

Abstract
TRPM6 is crucial for human Mg2+ homeostasis as patients carrying TRPM6 mutations develop hypomagnesemia and secondary hypocalcemia (HSH). However, the activation mechanism of TRPM6 has remained unknown. Here we demonstrate that phosphatidylinositol-4,5-bisphophate (PIP2) controls TRPM6 activation and Mg2+ influx. Stimulation of PLC-coupled M1-receptors to deplete PIP2 potently inactivates TRPM6. Translocation of over-expressed 5-phosphatase to cell membrane to specifically hydrolyze PIP2 also completely inhibits TRPM6. Moreover, depolarization-induced-activation of the voltage-sensitive-phosphatase (Ci-VSP) simultaneously depletes PIP2 and inhibits TRPM6. PLC-activation induced PIP2-depletion not only inhibits TRPM6, but also abolishes TRPM6-mediated Mg2+ influx. Furthermore, neutralization of basic residues in the TRP domain leads to nonfunctional or dysfunctional mutants with reduced activity by PIP2, suggesting that they are likely to participate in interactions with PIP2. Our data indicate that PIP2 is required for TRPM6 channel function; hydrolysis of PIP2 by PLC-coupled hormones/agonists may constitute an important pathway for TRPM6 gating, and perhaps Mg2+ homeostasis.
Magnesium (Mg2+) is the most abundant divalent cation in the mammalian cell and is essential for numerous fundamental cellular processes, including cell cycle, channel regulation, ATPase activity, metabolic regulation, and various signaling pathways1. Mg2+ deficiency has been implicated in many diseases, ranging from neurological to cardiovascular diseases2,3. Mg2+ homeostasis is therefore tightly controlled by maintaining the equilibrium between intestinal Mg2+ absorption and renal Mg2+ excretion/re-absorption.
Several Mg2+ transporters and channels have been implicated to be important in Mg2+ absorption and/or reabsorption4,5,6,7,8,9,10,11. Most notable, loss of function of TRPM6 causes familial hypomagnesemia and secondary hypocalcemia (HSH)9,10. TRPM7, the closest homologue of TRPM6, was demonstrated to be essential for cellular Mg2+ homeostasis in DT-40 lymphocytes11. However, it's role in Mg2+ homeostasis in mice remains disputed12,13. TRPM6 and TRPM7 are unique bi-functional channel proteins with protein kinase function14,15,16,17,18. TRPM7 is ubiquitously expressed in various cells and tissues and has well defined functions in embryonic development12,13,19, neuronal cell death20, and a variety of other functions21,22,23,24. Different from TRPM7, TRPM6 expression is restrained in the epithelial cells in intestine and distal convoluted tubule (DCT) of nephron9,10, consistent with its central role in controlling Mg2+ homeostasis.
Mg2+ homeostasis is regulated by a variety of hormonal and pathological conditions1. As a gatekeeper of human Mg2+ homeostasis25, TRPM6 has been demonstrated to be regulated at expression levels2,26 by hormones such as estrogen26 and AngII27, metabolic acidosis/alkalosis28, immunosuppressant tacrolimus29, diuretics Thiazide30, and EGF31. However, the gating mechanism of TRPM6, the key property which controls Mg2+ influx, has remained elusive. Like TRPM7, TRPM6 is inhibited by millimolar concentration of intracellular Mg2+ ([Mg2+]i); therefore, it only constitutively opens to a small degree under physiological [Mg2+]i18. Both TRPM6 and TRPM7 are permeable to Ca2+ and Mg2+ under physiological pH, and conduct monovalent Na+ currents at acidic extracellular pH32,33; however, they display significant differences in single channel conductance, pharmacological profiles32,33, and kinase activity17. Unlike TRPM7 whose channel activity is known to be controlled by PIP234, how TRPM6 is gated is not clear.
Here, we show that TRPM6 channel activity and TRPM6-mediated Mg2+ influx are controlled by PIP2 levels. Depletion of PIP2 by Gq-linked receptor activation, by depolarization-induced activation of voltage-dependent phosphatase (Ci-VSP), and by chemical translocation of 5-phosphatase, can all efficiently inactivate TRPM6. Neutralization of the positively charged residues in the TRP domain leads to dysfunctional or nonfunctional mutants with reduced single channel activity by PIP2, suggesting that these positively charged residues are likely to be the putative PIP2 binding sites. Furthermore, we demonstrate that the kinase domain of TRPM6 interacts with PLC isoforms, although the interaction is not necessary for PLC-induced TRPM6 channel inactivation. These results indicate that PIP2 controls TRPM6 gating, and perhaps Mg2+ homeostasis, under various physiological/pathological conditions.
Results
Phospholipase C stimulation inactivates the TRPM6 channel
Magnesium homoestasis is tightly controlled by many hormone/receptor interactions. In order to understand how TRPM6 gating is regulated, we first determined whether activation of G protein coupled receptor induced PIP2 hydrolysis has any effect on TRPM6 activation. TRPM6 was transfected to HEK-293 cells stably expressing the M1 receptor (HM1). Under whole cell configuration, TRPM6 current was small right after rupture, and ran up with time when intracellular free Mg2+ concentration was decreased after pipette solution dialyzed into the cell. Upon TRPM6 current reaching a steady-state, 200 μM carbachol (CCh) was applied to the cell (Fig. 1A). TRPM6 current was rapidly and almost completely inhibited by CCh application (Fig. 1A–B), suggesting that activation of the M1 receptor by CCh inactivates TRPM6 channel activity. In agreement with this notion, CCh failed to inhibit TRPM6 current in HEK-293 cells transfected with TRPM6 but without over-expression of M1 receptor (sFig. 1A). Ca2+ release induced by CCh was detected in HM1 cells but not in HEK-293 cells, further suggesting that HEK-293 cells lack M1 receptor (sFig. 1A). In order to determine whether M1 receptor stimulation induced inactivation of TRPM6 is through the PLC pathway, we tested the effect of PLC inhibitor U-73122. However, we found that both the PLC inhibitor U-73122 and its inactive isoform U-73343 inhibited TRPM6 and TRPM7 currents (sFig. 2). The inhibitory effects of U-73122 could be caused by direct inhibition of the channels, or through the partial agonist effect of U-73122 on PLC35.
Figure 1. TRPM6 current is inhibited by CCh stimulation of the M1 receptor.
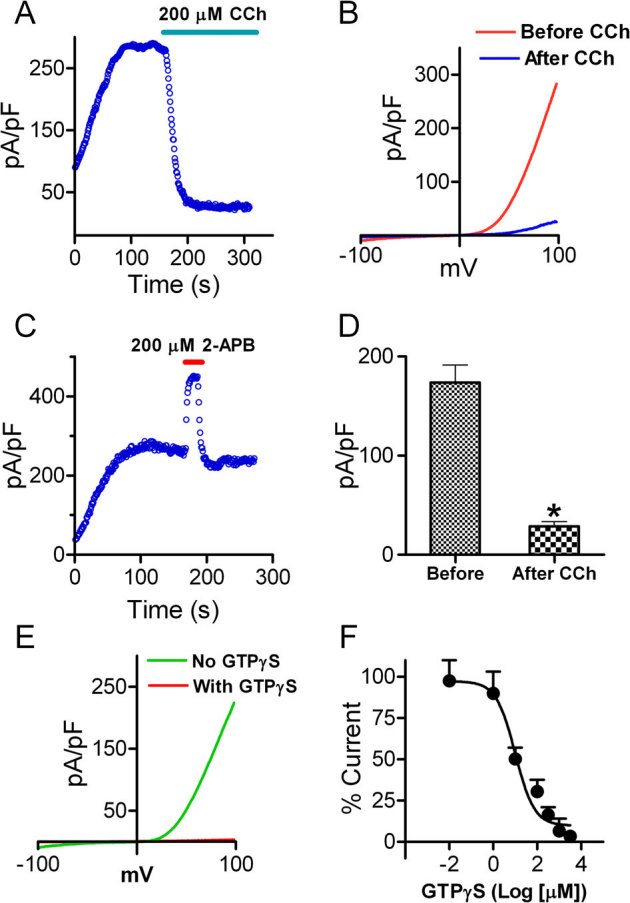
(A) A representative recording in HM1 cells transfected with TRPM6. Currents were elicited by 100 ms voltage ramps ranging from −100 to +100 mV. Application of 200 μM CCh to activate M1 receptor completely inhibited TRPM6 current. (B) Time-dependent changes of outward TRPM6 current measured at +100 mV before and after 200 μM CCh application. (C) Potentiation of the currents by 2-APB was used to confirm that the recordings were TRPM6 currents. (D) Mean current densities of TRPM6 before and after CCh (n = 10, * p<0.05). (E) Representative traces of TRPM6 currents recorded from different cells using pipette solution with or without 1 mM GTPγS. (F) Concentration-dependent effects of GTPγS on TRPM6. Currents recorded at various GTPγS concentrations were normalized to the current recorded without GTPγS in the pipette solution. Best fit of the dose-response curve yielded IC50 = 10.1 μM (n = 5–9 cells for each GTPγS concentration).
