

Abstract
Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, with a poor response to chemotherapy and low survival rate. This unfavorable treatment response is likely to derive from both late diagnosis and from complex, incompletely understood biology, and heterogeneity among NSCLC subtypes. To define the relative contributions of major cellular pathways to the biogenesis of NSCLC and highlight major differences between NSCLC subtypes, we studied the molecular signatures of lung adenocarcinoma (ADC) and squamous cell carcinoma (SCC), based on analysis of gene expression and comparison of tumor samples with normal lung tissue. Our results suggest the existence of specific molecular networks and subtype-specific differences between lung ADC and SCC subtypes, mostly found in cell cycle, DNA repair, and metabolic pathways. However, we also observed similarities across major gene interaction networks and pathways in ADC and SCC. These data provide a new insight into the biology of ADC and SCC and can be used to explore novel therapeutic interventions in lung cancer chemoprevention and treatment.
Keywords: NSCLC, adenocarcinoma, squamous cell carcinoma, molecular signature, gene expression, pathway
Introduction
Worldwide, over 1.3 million people are diagnosed each year with lung cancer, with over 1.1 million deaths [1, 2]. Lung cancer is the most common global cause of cancer death in men and second only to breast cancer in women (17.6% of cancer-related deaths in both sexes) [1-3]. There is a high fatality rate with this disease, with only 15% of patients still alive 5 years after diagnosis [4]. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for 85% of cases, and can be divided into three main subgroups: adenocarcinoma (ADC; 30-50% of cases), squamous cell carcinoma (SCC; ∼30%), and large cell carcinoma (LCC; ∼10%), according to the predominant morphology of the tumor cells as determined by light microscopy [4-7]. NSCLC is associated with high rates of proliferation and metastases, as well as poor prognosis for advanced-stage disease compared with other cancers. There are several potential explanations for the disparity between lung cancer survival and other common tumors, including late detection and histologic heterogeneity. This heterogeneity is reflected by the fact that the majority of prostate, breast, and colorectal carcinomas are ADC, while only 30% of NSCLC cases are of this subtype [6, 8]. For the most part, and until recently, NSCLC subtypes (SCC, LCC, and ADC) were treated similarly, regardless of the biologic heterogeneity associated with histology. It is likely that poor historic lung cancer response rates may be attributable in part to a relatively homogenous approach to treat a heterogeneous disease. Therefore, a better understanding and molecular characterization of the NSCLC subtypes could contribute to improved design of treatment schedules and management of lung cancer.
Gene expression profiling using microarrays is a robust and straightforward way to study the molecular features of different types and subtypes of cancer at a systems level. Although it is possible to distinguish the different NSCLC subgroups histologically, genomic profiling demonstrates that there are significant and distinct differences in the molecular signature associated with each subtype. The objective of this study was to enhance the understanding of NSCLC pathogenesis through characterization of molecular and pathway signatures in primary lung ADC and SCC samples, and to compare these signatures with those of normal lung tissue.
Materials and methods
Isolation of RNA
Sets of syngeneic normal and tumor samples (lung ADC and SCC) were obtained from Cure-line Biobank (Cureline Inc., San Francisco, CA). Eighty formalin-fixed, paraffin-embedded (FFPE) tissue samples from 20 patients with NSCLC were analyzed (20 FFPE tissue samples for each group). For RNA isolation, five 10-μm sections were sliced via microtome, placed in 1.7 ml tubes, and deparaffinized with 1 ml of xylene (EMD Chemicals, Gibbstown, NJ). The samples were digested for 16 hours with Proteinase K and total RNA isolation was performed using the Epicentre Biotechnologies MasterPure™ Complete DNA and RNA Purification Kit (Epicentre Biotechnologies, Madison, Wl) following the manufacturer's instructions. All samples were subsequently treated with RNase-free DNase I (Ambion, Austin, TX).
