An approach to experimental synaptic pathology using green fluorescent protein-transgenic mice and gene knockout mice: excitotoxic vulnerability of interneurons and motoneurons is associated with mitochondrial accumulation and mediated by the mitochondrial permeability transition pore. Toxicol Pathol. 2011;39:220–233. doi: 10.1177/0192623310389475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and bax-dependent. J Neurobiol. 2002;50:181–197. doi: 10.1002/neu.10026. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Kaiser A, Price AC. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J Neurobiol. 1999;40:185–201. [PubMed] [Google Scholar]
- Martin LJ, Sieber FE, Traystman RJ. Apoptosis and necrosis occur in separate neuronal populations in hippocampus and cerebellum after ischemia and are associated with alterations in metabotropic glutamate receptor signaling pathways. J Cereb Blood Flow Metab. 2000;20:153–167. doi: 10.1097/00004647-200001000-00020. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR, Wong PC, Liu Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem. 2003;85:234–247. doi: 10.1046/j.1471-4159.2003.01659.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci. 2005;25:6449–6459. doi: 10.1523/JNEUROSCI.0911-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500:20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Pipino J, Chestnut B, Landek MA. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb Cortex. 2009a;19:1273–1293. doi: 10.1093/cercor/bhn167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009b;218:333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Morahan JM, Yu B, Trent RJ, Pamphlett R. Are metallothionein genes silenced in ALS? Toxicol Lett. 2007;168:83–87. doi: 10.1016/j.toxlet.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Morahan JM, Yu B, Trent RJ, Pamphlett R. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. 2009;10:418–429. doi: 10.3109/17482960802635397. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia ischemia. Ann Neurol. 2011;69:743–758. doi: 10.1002/ana.22419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates N, Pamphlett R. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph Lateral Scler. 2007;8:83–86. doi: 10.1080/17482960601149160. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984;12:4811–4824. doi: 10.1093/nar/12.12.4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase. I. expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- Puyal J, Vaslin A, Mottier V, Clarke PGH. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- Razin A. CpG methylation, chromatin structure and gene silencing—a three-way connection. EMBO J. 1998;17:4905–4908. doi: 10.1093/emboj/17.17.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene, 2001;20:3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- Schaefer M, Lyko F. Solving the Dnmt2 enigma. Chromosoma. 2010;119:35–40. doi: 10.1007/s00412-009-0240-6. [DOI] [PubMed] [Google Scholar]
- Shaikh AY, Martin LJ. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromol Med. 2002;2:47–60. doi: 10.1007/s12017-002-0038-7. [DOI] [PubMed] [Google Scholar]
- Shmookler Reis RJ, Goldstein S. Mitochondrial DNA in mortal and immortal cells: genome number, integrity, and methylation. J Biol Chem. 1983;258:9078–9085. [PubMed] [Google Scholar]
- Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011;108:3630–3635. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR. Putting the “HAT” back on survival signaling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18:573–584. doi: 10.1517/13543780902810345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyf M. DNA methylation and demethylation as targets for anticancer therapy. Biochemistry. 2005;70:533–549. doi: 10.1007/s10541-005-0147-7. [DOI] [PubMed] [Google Scholar]
- Tovar-y-Romo LB, Santa-Cruz LD, Tapia R. Experimental models for the study of neurodegeneration in amyotrophic lateral sclerosis. Mol Neurodegeneration. 2009;4:31. doi: 10.1186/1750-1326-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Wong M, Martin LJ. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet. 2010;19:2284–2302. doi: 10.1093/hmg/ddq106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Wang Z, Okano M, Nogami M, Li Y, He WW, Okumura K, Li E. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene. 1999;236:87–95. doi: 10.1016/s0378-1119(99)00252-8. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Vidensky S, Robinson MB, Rothstein JD. Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia. 2010;58:277–286. doi: 10.1002/glia.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW-L, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE, Ecker JR. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]