Although HM1 cells have much lower expression of endogenous TRPM7 (∼ 200 pA)36, to ensure that the CCh inhibited current is indeed TRPM6 current, we applied 200 μM 2-APB before application of CCh. We have previously demonstrated that 200 μM 2-APB can completely inhibit TRPM7 currents, but fully potentiate TRPM6 currents33. As shown in Fig. 1C, 2-APB at 200 μM markedly enhanced current amplitude by 100%, indicating that the currents recorded in HM1 cells that were largely inhibited by CCh were indeed TRPM6 currents.
Stimulation of the M1 receptor by CCh can activate PLC-β which hydrolyzes PIP2 and generates diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). We hypothesized that depletion of PIP2 inactivates TRPM6 channel activity, similar to the effects of PIP2 on TRPM734. Indeed, inclusion of DAG or IP3 in the pipette solution did not influence TRPM6 current amplitude (data not shown). Furthermore, we applied GTPγS in the pipette solution to mimic the effect of M1 receptor activation by CCh. Consistent with the effects of CCh on TRPM6 current in HM1 cells, GTPγS inhibited TRPM6 currents in a dose-dependent manner (Figure 1E–F). These results indicate that Gq linked receptor activation can inhibit TRPM6 channel activity. Since GTPγS activates several downstream pathways, we further tested the effects of activators of PKC and cAMP on TRPM6 currents. As shown in sFig. 3, application of 5 μM Foskolin to activate cAMP did not change TRPM6 current amplitude, consistent with previous report37. Perfusing the cells with PDBu to activate PKC could not produce significant changes on TRPM6 currents either. Furthermore, GTPγS included in the pipette solution could indeed cause PIP2 deletion, as detected by the translocation of the PH-GFP, an indicator of PIP2. (sFig. 3C–D). These results suggest that GTPγS elicited inactivation of TRPM6 is mainly through PLC activation induced PIP2 hydrolysis.
Direct removal of PIP2 by chemical translocation of 5-phosphatase for PtdIns(4,5)P2 to the plasma membrane inactivates TRPM6
To determine whether depletion of PIP2 underlies the mechanism by which TRPM6 is inhibited by M1 receptor activation, we first applied a chemical dimerization system induced by Rapamycin (Rap) to deplete PIP2. The membrane-localized Rap binding protein PM-FRB and the fluorescent cytoplasmic enzyme construct FKBP-5-phosphatase (5-ptase) were co-transfected with TRPM6 into HM1 cells. Application of the chemical inducer Rap will recruit 5-ptase to the plasma membrane, thereby rapidly and irreversibly convert PIP2 to PI(4)P by removing the 5′ position phosphate at the triphosphoinositol ring (Fig. 2A)38,39. As shown in Fig. 2, in the cells co-expressing TRPM6 with 5-ptase and the anchor protein FBR, application of 10 μM Rap substantially reduced TRPM6 current amplitude (Fig. 2B–C) with an average inhibition of 90%± 9% (Fig. 2D). Rap (10 μM) did not produce noticeable effects on TRPM6 current in the cells without over-expression of 5-ptase and anchor protein FRB (Fig. 2E–G). Moreover, when TRPM6 was co-transfected with FRB and the mutant 5-ptase (D281A)38, application of 10 μM Rap failed to reduce TRPM6 current amplitude (sFig. 4). Thus, the results in Fig. 2 demonstrate that activation of 5-ptase to deplete PIP2 can fully inactivate TRPM6 channels, providing strong evidence that PIP2 but not PI(4)P is required for TRPM6 channel function.
Figure 2. Membrane tethering of type IV 5-ptase reduces membrane PIP2 and suppresses TRPM6 current.
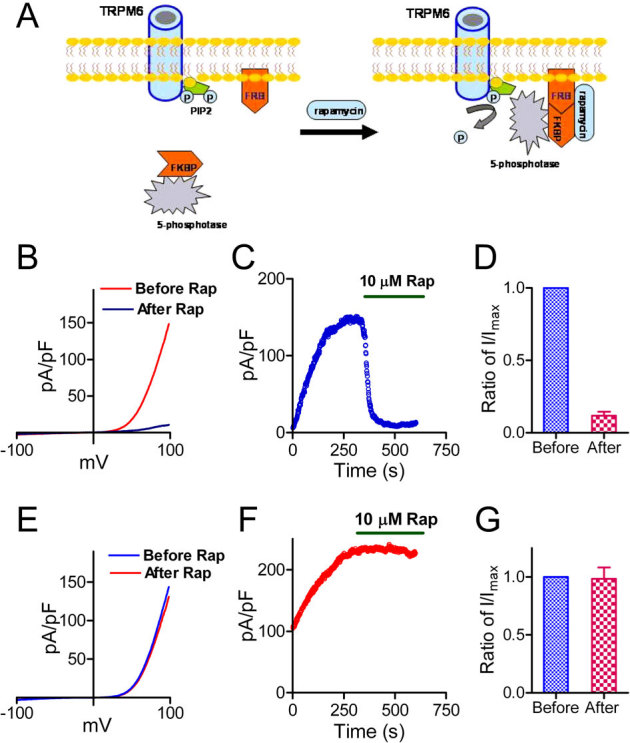
(A) Schematic diagram demonstrating how the rapamycin (Rap) inducible PIP2 specific phosphatase system works. (B) Representative recordings of TRPM6 before and after Rap application in cells transfected with GFP-TRPM6, FKBP-Inp54p and Lyn-FRB. (C) Time course of TRPM6 inhibition by perfusing 10 μM Rap. (D) Average inhibition of TRPM6 by 10 μM Rap (n = 12) in cells transfected with TRPM6, FKBP-Inp54p and Lyn-FRB. (E) Representative recordings of TRPM6 before and after Rap application in cells transfected with TRPM6 alone. (F) Time-dependent changes of TRPM6 before and after 10 μM Rap in cells shown in (E). (G) Mean current densities of TRPM6 before and after Rap. Rap did not produce inhibition on TRPM6 in cells transfected with TRPM6 alone.
Simultaneous changes of PIP2 level and TRPM6 channel activity
The membrane PIP2 levels can be modulated by a variety of stimuli and intracellular events. A recently identified voltage-sensitive phosphatase from Ciona intestinalis (Ci-VSP) possesses voltage-sensitive phosphatase activity40. Ci-VSP can convert PIP2 to PI(4)P upon depolarization, but does not have phosphatase activities at hyperpolarized voltage40,41. Co-transfection of TRPM6 and Ci-VSP provides us an opportunity to change membrane PIP2 levels using TRPM6 recording protocol (voltage ramp) without applying additional chemicals. Cells were held at −60 mV for 3 min to allow the pipette solution to fully dialyze into the cells. Then, a standard voltage ramp protocol ranging from −120 mV to +100 mV was used to activate Ci-VSP and to record TRPM6 currents (Fig. 3A). As shown in Fig. 3, TRPM6 current was rapidly decreased in the cells co-expressing TRPM6 and Ci-VSP (Fig. 3B, D–E); whereas, in cells transfected with TRPM6 alone, TRPM6 current remained unchanged (Fig. 3C–D, F), indicating that depolarization induced dephosphorylation of PIP2 by Ci-VSP inhibited TRPM6 channel activity. These results provide further evidence that PIP2 hydrolysis inactivates TRPM6.
Figure 3. Simultaneous monitoring of PIP2 depletion and TRPM6 inactivation.