Synthesis of cDNA, amplification, and labeling
RNA samples were amplified and converted to cDNA using the NuGEN WT-Ovation FFPE v2 RNA Amplification System (NuGEN Technologies, San Carlos, CA) [9, 10]. Briefly, 50 ng of RNA was reverse transcribed to antisense-cDNA, amplified using kit reagents, and purified using a QIAGEN PCR purification kit (QIAGEN, Valencia, CA). DNA concentration was assessed using a Nanodrop ND-100 spectrophotometer. Sense transcript cDNA (ST-cDNA) was generated from 2-4 μg of purified antisense-cDNA using the kit reagents according to manufacturer's instructions. ST-cDNA was purified by QIAGEN kit and final DNA concentration determined by absorption. Up to 5 μg of purified ST-cDNA was fragmented and biotin-labeled using the NuGEN Encore Biotin Module Kit (NuGEN Technologies, San Carlos, CA).
Hybridization, washing, and analysis
Biotin-labeled cDNA from each sample was directly hybridized to GeneChip Human Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA) along with GeneChip Eukaryotic Hybridization controls (Affymetrix, Santa Clara, CA). Samples were incubated at 45°C in an Affymetrix hybridization oven 640 at 60 rpm for 16 hours, and washed with an Affymetrix GeneChip Fluidics Station 450 according to the manufacturer's specifications. Scanning was performed using the Affymetrix GeneChip 7G scanner using manufacturer-recommended default settings.
Data analysis
An Affymetrix Expression Console was used to generate quality control parameters, process probe intensity files and CEL-format data files, and to normalize and summarize a gene expression measurement for each probe set on the array through a Robust Multiarray Averaging (RMA) algorithm [11]. All the clinical and control samples in the study and the expression values were log-transformed with a base of 2 for downstream data analysis. For each individual sample, differential expression profiles of cancer versus normal syngeneic tissue were calculated. In addition, differential profiles of all cancer samples versus all normal breast samples were calculated using an unpaired t-test.
All gene expression analyses were performed in Pathway Studio 7 (Ariadne Genomics, Inc., Rockville, MD) [9, 11-15], using the ResNet 7 database (Ariadne Genomics, Inc.) [9, 10]. Enrichment analysis in Pathway Studio 7 was performed by Gene Set Enrichment Analysis (GSEA) [16] and Sub-Network Enrichment Analysis (SNEA) algorithms (Supplementary Figure 1S) [11]. Functional enrichment was performed using Fisher's Exact Test.
SNEA enrichment in Pathway Studio was calculated using the Mann-Whitney test, a non-parametric method that compares the medians of non-normal distributions X and Y. Both samples (having sizes N and M) are combined into one array in ascending order with each element then replaced by its rank in the array from 1 to N+M. The ranks of the first sample elements are summarized and a Mann-Whitney U-value calculated from Equation 1:
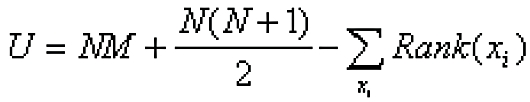 |
Eq. 1 |
If U is close to the mean of U (i.e. 0.5NM) then the medians of X and Y are similar. The significance level of the U statistic can be derived from the distribution quantiles. When applied to gene expression data, two distributions are typically derived from the gene set or sub-network, and from the entire gene expression profile measured on the chip. The following steps describe the computational steps performed by the SNEA algorithm.
Preparation of sub-networks
SNEA was used to build sub-networks from the relationships in a database based on criteria specified by the user. A central ‘seed’ is initially created from all relevant entities in the database, and associated entities retrieved based on their relationship with the seed (binding partners, expression targets, protein modification targets, etc.).
Calculation of background distribution
This algorithm was used to calculate a background distribution of all expression values for the selected sample in the experiment, typically from a differential measurement such as that resulting from the ‘Find Differentially Expressed Genes’ tool.
Calculation of sub-network distribution
This algorithm was used to create a ‘subnetwork’ distribution of the expression values in a similar manner for all sub-networks constructed in the previous step. More importantly, during distribution calculation, the expression value for each entity connected to a seed is accounted for as many times as the connectivity of that entity in ResNet. The purpose of this correction is to correct the bias introduced by the different connectivity of entities in ResNet.
Statistical comparison of sub-network distribution with background distribution
This algorithm is used to compare the subnetwork distribution with the background distribution using a one-sided Mann-Whitney U-test, and calculates a p-value indicating the statistical significance of difference between two distributions. Presentation and prioritization of results were done with Pathway Studio, which presents the ‘seed’ entity for each sub-network along with the sub-networks themselves in the user interface, ranked from the lowest (best) to the highest (worst) p-value. Note: the percentage overlap is also presented in order to provide an adequate measurement of significance and confidence in various statistical tests of overlap.
Analysis of key regulators of differential gene expression
The key regulators of differential response are those components of signal transduction pathways and expression regulators that are most likely to be involved in the regulation of genes differentially expressed between tumor and normal samples. Such key regulatory signaling pathways are assumed to be deregulated (e.g. abnormally activated or suppressed) in a disease state and provide insights into the mechanisms and molecular features of a disease. Our analysis implements a proprietary SNEA algorithm, which utilizes a gene expression regulatory network built from facts extracted from the literature. The network is used to generate a comprehensive collection of gene sets, each representing immediate downstream neighbors of each individual protein in the network. It is assumed that if the downstream expression targets of the central seed protein are enriched with differentially expressed genes (i.e. the subnetwork is found to be statistically significant in enrichment analysis), then the seed protein is one of the key regulators of the observed differential response. As sub-networks are constructed from all the proteins in the entire expression network, including ligands, receptors, signaling proteins, and transcription factors, the seed proteins of statistically significant subnetworks presumably constitute the components of a regulatory network involved in the modulation of the observed differential response.
Results
Significant regulators of ADC and SCC
We focused our analysis on the regulation of major cellular pathways. The key regulators of differential response were identified by searching for all expression sub-networks in the Res-Net 7 database enriched with differentially changed genes using a Mann-Whitney test with a p-value cut-off of 0.001. We performed analyses separately for lung SCC and ADC, and all identified significant regulators are shown in Table 1. We found that transcription factors associated with differential expression in both ADC and SCC, in comparison with normal lung tissue, were representatives of the E2F family (E2F3 and E2F4). The connective tissue growth factor (CTGF) and platelet-derived growth factor (PDGF) pathways are also significantly changed in both subtypes of NSCLC. The retinoblastoma (Rb1) pathway is up-regulated specifically in ADC, whereas the epidermal growth factor (EGF) pathway is significantly affected (down-regulated) only in SCC. Lung ADC samples show significant and specific down-regulation of the miR-200 molecular network and the epithelial membrane protein 2 (EMP2). The miR-200 family of microRNAs plays a major role in specifying the epithelial phenotype by preventing expression of the transcription repressors ZEB1/deltaEF1 and SIP1/ZEB2, and regulates epithelial-mesenchymal transition [17]. EMP2 controls surface levels of several classes of integrin and other cell-interaction molecules, and their trafficking to glycolipid-enriched lipid raft domains is important in receptor signaling [18]. Lung SCC samples show down-regulation of chemokine (C-X3-C motif) ligand 1 (CX3CL1), interleukin-1 family member 8 (IL1F8), and protein kinase C beta (PRKCB) pathways.
Table 1.