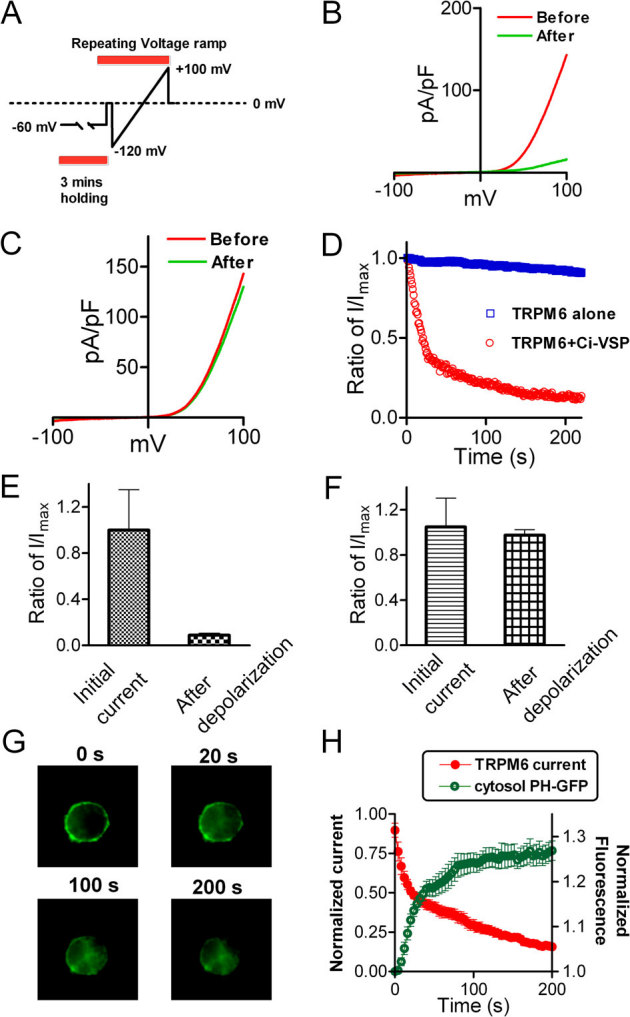
(A) The protocol for activation of Ci-VSP and recording of TRPM6 currents. Cells were held at −60 mV to allow dialysis of pipette solution without activation of Ci-VSP. Voltage Ramp ranging from −120 to +100 mV was applied to activate Ci-VSP and record TRPM6 currents. (B–C) Representative traces recorded before and after activation of Ci-VSP in cells transfected with TRPM6 and Ci-VSP (B) and TRPM6 alone (C). (D) Time-dependent changes of TRPM6 outward current measured at +100 mV in cells transfected with TRPM6+Ci-VSP and TRPM6 alone. (E–F) Normalized currents before and after depolarization from cells with TRPM6+Ci-VSP transfection (E) and with TRPM6 transfection alone (F) (n = 8 for each group). (G) Representative GFP-PH domain sub-cellular location at different time points upon depolarization. Cells were co-transfected with GFP-PH, Ci-VSP and TRPM6. (H) Kinetics of cytosol fluorescence increment and TRPM6 current inhibition. The intensity of fluorescence and current amplitude of TRPM6 at each time-point were normalized to the initial values, respectively. The time constants obtained by mono-exponential fit were 24.2± 2.5 s for PH-GFP translocation, and 36.5±6.9 s for TRPM6 inactivation.
To determine the correlation of PIP2 hydrolysis and TRPM6 channel activity, we simultaneously monitored the changes of membrane PIP2 levels and TRPM6 current amplitude. A PtdIns(4,5)P2 reporter, PLCδ1-PH-GFP construct, was co-transfected with TRPM6 and Ci-VSP to the cells. PH-GFP is localized in the cell membrane by binding to PIP2, and can be released to the cytosol when PIP2 is hydrolyzed. We monitored the translocation of PH-GFP and recorded TRPM6 current simultaneously in the same cells. In the co-transfected cells, TRPM6 current was rapidly inhibited in the first 20 s, and almost completely inhibited at 200 s; whereas, in the TRPM6 only transfected cells, current amplitude was barely changed over the time course of 200 s recording (Fig. 3D). Meanwhile, the membrane fluorescence representing translocation of PH-GFP significantly decreased in the first 20 s, and slowly disappeared over the time course of 200 s (Fig. 3G–H). The time-dependent inhibition of TRPM6 current amplitude exhibited a similar kinetics to that of translocation of PH-GFP. The time constant was 36.5±6.9 s for TRPM6 inhibition, and for PH-GFP translocation was 24.2± 2.5 s (Fig. 3H). The kinetics of PIP2 hydrolysis by activation of Ci-VSP was slower than previously reported42, probably due to the ramp protocols used in our experiments. Nevertheless, the well-correlated kinetics of TRPM6 inactivation and PIP2 hydrolysis (Fig. 3H) indicates that TRPM6 channel inactivation is caused by the decrease of PIP2 levels. Indeed, including water soluble PIP2 (DiC8-PIP2) in the pipette solution significantly reduced the inactivation speed of TRPM6 caused by PIP2 hydrolysis in the TRPM6 and Ci-VSP co-transfected cells (sFig. 5), supporting the notion that PIP2 is necessary for TRPM6 channel activation.
Depletion of PIP2 inactivates TRPM6 channel activity in perforated-patch
Since the above results were obtained by whole-cell configuration, to eliminate any interference of pipette solution to the effects of PIP2 on TRPM6 activity, we applied perforated-patch to determine the effects of PIP2 hydrolysis on TRPM6. As shown in sFig. 6, in the cells co-transfected with TRPM6 and Ci-VSP, there was substantial TRPM6 current recorded by perforated-patch, albeit the current amplitude being smaller than that of whole-cell current amplitude. TRPM6 current was readily reduced by depolarization which activated Ci-VSP to hydrolyze PIP2, further confirming that PIP2 is essential for TRPM6 channel activity. The effects of deletion of PIP2 in perforated patches were also observed in TRPM7 transfected cells (sFig. 7).
Putative PIP2 interacting residues of TRPM6
PIP2 regulates ion channel function by binding to specific residues43. To understand the mechanism by which PIP2 activates TRPM6, we mutated the positively charged residues equivalent to those of TRPM8, which have been shown to be responsible for PIP2 modulation44. We created single and double mutants as shown in Fig. 4, and tested channel function of these mutants. The current amplitude of K1085Q and R1088Q mutants was much smaller than that of WT TRPM6 (Fig. 4B, E), yet they can be potentiated by 2-APB (Fig. 4C–D), indicating that the recorded currents from K1085Q and R1088Q transfected cells were TRPM6. The reduced current amplitude of the mutants could be the consequence of disrupted interaction of PIP2 with the putative binding residues. Interestingly, the mutation of Lys to Glu (K1098Q) completely abolished TRPM6 channel activity (Fig. 4E), suggesting that Lys1098 may be more crucial for TRPM6 channel activity than Lys1085 and Arg1088. The loss-of-function mutant K1098Q has no response to 2-APB stimulation (Fig. 4F). Furthermore, although both K1085Q and R1088Q mutants produced small currents, double mutation by neutralizing both Lys1085 and Arg1088 abolished channel function completely (Fig. 4E–F). Like K1098Q, the non-functional double mutant K1085Q/R1088Q could not be reactivated or potentiated by 2-APB, suggesting that the effects of 2-APB on TRPM6 depend on the presence of PIP2 and the availability of the interaction between PIP2 and TRPM6. This hypothesis is supported by the results that 2-APB could not potentiate TRPM6 after the channel is completely inactivated by PIP2 depletion induced by Ci-VSP activation (sFig. 8).
Figure 4. PIP2 binding sites of TRPM6.
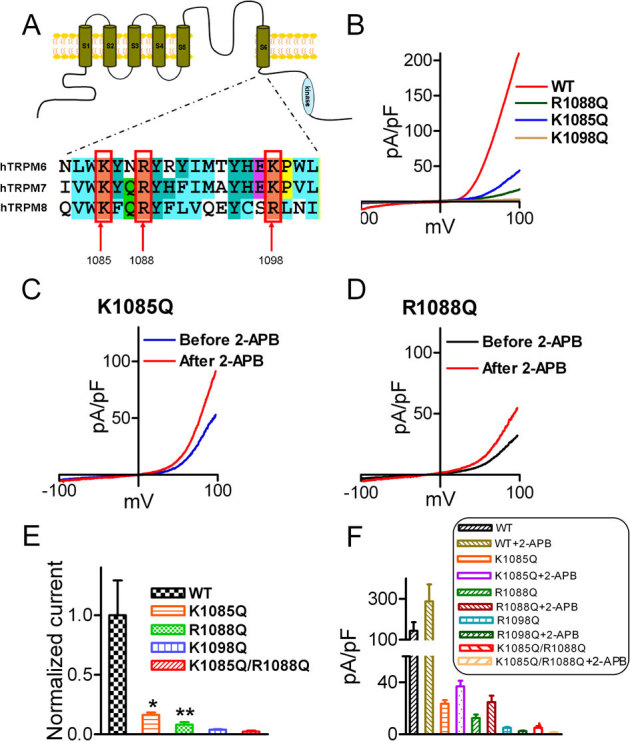
(A) Alignment of the TRP domain of TRPM6, TRPM7 and TRPM8. The highlighted residues in TRPM8 are the PIP2 binding sites. (B) Representative traces of WT-TRPM6 and mutants K1085Q, R1088Q, and K1098Q. (C–D) Effects of 2-APB on the mutants K1085Q and R1088Q. (E) Normalized mean current density of TRPM6 mutants in comparison with WT TRPM6 (n = 10 *p<0.05; ** P<0.01). Mutants K1098Q and the double mutants K1085Q/R1088Q did not produce any current. (F) Average changes of current amplitude by 2-APB (200 μM). 2-APB did not have any effect on the non-functional mutants K1098Q and K1085Q/R1088Q.
To determine whether mutations disrupt plasma membrane localization leading to dysfunctional channels, we measured plasma membrane protein and total protein levels of TRPM6 and the mutants (Fig. 5). GADPH was used as a negative control for the plasma membrane protein. The ratios of plasma membrane/total protein of K1085Q, R1088Q, K1098Q, and K1085Q/R1088Q were not significantly different from that of WT TRPM6 (Fig. 5B), indicating that mutations of the positively charged residues in the TRP domain, but not the expression or trafficking of the channel proteins, disrupted TRPM6 channel function.
Figure 5. Plasma membrane expression of WT TRPM6 and the putative PIP2 binding-site mutants.
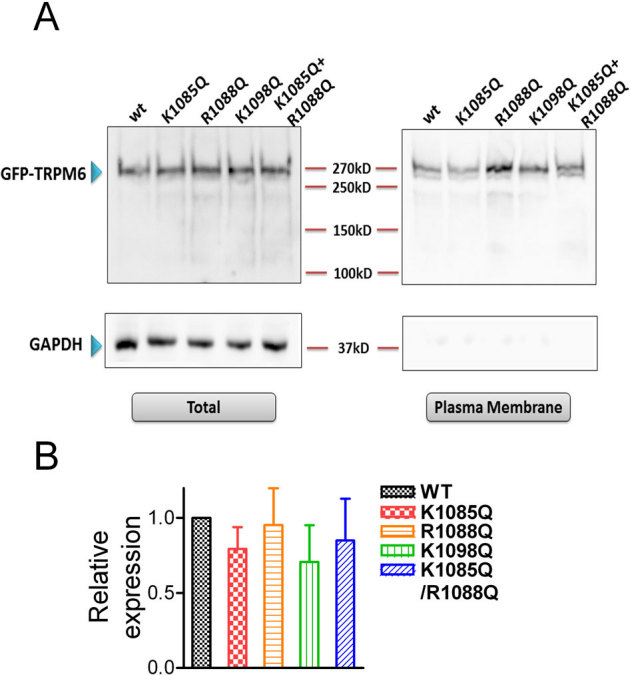
(A) Western-blot (with GFP antibody) detection of proteins of the WT and mutants in the plasma membrane portion and in total lysate. GADPH was used as a negative control for the plasma membrane protein. (B) Mean relative expression level of WT TRPM6 and the mutants in the plasma membrane versus total protein. No statistically significant difference was observed (n = 3).
PIP2 regulates single channel activities in excised patches of TRPM6 and its mutant
To examine directly the effects of PIP2 on channel activity, TRPM6 single channel currents were recorded at −50 mV in inside-out patches in the DVF solution as shown in Fig. 6A. Similar single channel currents were not observed in mock-transfected cells. The slope conductance calculated based on single channel current amplitude at −50 mV (3.99±0.3pA), −80 mV (6.7±0.8pA), and −120 mV (9.8±1.1pA) was 82.7 pS (n = 4), which is indistinguishable from the signature conductance of TRPM6 (83.6 pS)33. To further confirm that the recorded currents were TRPM6 currents, we applied 2-APB to the excised patches. Similar to the effects of 2-APB on macroscopic TRPM6 currents, we observed that 2-APB increased TRPM6 channel activity (Data not shown). Like other PIP2 activated channels such as TRPM734 and TRPM844, single channel activities of TRPM6 rundown in excised patches (Fig. 6A), presumably caused by depletion of PIP234,44. After rundown, application of PIP2 (DiC8-PIP2) recovered TRPM6 channel activity by almost 80% (Fig. 6A, D), indicating that PIP2 is sufficient to activate TRPM6. In contrast to the WT TRPM6 single channel activity, the putative PIP2 binding residue mutant R1088Q exhibited extremely low open probability (Fig. 6B–C), suggesting that neutralization of the positive charges of the putative PIP2 binding sites might have impaired channel activity. Moreover, after rundown, application of 10 or 50 μM PIP2 to the excised patches of R1088Q only recovered the channel activity by 10%, suggesting that the activation of R1088Q by PIP2 is significantly decreased in comparison with WT TRPM6. These results imply that R1088 is likely to be involved in the interaction of TRPM6 and PIP2.
Figure 6. Effects of PIP2 on channel activities of TRPM6 and its mutant in the inside-out patches.
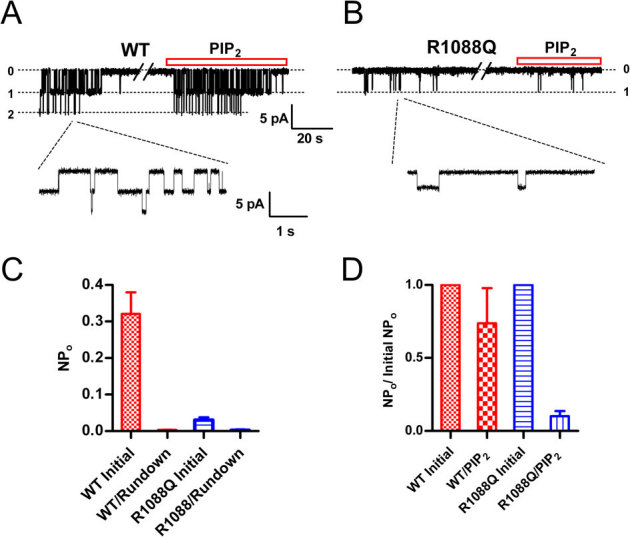
(A) Representative recordings of WT TRPM6 single channel currents in an inside-out patch at -50 mV recorded in divalent free solution (DVF). PIP2 (DiC8-PIP2) at 10 μM was applied to the patch after completely rundown of channel opening. (B) Single channel currents of R1088Q recorded at −50 mV right after formation of excised patch, after rundown, and upon application of 10 μM PIP2. (C) Average open probability of TRPM6 (n = 8) and R1088Q (n = 7) before and after completely rundown. (D) Percentage of channel open probabilities rescued by 10 μM PIP2 after rundown of TRPM6 and R1088Q. The effect of DiC8-PIP2 was reversible and reproducible in the same patches.
The putative PIP2 binding residues are essential TRPM7 channel activity
TRPM6 and TRPM7 are the closest homologues with a number of similar and distinctive properties17,18,33,45. Previous studies have shown that depletion of PIP2 inactivates TRPM7, yet the PIP2 binding sites were not determined34,46. Thus, we mutated the positively charged residues equivalent to those of TRPM6 in the TRP domain (Fig. 4A). Unlike TRPM6, all the single mutants of TRPM7 exhibited channel activity (Fig. 7A–B), although the current amplitude of K1112Q was significantly smaller than that of WT TRPM7. The double-mutant K1112Q/R1115Q and the triple mutant K1112Q/R1115Q/K1125Q, however, completely lost channel function. Moreover, current amplitude of the cells transfected with K1112Q/R1115Q or K1112Q/R1115Q/K1125Q (Fig. 7A) was even smaller than that of mock-transfected cells, suggesting that the loss-of-function mutants K1112Q/R1115Q and K1112Q/R1115Q/K1125Q behave as a dominant-negative to the endogenous TRPM7 channel. Given that PIP2 has been shown to be necessary and sufficient for TRPM7 channel activation, it is likely that the conserved positively charged residues in the proximity of the TRP domain are essential for PIP2 and TRPM7 interaction.
Figure 7. Mutation of the putative PIP2 binding residues of TRPM7 disrupts channel function.