Significant regulators in lung adenocarcinoma (ADC) and squamous cell carcinoma (SCC) by SNEA (Mann-Whitney p<0.001)
| Name | Sub-network size | Median fold change | p-value |
|---|---|---|---|
| Adenocarcinoma | |||
| CTGF | 47 | 1.02072 | 8.98 × 10-5 |
| S100A4 | 15 | 1.28985 | 1.52 × 10-4 |
| Vitronectin receptor | 21 | 1.14296 | 2.38 × 10-4 |
| E2F3 | 34 | 1.20872 | 3.45 × 10-4 |
| PDGF | 285 | 1.01231 | 4.48 × 10-4 |
| miR-200 | 5 | -1.52286 | 4.74 × 10-4 |
| E2F4 | 40 | 1.18259 | 8.11 × 10-4 |
| EMP2 | 5 | -1.48652 | 8.50 × 10-4 |
| RB1 | 75 | 1.10424 | 8.61 × 10-4 |
| Squamous cell carcinoma | |||
| PDGF | 285 | -1.03133 | 3.05 × 10-5 |
| EGF | 393 | -1.01802 | 3.33 × 10-5 |
| CTGF | 47 | -1.03683 | 2.61 × 10-4 |
| CX3CL1 | 11 | -1.45216 | 4.38 × 10-4 |
| E2F4 | 40 | 1.3792 | 4.98 × 10-4 |
| IL1F8 | 34 | -1.11308 | 5.47 × 10-4 |
| E2F3 | 34 | 1.3792 | 8.09 × 10-4 |
| PRKCB | 33 | -1.17599 | 8.97 × 10-4 |
| COMP | 9 | -1.45216 | 9.25 × 10-4 |
DNA repair, cell proliferation, and apoptotic pathways
DNA damage repair is a complex and multifaceted process that is critical to cancer cell survival and response to DNA-damaging chemotherapy. To define the relative contributions of DNA repair to ADC and SCC, we investigated the differential changes in all known DNA repair pathways. The record of genes and proteins involved in the regulation of DNA repair pathways were assembled using the ResNet 7 database and then verified as described previously [19]. Figures 1, 2, and 3 show graphically the changes in differential gene expression between all lung cancer samples and normal tissue among DNA-repair pathways, cell-cycle pathways, and apoptosis pathways, respectively, and defined by differential changes of the pathway proteins. Individual gene expression differences and p-values for components of each of these pathway types are provided in Supplementary Tables S1, S2, and S3, respectively.
Figure 1.

Gene expression changes in DNA repair pathways.
Figure 2.

Gene expression changes in cell cycle pathways.
Figure 3.

Gene expression changes in apoptotic pathways.
DNA repair components significantly up-regulated (2-fold or more with p<0.001) in lung SCC included retinoblastoma-binding protein 8 (RBBP8), protein kinase DNA-activated catalytic polypeptide (PRKDC), split hand/foot malformation (ectrodactyly) Type 1 (SHFM1), and proliferating cell nuclear antigen (PCNA). By contrast, there were no significant changes in expression of DNA-repair genes in lung ADC.
Cell-cycle genes were more significantly up-regulated in SCC than in ADC. Topoisomerase (DNA) II alpha (170 kD) (TOP2A), cyclin B1 (CCNB1), maternal embryonic leucine-zipper kinase (MELK), mitotic arrest deficient-like 2 (yeast), human homolog like-1 (MAD2L1), stratifin (SFN), cell division cycle 6 homolog (CDC6), abnormal spindle-like microcephaly-associated protein (ASPM), and DNA topoisomerase binding protein 1 (TOPBP1) were all significantly up-regulated in SCC. By contrast, only TOP2A was significantly up-regulated in ADC.
Among apoptotic pathways, CASP8 and FADD-like apoptosis regulator (CFLAR) was significantly down-regulated (-2.34) in SCC. No other apoptotic proteins were significantly changed in any of the ADC or SCC samples.
The SNEA approach was also applied to identify key regulators and detect global cell proliferation processes significantly affected in NSCLC.
In this approach, sub-networks were built around each cell process in the ResNet 7 database and contained all proteins known to be involved in the regulation of the process (SNEA was applied with a Mann-Whitney p-value cut-off value of 0.001). Significantly affected processes are documented in Table 2. Most of the significantly affected processes in both lung cancer subtypes are related to cellular proliferation (spindle assembly, chromosome segregation, cytokinesis, kinetochore assembly, mitotic checkpoint, etc.), as would be expected in actively proliferating lung tumor cells. Other cellular processes are related to metastasis, including extracellular matrix remodeling, cell invasion, and cell-cell contact. In general, changes were similar across subtypes.