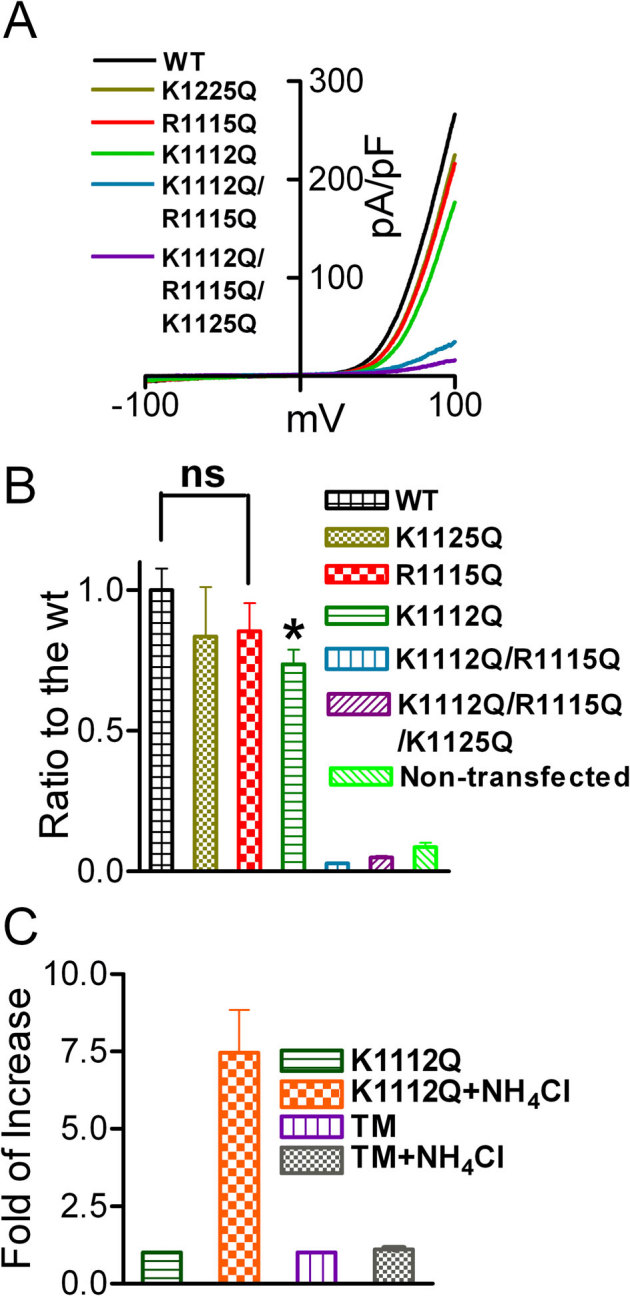
(A) Representative traces of WT TRPM7 and its mutants. The double mutant K1112Q/R1115Q (DM) and the triple mutant K1112Q/R1115Q/K1125Q did not produce currents. (B) Mean current densities of the WT and its mutants (n = 8–13, *p<0.05). (C) Effects of NH4Cl on K1112Q, and triple mutant K1112Q/R1115Q/K1125Q (TM) were tested by using pipette solution containing 1 mM free Mg2+. Perfusion of 30 mM NH4Cl can increase intracellular pH thereby releasing PIP2 sequestered by Mg2+, thus, the effects of PIP2 on the channel activity is manifested. NH4Cl significantly increase the current amplitude of K1112Q, but failed to induce any change in the DM and TM transfected cells, indicating that the double mutant K1112Q/R1115Q and the triple mutant K1112Q/R1115Q/K1125Q have no ability to sense the changes of PIP2 levels.
Since PIP2 is the only activator for TRPM7, to further confirm that double mutant and the triple mutant of TRPM7 are indeed loss-of-function mutants, we tested the effects of NH4Cl on these mutants. Perfusion of NH4Cl has been proposed to increase intracellular pH thereby releasing PIP2 sequestered by other cations such as H+, Mg2+, or other polyvalent cations46. For these experiments, we included 1 mM free Mg2+ in the pipette solution. As shown in Fig. 7C, after perfusion with 30 mM NH4Cl, K1112Q current amplitude was increased by 5-fold; whereas the triple mutant had no response to NH4Cl, suggesting that the double mutant (data not shown) and the triple mutant are nonfunctional mutants.
PIP2 depletion abolishes Mg2+ currents and Mg2+ influx through TRPM6
Next we examined whether PIP2 depletion affects Mg2+ influx through TRPM6. As shown in Fig. 8A, Mg2+ currents recorded using isotonic Mg2+ external solution were significantly increased by 200 μM 2-APB. After depletion of PIP2 in cells co-transfected with TRPM6 and Ci-VSP, the Mg2+ current was fully inhibited (Fig. 8A–B). Similar results were observed using CCh and GTPγs to delete PIP2 (data not shown). We further measured Mg2+ influx through TRPM6 before and after PIP2 depletion. The membrane permeable Mg2+ indicator KMG104-AM was used to measure changes of intracellular Mg2+ induced by perfusing cells with isotonic Mg2+ solution. As shown in Fig. 8 C, significant increase in the normalized fluorescence intensity (F/F0) was induced by perfusion of isotonic Mg2+ in TRPM6-transfected cells in comparison with the non-transfected cells. Moreover, the change of Mg2+ influx was largely diminished in the cells perfused with 200 μM CCh in DVF and isotonic Mg2+ solutions to hydrolyze PIP2. These results indicate that hydrolysis of PIP2 can induce TRPM6 inactivation and abolish TRPM6 mediated Mg2+ influx, suggesting that activation of PLC by Gq-linked receptors may play an pivotal role in regulating Mg2+ homeostasis by controlling TRPM6 channel activity.
Figure 8. Depletion of PIP2 inhibits Mg2+ currents and eliminates Mg2+ influx through TRPM6.
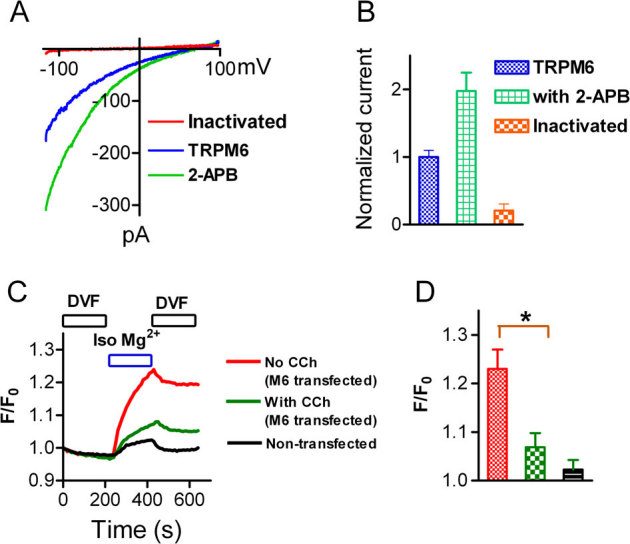
(A) Mg2+ currents recorded using isotonic extracellular Mg2+ in cells transfected with either TRPM6 alone (blue) or TRPM6+Ci-VSP (red). Mg2+ current (blue) was significantly increased by 2-APB (green), and completely inhibited after PIP2 depletion induced by activation of Ci-VSP. (B) Normalized Mg2+ current amplitude (n = 9). (C) Mg2+ influx through TRPM6 under control conditions and after PIP2 hydrolysis by 200 μM CCh. Changes of Mg2+ influx were measured in isotonic Mg2+ extracellular solution. Note the normalized fluorescence intensity (F/F0) induced by isotonic Mg2+ was largely diminished by CCH. (D) Average changes of F/F0 (n = 33∼36 in each group).
TRPM6 kinase domain interacts with PLC, but the interaction is not required for PLC induced TRPM6 inactivation
As shown in previous studies, the TRPM7 channel kinase domain interacts with the C2 domain of PLC, especially PLC-β2 isoform34. Since TRPM6 and TRPM7 share high homology, we tested whether the TRPM6 kinase domain binds to the PLC isoforms. As shown in sFig. 9, the TRPM6 kinase domain primarily binds to the PLC β isoforms, including PLCβ1-3. Unlike GST-M7 KIN, GST-M6 KIN did not pull-down the PLC-δ1. This result suggests that the kinase domain of TRPM6 and TRPM7 preferentially interact with various PLC isoforms.
To understand whether interaction of the kinase domain with PLC is necessary for PLC activation induced inhibition of TRPM6 and TRPM7, we made truncation mutants of TRPM6 (M6-Δkinase) and TRPM7 (M6-Δkinase) by deleting the kinase domain at the C-terminus. Currents recorded from M6-Δkinase were verified by response to 2-APB (sFig. 9 C). Since we have shown that GTPγS inactivates TRPM6 mainly through activation of PLC pathway, we used GTPγS to activate PLC. As shown in sFig. 9 C–F, inclusion of GTPγS in the pipette solution markedly inhibited M6-Δkinase and M7-Δkinase currents, indicating that the interaction between kinase domain and the PLC is not necessary for the PLC activation induced TRPM6 and TRPM7 channel inactivation. Similar results were also obtained using CCh to induce inactivation of M6-Δkinase and M7-Δkinase expressed in the HM1 cells. CCh (200 μM) completely inhibited M6-Δkinase and M7-Δkinase currents (n = 5 for each group).
Discussion
We reported several new findings here. First, we demonstrated that PIP2 is necessary and sufficient for TRPM6 activation. Second, we identified that the positively charged residues in the TRP domain are likely to participate in TRPM6 and PIP2 interactions. Third, we found that TRPM6 interacts with PLC isoforms, but the interaction is not necessary for PLC induced TRPM6 or TRPM7 inactivation. Fourth, inactivation of TRPM6 by PIP2 depletion eliminates Mg2+ currents and Mg2+ influx, suggesting that PIP2 regulates Mg2+ homeostasis by controlling TRPM6 gating. Our results indicate that regulation of TRPM6 by PIP2 through PLC activation pathway may serve as an important pathway to gate TRPM6 and control Mg2+ homeostasis.
TRPM6 plays a vital role in Mg2+ homeostasis, as demonstrated by the mutations of TRPM6 that cause HSH9,10. Although it has been reported that TRPM6 expression can be regulated by various factors, including estrogen26,47, AngII27, acidosis/alkalosis conditions28, immunosuppressant tacrolimus29, diuretics Thiazide30, and EGF31, how TRPM6 is gated has remained elusive. Here we provided several lines of evidence demonstrating that TRPM6 gating is controlled by cellular PIP2 levels. Activation of PLC-coupled M1 receptor by CCh completely suppressed TRPM6 current amplitude (Fig. 1). This result is supported by pharmacological activation of PLC induced suppression of TRPM6 (Fig. 1E–F). Using the recently developed approach for the study of phosphoinositide signaling, which is based on the rapamycin-induced heterodimerization of the rapamycin (Rap) binding domain of mTOR (FRB) and FKBP12 (Fig. 2)38,39, we showed that application of Rap rapidly and fully inactivated TRPM6 channel activity (Fig. 2). Rap induced translocation of over-expressed type-IV 5-phosphatase (5-Ptase) specifically hydrolyzes PIP2 without involvement of other signaling factors. Thus, Rap induced inactivation of TRPM6 strongly indicates that PIP2 is required for TRPM6 channel function. This conclusion was further supported by the fact that TRPM6 can be inactivated by Ci-VSP elicited PIP2 depletion (Fig. 3). Furthermore, application of PIP2 in excised patches can reactivate TRPM6 after rundown (Fig. 6). Thus, it appears that PIP2 is necessary and sufficiently for TRPM6 channel activity.
PIP2 regulates a large number of ion channel functions by interacting with specific residues or domains43. However, due to the lack of crystal structure information of most of ion channels, putative PIP2 interacting sites are usually predicted based on direct binding experiments with PIP2, or changes of PIP2 affinity evaluated by channel functions between Wide-Type and mutants43. Positively charged residues at both N- and C-termini have been shown to be putative PIP2 interacting domains43,48,49,50. Rohacs and colleagues demonstrate that the basic residues within the TRP domain are likely the putative PIP2 binding sites for TRPM8, TRPV5 and TRPM544. Nilius and colleagues found that a PH-like domain at the more distal C-terminal region of TRPM4 may be involved in PIP2-dependent regulation51. We mutated the conserved basic residues at the TRP domain of TRPM6, which are equivalent to the putative PIP2-binding sites of TRPM844. The mutants K1085Q and R1088Q displayed largely diminished channel activity, whereas K1098Q and the double mutant K1085Q/R1088Q completely lost channel function. Since PIP2 is the only activator of TRPM6, the decreased channel function of the mutants suggests that the mutations disrupt TRPM6 activation by PIP2. Moreover, it seems that mutant R1088Q has reduced activity by PIP2, as evidenced that PIP2 at 10 μM recovered channel activity by 80% of WT, but only 10% of R1088 (Fig. 6). Mutation of the conserved residues of TRPM7 also markedly reduced channel activity. Therefore, it is likely that the basic residues at the TRP domain may participate in channel-PIP2 interactions. Further studies are required to confirm whether the positively charged residues at the TRP domain are PIP2 binding sites. Excitingly, Hansen and colleagues have recently discovered the structure basis of interaction between PIP2 and Kir2.252. Putative PIP2 binding residues at the cytoplasmic domain predicted by previous mutagenesis studies43 are proven to interact with PIP2. A conserved non-specific phospholipid binding region is also shown to constitute the PIP2-binding interface with the cyloplasmic domain for PIP2 binding. Thus, it is conceivable that the predicted PIP2 binding residues at the TRP domain may be proven to be the PIP2 binding sites in the future.
Like TRPM734, TRPM6 kinase domain interacts with different PLC isoforms (sFig. 9). However, we found that the interaction of M6-KIN and M7-KIN with PLCs is not required for modulation of TRPM6 and TRPM7 by PLC activation, as evidenced by the fact that the truncation mutants of TRPM6 and TRPM7 without the kinase domain can also be inactivated by PLC activation (sFig. 9). Although it is not necessary for PLC-induced regulation on TRPM6 and TRPM7, given the different distribution profiles of the PLC isoforms53,54, the interaction or co-localization of TRPM6 and TRPM7 with different isoforms of PLC may underlie different, yet to be determined physiological/pathological functions.
Our results seem to indicate that TRPM6 is exclusively gated by PIP2. The loss-of-function mutants of putative PIP2 binding sites cannot be potentiated by 2-APB (Fig 4). Similarly, inactivated TRPM6 by completely deletion of PIP2 has no response to 2-APB (sFig. 8). PIP2 may also be the exclusive activator of TRPM734. The important role of PIP2 on TRPM7 activation was demonstrated in 200234, and the conclusion was supported by other studies46,55. However, Takezawa and colleagues reported that activation of PLC through endogenous M1-muscarinic receptors has little effect on TRPM7 over-expressed in HEK cells56. Rather, they suggested that cAMP enhances TRPM7 activity. It was reported later that failure of inhibition of TRPM7 by CCh using endogenous M1 receptor56 was probably due to the minor PIP2 breakdown by limited endogenous M1 receptor57. The effects of cAMP on TRPM7 activation56 was not observed by Langeslag and colleagues57. Indeed, Langeslag and colleagues elegantly demonstrated a positive correlation between TRPM7 channel activity and PIP2 level57, in agreement with the results that TRPM7 activity requires PIP2 as shown in Fig. 7 and as previous reported34. However, under perforated-patch configuration, Langeslag and colleagues observed that Bradykinin induced a transient inhibition of TRPM7 currents57, because PIP2 levels were quickly recovered in N1E-115 cells. They therefore suggested that PLC-coupled agonists activates TRPM757. Although it was unclear why PIP2 levels were quickly recovered under the perforated-patch configuration57, it is plausible that other signaling pathways were involved in PIP2 synthesis under their experimental conditions. Nevertheless, using perforated patch configuration, we demonstrated that activation of the voltage-sensitive Ci-VSP by depolarization can completely inhibit both TRPM6 (sFig. 6) and TRPM7 (sFig. 7) channel activities. Therefore, it seems that PIP2 is a strong and exclusive activator of TRPM6 and TRPM7.
Mg2+ permeation through TRPM6 was substantially enhanced when TRPM6 channel activity was increased by 2-APB. However, after depletion of PIP2 by CCh, Mg2+ current and Mg2+ influx were largely blocked (Fig. 8). This PLC-activation induced Mg2+ regulation may produce physiological/pathological significance. In the distal convoluted tubule (DCT) where TRPM6 is highly expressed, Ca2+/Mg2+-sensing receptors (CaSR) are also abundantly expressed58. Stimulation of CaSR activates PLC, especially the β and γ isoforms59, which therefore may influence the magnesium re-absorption by regulation of TRPM6 channel function. Several peptide hormones such as PTH, calcitonin, glucagon, and AVP enhance magnesium reabsorption in the DCT60. Groenestege and colleagues very nicely showed that EGF regulates Mg2+ re-absorption by regulation of TRPM6 trafficking to the cell membrane37,47. A mutation of the EGF gene encoding pro-EGF causes renal Mg2+ wasting37,47. As EGF can also activate PLC-γ, leading to PIP2 depletion, there might a fine-tuned regulation regarding how EGF modulates TRPM6. Nonetheless, it will be of great interest to investigate whether and how the other Mg2+ regulating hormones may regulate the Mg2+ homeostasis through Mg2+ gate-keeper TRPM6.
In conclusion, we have demonstrated at various levels that PIP2 is required for TRPM6 channel activity, and that disruption of the putative PIP2 binding sites results in dysfunctional or non-functional TRPM6 channels. As TRPM6 is considered as a Mg2+ gate-keeper, understanding the critical role of PIP2 in TRPM6 channel function may facilitate our understanding of how Mg2+ contributes to physiological/pathological processes, as well as the development of therapeutic approaches for hypomagnesemia treatment.
Methods
Molecular biology
TRPM6 plasmid (pCINeo/IRES-GFP) was kindly provided by Dr. Joost G. J. Hoenderop. Mutations of TRPM6 and TRPM7 were made by using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) following the manufacture's instruction. The primers sequences will be available upon request.
Electrophysiology
Whole-cell and single-channel currents were recorded using an Axopatch 200B amplifier. Data were digitized at 5 or 10 kHz, and digitally filtered off-line at 1 kHz. Patch electrodes were pulled from borosilicate glass and fire-polished to a resistance of ∼3 MΩ when filled with internal solutions. Series resistance (Rs) was compensated up to 90% to reduce series resistance errors to <5 mV. Cells with Rs bigger than 10 MΩ were discarded21.