Table 2.
Cellular processes significantly changed in lung adenocarcinoma (ADC) and squamous cell carcinoma (SCC) by SNEA (Mann-Whitney p<0.001)
| Cellular process | Sub-network size | Median fold change | p-value |
|---|---|---|---|
| Adenocarcinoma | |||
| Wound healing | 385 | 1.02072 | 2.19 × 10-6 |
| ECM proteins | 571 | 1.0277 | 2.38 × 10-6 |
| Chromosome segregation | 149 | 1.10384 | 7.32 × 10-6 |
| Tissue remodeling | 168 | 1.05076 | 1.16 × 10-5 |
| Cell survival | 1251 | 1.03351 | 6.44 × 10-5 |
| Cell invasion | 486 | 1.02405 | 7.79 × 10-5 |
| Tissue invasion | 40 | 1.11402 | 1.23 × 10-4 |
| Mitotic cell cycle | 37 | 1.10189 | 2.31 × 10-4 |
| G2/M transition | 465 | 1.05898 | 2.37 × 10-4 |
| Abscission | 25 | 1.00683 | 2.82 × 10-4 |
| Oncogenesis | 347 | 1.02691 | 4.62 × 10-4 |
| Mitotic entry | 123 | 1.07766 | 4.95 × 10-4 |
| Centrosome separation | 38 | 1.13508 | 5.83 × 10-4 |
| Drug resistance | 244 | 1.04375 | 7.66 × 10-4 |
| Cell redox homeostasis | 18 | 1.12308 | 7.79 × 10-4 |
| Cell motility | 691 | 1.02671 | 7.94 × 10-4 |
| Mitotic checkpoint | 57 | 1.11636 | 8.92 × 10-4 |
| Cell contact | 929 | 1.02568 | 9.04 × 10-4 |
| Squamous cell carcinoma | |||
| Chromosome segregation | 149 | 1.1897 | 4.58 × 10-10 |
| Kinetochore assembly | 126 | 1.23986 | 7.28 × 10-10 |
| Mitosis | 924 | 1.05925 | 1.35 × 10-8 |
| Spindle assembly | 386 | 1.08566 | 6.70 × 10-8 |
| Cytokinesis | 291 | 1.05385 | 1.03 × 10-7 |
| ECM proteins | 571 | -1.02692 | 2.90 × 10-7 |
| Wound healing | 385 | -1.05074 | 2.44 × 10-6 |
| DNA replication initiation | 41 | 1.3792 | 1.02 × 10-5 |
| Mitotic checkpoint | 57 | 1.24069 | 1.03 × 10-5 |
| Cell invasion | 486 | -1.01081 | 1.20 × 10-5 |
| Mitotic spindle assembly | 41 | 1.23754 | 2.72 × 10-5 |
| G2/M transition | 465 | 1.05662 | 2.81 × 10-5 |
| Premeiotic DNA synthesis | 17 | 1.44176 | 3.11 × 10-5 |
| Cell motility | 691 | -1.01698 | 3.41 × 10-5 |
| Mitotic entry | 123 | 1.1247 | 3.64 × 10-5 |
| Drug resistance | 244 | 1.01383 | 4.29 × 10-5 |
| DNA replication checkpoint | 40 | 1.35065 | 5.73 × 10-5 |
| Genome instability | 178 | 1.13299 | 7.80 × 10-5 |
| S phase | 737 | 1.02773 | 2.19 × 10-4 |
| DNA unwinding | 117 | 1.15665 | 2.39 × 10-4 |
| Tissue remodeling | 168 | -1.03451 | 3.11 × 10-4 |
| Chromosome condensation | 174 | 1.05662 | 3.14 × 10-4 |
| Cell growth | 2072 | 1.00691 | 3.18 × 10-4 |
| Centrosome separation | 38 | 1.30654 | 4.21 × 10-4 |
| Translation | 689 | -1.01232 | 4.53 × 10-4 |
| Cell division | 598 | 1.00883 | 4.69 × 10-4 |
| Chemosensitivity | 129 | 1.03154 | 4.94 × 10-4 |
| Cell–cell contact | 307 | -1.04502 | 5.89 × 10-4 |
| rRNA processing | 69 | 1.21023 | 6.52 × 10-4 |
Metabolic pathways
Because the cell cycle is functionally linked to cellular metabolism and energy production, we next analyzed the differential gene expression of ADC and SCC versus pathologically normal lung tissue and all metabolic pathways in the ResNet 7 database, using the GSEA algorithm, and a Mann-Whitney test with p-value cut-off at 0.05. Significantly changed metabolic processes for each lung cancer subtype are documented in Table 3. Purine and pyrimidine synthesis pathways were significantly up-regulated in both subtypes. The SCC type also demonstrated up-regulated energy production pathways for oxidative phosphorylation, glucose metabolism, and the tricarboxylic acid cycle. These findings are consistent with active DNA synthesis and active proliferation of lung cancer cells.