For whole cell current recordings, voltage stimuli lasting 250 ms were delivered at 1 s intervals, with either voltage ramps or voltage steps ranging from −120 to +100 mV. A fast perfusion system was used to exchange extracellular solutions, with complete solution exchange achieved in about 1 to 3 s32. The internal pipette solution for TRPM6 or TRPM7 whole cell current recordings contained (in mM) 145 Cs-methanesulfonate (CsSO3CH3), 8 NaCl, 10 EGTA, and 10 HEPES, with pH adjusted to 7.2 with CsOH. Ca2+ was adjusted to various concentrations based on calculation using MaxChelator32. The standard extracellular Tyrode's solution for whole cell recording contained (mM): 145 NaCl, 5 KCl, 2 CaCl2, 10 HEPES and 10 glucose; pH was adjusted to 7.4 with NaOH. Divalent-free solution (DVF) contained (mM) 145 NaCl, 20 HEPES, 5 EGTA, 2 EDTA and 10 glucose, with estimated free [Ca2+]< 1 nM and free [Mg2+] ≈10 nM at pH 7.4. MaxChelator was used to calculate free Ca2+ and free Mg2+ concentrations.
Single channel currents were recorded in inside-out patches. The extracellular DVF solution contained (mM) 145 Na-methanesulfonate (NaSO3CH3), 20 HEPES, 5 EGTA, 2 EDTA, and 10 glucose, with pH adjusted to 7.4; internal solution contained (mM) 145 CsSO3CH3, 8 NaCl, 10 EGTA, and 10 HEPES, with pH adjusted to 7.2.
Rapamycin induced PIP2 depletion
The two plasmids PM-FRB-CFP and mRFP-FKBP-5-ptase were kindly provided by Tamas Balla. The phosphatase dead mutant CF-InP-D281A was kindly provided by Dr. Thomas Mayer. PM-FRB-CFP and mRFP-FKBP-5-ptase were co-transfected with TRPM6 to the cells, and 10 μM rapamycin was applied to induce 5-ptase translocation to deplete PIP2. In the case of testing phosphatase dead mutant, PM-FRB-CFP and CF-InP-D281A were co-transfected with the channel to HEK293 cells.
Western blot
The HEK 293 cells transfected with TRPM6 were allowed 48 hours to express protein. Cell lysate was used to blot with anti-GFP antibody (Neuromab). The ECL chemiluminescence was detected and captured by FUJI LAS-3000 Imaging System.
Membrane protein detection
HEK 293T cells were transfected with WT TRPM6 (GFP-tagged) or mutants. After 48 h, cell membrane protein biotinylation was performed with Pierce Cell Surface Protein Isolation Kit (catalogue no. 89881). Briefly, monolayers of cells were washed with cold PBS and then incubated with Sulfo-NHS-SS-Biotin solution at 4°C for 30 min. After incubation, the reaction was quenched, and the cells were washed 3 times with TBS solution (50 mM tris, 150 mM Nacl, pH 7.6), followed by lysing procedure for 30 min on ice. After centrifugation for 10 min at 16,000 g, supernatant passed a sufficient amount of NeutrAvidin Agarose (Pierce Biotechnology), and the flow-through was kept as cytosol portion. Agarose beads with protein were washed 3 times and then boiled with 1X SDS buffer containing 50 mM DTT at 95°C, and supernatant containing released membrane protein was kept for analysis by Western blotting.
Ratio Ca2+ imaging experiments
Cells were plated on 25 mm glass coverslips loaded with 1 μM Fura-2 for 20 minutes. Non-incorporated dye was washed away using a HEPES-buffered Saline Solution (HBSS) containing (in mM) 20 HEPES, 10 glucose, 1.2 MgCl2, 1.2 KH2PO4, 4.7 KCl, 140 NaCl, 1.3 Ca2+ (pH 7.4). Ca2+ influx was measured by perfusing the cells with Tyrode's solution containing 20 mM Ca2+ after the cells were perfused with nominal Ca2+ and Mg2+ free solution for 2 minutes. Ionomycin (Iono) at 1 mM was applied as internal control. Fluorescence intensities at 510 nm with 340 nm and 380 nm excitation were collected at a rate of 1 Hz using CoolSNAP HQ2 (Photometrics) and data were analyzed using NIS-Elements (Nikon)21. The 340∶380 nm ratio in the presence of 20 mM Ca2+ was normalized to that of 1 μM Ionomycin (Iono) elicited Ca2+ signal.
Detection of Mg2+ influx
HM1 cells were cultured on poly-L-lysine-coated cover slips in DMEM-F12 medium at 37°C. Cells were then loaded with KMG104-AM dye at a final concentration of 20 μM for 30 min in standard tyrode solution without added magnesium. Tyrode solution contained: 135 mM NaCl, 4 mM KCl, 10 mM glucose, 10 mM HEPES, 2 mM CaCl2, pH 7.4. Fluorescence intensities at 510 nm with 488 nm excitation were collected very 10 s using CoolSNAP HQ2 (Photometrics) and data were analyzed using NIS-Elements (Nikon). F0 refers to the initial fluorescence recorded at t = 0 s. Mg2+ influx was measured by perfusing the cells with isotonic Mg2+ solution containing (mM) 120 MgCl2, 10 Hepes, 10 glucose, and pH was adjusted to 7.4.
GST-pulldown purification assay
HEK-293T cells in 10-cm dishes were transfected using LipofectAMINE 2000 (Life Technologies, Rockville, MD) with 10 μg expression vector containing either PLC-β1–4, PLC-γ1, or GFP–PLC-δ1 cDNAs. After 48 h, cells were lysed in 2 ml of RIPA buffer (50 mM Tris-HCl at pH 7.4, 150 mM sodium chloride, 1 mM EDTA, 1% IGEPAL CA-630, 0.5% (w/v) deoxycholate, 0.1% (w/v) SDS and 10 mM iodoacetamide). GST–TRPM6 kinase, GST-TRPM7 kinase or GST alone was expressed and purified with glutathione-4B Sepharose beads. Bound GST–kinases, GST or beads were incubated with lysates containing various PLCs for 12 h. Bound proteins were washed three times in RIPA buffer, eluted by boiling in 2×SDS–polyacrylamide gel electrophoresis (PAGE) sample buffer and then separated by SDS–PAGE. PLC-β1–4 and PLC-γ1 were detected by western blotting with the following specific anti-PLC antibodies from Santa Cruz Biotechnology (Santa Cruz, CA): PLCβ1(G-12), PLCβ2(Q-15), PLCβ3(C-20), PLCβ4(C-18), PLCγ1(E-12). An anti-GFP antibody (Clontech) was used to detect GFP-tagged PLC-δ1. A horseradish peroxidase-linked antibody to rabbit Ig (Amersham Pharmacia Biotech, Piscataway, NJ) and SuperSignal West Dura substrate was used for all chemiluminescent detection (Pierce, Rockford, IL).
Data analysis
Pooled data are presented as mean ± SEM. Dose–response curves were fitted by an equation of the form E = Emax{1/[1 + (EC50/C)n]}, where E is the effect at concentration C, Emax is maximal effect, EC50 is the concentration for half-maximal effect and n is the Hill coefficient33. Statistical comparisons were made using ANALYSIS of variance (ANOVA) and two-tailed t-test with Bonferroni correction. P < 0.05 indicated statistical significance.
Author Contributions
JX designed and carried out most of the experiments, analyzed data, and co-wrote the paper. BS, JD, and WY performed electrophysiological experiments. JO and HC carried out the pull-down experiments and created reagents. LR designed and supervised the pull-down experiments and advised some other experiments. LY conceived and supervised the work and co-wrote the paper.
Supplementary Material
Supplementary Figures_Legends
Acknowledgments
We thank Drs. Joost G. J. Hoenderop, Tamas Balla and Thomas Mayer for kindly providing TRPM6 plasmid (pCINeo/IRES-GFP vector), PM-FRB-CFP and mRFP-FKBP-5-ptase constructs, and the phosphatase dead mutant CF-InP-D281A plasmids. The Ci-VSP construct was kindly provided by Dr. Yasushi Okamura. We'd also like to thank Dr. Kotaro Oka for providing us with the Mg2+ indicator (KMG-104). This work was generously supported by the National Institutes of Health, National Heart, Lung and Blood Institute (NHLBI) (www.nhlbi.nih.gov, grant number 2R01HL078960) to LY, National Institute of General Medical Science (NIGMS) (www.nigms.nih.gov, grant number 1R01GM080753) to LR, and a bio-medical grant (grant number 2009-0099) from the Department of Public Health of Connecticut to LY.
References
- Romani A. A. CELLULAR MAGNESIUM HOMEOSTASIS. Archives of Biochemistry and Biophysics 512, 1–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quamme G. A. Molecular identification of ancient and modern mammalian magnesium transporters. Am J Physiol Cell Physiol 298, C407–429 (2010). [DOI] [PubMed] [Google Scholar]
- Touyz R. M. Magnesium in clinical medicine. Front Biosci 9, 1278–1293 (2004). [DOI] [PubMed] [Google Scholar]
- Goytain A. & Quamme G. A. Identification and characterization of a novel mammalian Mg2+ transporter with channel-like properties. BMC Genomics 6, 48 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. & Clapham D. E. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci U S A 106, 15750–15755 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni J., Nelson B. & Scharenberg A. M. SLC41A2 encodes a plasma-membrane Mg2+ transporter. Biochem J 401, 505–513 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goytain A. & Quamme G. A. Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol. Genomics 22, 382–389 (2005). [DOI] [PubMed] [Google Scholar]
- Goytain A. & Quamme G. A. Functional characterization of human SLC41A1, a Mg2+ transporter with similarity to prokaryotic MgtE Mg2+ transporters. Physiol Genomics 21, 337–342 (2005). [DOI] [PubMed] [Google Scholar]
- Schlingmann K. P. et al.. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet 31, 166–170 (2002). [DOI] [PubMed] [Google Scholar]
- Walder R. Y. et al.. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet 31, 171–174 (2002). [DOI] [PubMed] [Google Scholar]
- Schmitz C. et al.. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114, 191–200 (2003). [DOI] [PubMed] [Google Scholar]
- Jin J. et al.. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazanova L. V. et al.. TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat Commun 1, 109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler M. J. et al.. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 411, 590–595 (2001). [DOI] [PubMed] [Google Scholar]
- Runnels L. W., Yue L. & Clapham D. E. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291, 1043–1047 (2001). [DOI] [PubMed] [Google Scholar]
- Chubanov V. et al.. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A 101, 2894–2899 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C. et al.. The Channel Kinases TRPM6 and TRPM7 Are Functionally Nonredundant. J. Biol. Chem. 280, 37763–37771 (2005). [DOI] [PubMed] [Google Scholar]
- Voets T. et al.. TRPM6 Forms the Mg2+ Influx Channel Involved in Intestinal and Renal Mg2+ Absorption. J. Biol. Chem. 279, 19–25 (2004). [DOI] [PubMed] [Google Scholar]
- Liu W. et al.. TRPM7 regulates gastrulation during vertebrate embryogenesis. Dev Biol 350, 348–357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarts M. et al.. A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877 (2003). [DOI] [PubMed] [Google Scholar]
- Du J. et al.. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res 106, 992–1003 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni J., Tamura R., Sweet I. R. & Scharenberg A. M. TRPM7 regulates quiescent/proliferative metabolic transitions in lymphocytes. Cell Cycle 9, 3565–3574 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill M. S. et al.. Cell death of melanophores in zebrafish trpm7 mutant embryos depends on melanin synthesis. J Invest Dermatol 127, 2020–2030 (2007). [DOI] [PubMed] [Google Scholar]
- Elizondo M. R. et al.. Defective skeletogenesis with kidney stone formation in dwarf zebrafish mutant for trpm7. Curr Biol 15, 667–671 (2005). [DOI] [PubMed] [Google Scholar]
- Schlingmann K. P., Waldegger S., Konrad M., Chubanov V. & Gudermann T. TRPM6 and TRPM7--Gatekeepers of human magnesium metabolism. Biochim Biophys Acta 1772, 813–821 (2007). [DOI] [PubMed] [Google Scholar]
- van der Wijst J., Hoenderop J. G. & Bindels R. J. Epithelial Mg2+ channel TRPM6: insight into the molecular regulation. Magnes Res 22, 127–132 (2009). [PubMed] [Google Scholar]
- Touyz R. M. et al.. Differential Regulation of TRPM6/7 Cation Channels by Ang II in Vascular Smooth Muscle Cells from Spontaneously Hypertensive Rats. Am J Physiol Regul Integr Comp Physiol 290, R73–78 (2005). [DOI] [PubMed] [Google Scholar]
- Nijenhuis T., Renkema K. Y., Hoenderop J. G. J. & Bindels R. J. M. Acid-Base Status Determines the Renal Expression of Ca2+ and Mg2+ Transport Proteins. J Am Soc Nephrol 17, 617-626 (2006). [DOI] [PubMed] [Google Scholar]
- Nijenhuis T., Hoenderop J. G. & Bindels R. J. Downregulation of Ca(2+) and Mg(2+) transport proteins in the kidney explains tacrolimus (FK506)-induced hypercalciuria and hypomagnesemia. J Am Soc Nephrol 15, 549–557 (2004). [DOI] [PubMed] [Google Scholar]
- Nijenhuis T. et al.. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115, 1651–1658 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenestege W. M. et al.. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest 117, 2260–2267 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Li M. & Yue L. Potentiation of TRPM7 Inward Currents by Protons. J. Gen. Physiol. 126, 137–150 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Jiang J. & Yue L. Functional Characterization of Homo- and Heteromeric Channel Kinases TRPM6 and TRPM7. J Gen Physiol 127, 525–537 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runnels L. W., Yue L. & Clapham D. E. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol 4, 329–336 (2002). [DOI] [PubMed] [Google Scholar]
- Horowitz L. F. et al.. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol 126, 243–262 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al.. Molecular Determinants of Mg2+ and Ca2+ Permeability and pH Sensitivity in TRPM6 and TRPM7. J. Biol. Chem. 282, 25817–25830 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thebault S., Alexander R. T., Tiel Groenestege W. M., Hoenderop J. G. & Bindels R. J. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol 20, 78–85 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B. C., Inoue T., Meyer T. & Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314, 1454–1457 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P., Thyagarajan B., Rohacs T. & Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J Cell Biol 175, 377–382 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y., Iwasaki H., Sasaki M., Inaba K. & Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243 (2005). [DOI] [PubMed] [Google Scholar]
- Murata Y. & Okamura Y. Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J Physiol 583, 875–889 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenburger B. H., Jensen J. B. & Hille B. Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol 135, 99–114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B. C. & Hille B. PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 37, 175–195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs T., Lopes C. M., Michailidis I. & Logothetis D. E. PI(4,5)P(2) regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci 8, 626–634 (2005). [DOI] [PubMed] [Google Scholar]
- Thebault S. et al.. Role of the alpha-kinase domain in transient receptor potential melastatin 6 channel and regulation by intracellular ATP. J Biol Chem 283, 19999–20007 (2008). [DOI] [PubMed] [Google Scholar]
- Kozak J. A., Matsushita M., Nairn A. C. & Cahalan M. D. Charge Screening by Internal pH and Polyvalent Cations as a Mechanism for Activation, Inhibition, and Rundown of TRPM7/MIC Channels. J Gen Physiol 126, 499–514 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenestege W. M., Hoenderop J. G., van den Heuvel L., Knoers N. & Bindels R. J. The epithelial Mg2+ channel transient receptor potential melastatin 6 is regulated by dietary Mg2+ content and estrogens. J Am Soc Nephrol 17, 1035–1043 (2006). [DOI] [PubMed] [Google Scholar]
- Dong X. P. et al.. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun 1, 38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B., Owsianik G. & Voets T. Transient receptor potential channels meet phosphoinositides. EMBO J 27, 2809–2816 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs T. Regulation of TRP channels by PIP(2). Pflugers Arch 453, 753–762 (2007). [DOI] [PubMed] [Google Scholar]
- Nilius B. et al.. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J 25, 467–478 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S. B., Tao X. & Mackinnon R. Structural basis of PIP(2) activation of the classical inward rectifier K(+) channel Kir2.2. Nature (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea J. P., Ertoy D., Hollis J. L., Marrero M. B. & Sands J. M. Immunolocalization of phospholipase C isoforms in rat kidney. Kidney Int 54, 1484–1490 (1998). [DOI] [PubMed] [Google Scholar]
- Rebecchi M. J. & Pentyala S. N. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev 80, 1291–1335 (2000). [DOI] [PubMed] [Google Scholar]
- Gwanyanya A., Sipido K. R., Vereecke J. & Mubagwa K. ATP and PIP2 dependence of the magnesium-inhibited, TRPM7-like cation channel in cardiac myocytes. Am J Physiol Cell Physiol 291, C627–635 (2006). [DOI] [PubMed] [Google Scholar]
- Takezawa R. et al.. Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain. Proc Natl Acad Sci U S A 101, 6009–6014 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeslag M., Clark K., Moolenaar W. H., van Leeuwen F. N. & Jalink K. Activation of TRPM7 channels by phospholipase C-coupled receptor agonists. J Biol Chem 282, 232–239 (2007). [DOI] [PubMed] [Google Scholar]
- Houillier P. & Paillard M. Calcium-sensing receptor and renal cation handling. Nephrol Dial Transplant 18, 2467–2470 (2003). [DOI] [PubMed] [Google Scholar]
- Godwin S. L. & Soltoff S. P. Calcium-sensing receptor-mediated activation of phospholipase C-gamma1 is downstream of phospholipase C-beta and protein kinase C in MC3T3-E1 osteoblasts. Bone 30, 559–566 (2002). [DOI] [PubMed] [Google Scholar]
- Cole D. E. C. & Quamme G. A. Inherited Disorders of Renal Magnesium Handling. Journal of the American Society of Nephrology 11, 1937–1947 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures_Legends