Table 3.
Metabolic pathways significantly changed in lung adenocarcinoma (ADC) and squamous cell carcinoma (SCC) by GSEA (Mann-Whitney test, p<0.05)
| Name | Sub-network size | Median fold change | p-value |
|---|---|---|---|
| Adenocarcinoma | |||
| Nicotinate and nicotinamide metabolism | 79 | 1.05861 | 2.953 × 10-3 |
| Glut/Gln/Pro metabolism | 38 | 1.11358 | 1.0798 × 10-2 |
| Bile acids metabolism | 41 | 1.04722 | 1.1389 × 10-2 |
| Pyrimidine metabolism | 100 | 1.08111 | 1.784 × 10-2 |
| Purine metabolism | 154 | 1.06749 | 1.8009 × 10-2 |
| Squamous cell carcinoma | |||
| Respiratory chain and oxidative phosphorylation | 74 | 1.16544 | 4.60 × 10-6 |
| Purine metabolism | 154 | 1.03009 | 7.87 × 10-4 |
| Tricarboxylic acid cycle | 27 | 1.13055 | 1.177 × 10-3 |
| Branched chain amino acids metabolism | 55 | 1.06616 | 3.688 × 10-3 |
| Glucose metabolism | 53 | 1.08078 | 7.449 × 10-3 |
| Aspartate metabolism | 26 | 1.14841 | 8.051 × 10-3 |
| Folate biosynthesis | 20 | 1.17777 | 1.2205 × 10-2 |
| Pyrimidine metabolism | 100 | 1.09129 | 2.0488 × 10-2 |
| Mannose metabolism | 33 | 1.0791 | 2.3137 × 10-2 |
| Amino sugars synthesis | 19 | 1.13317 | 2.5646 × 10-2 |
Oncogenes and tumor suppressors
Because oncogenes and tumor suppressors play a significant role in the regulation of cell proliferation, we next investigated a major molecular network of 273 oncogenes and 92 tumor suppressors in ADC and SCC using the Res-Net 7 database. The oncogenes with at least 2-fold change (p<0.001) are documented in Table 4. All oncogenes in ADC, and most in SCC, with the exception of ECT2 (elevated 3.6-fold) and DCUN1D1 (elevated 2.7-fold), were significantly down-regulated. The oncogenes down-regulated in both types of lung cancer included FOS and FOSB. Changes in tumor suppressors were examined (Table 4) for both lung cancer subtypes. Tumor suppressors were not changed significantly in ADC, whereas in SCC, DLG1 and DLGAP5 were up-regulated, and TGFBR2 was down-regulated. Full details of differential changes in oncogenes and tumor suppressors (including p-values) are provided in Supplementary Tables S4 and S5, respectively.
Table 4.
Oncogenes and tumor suppressors significantly affected in lung adenocarcinoma (ADC) and squamous cell carcinoma (SCC)
| Name | Description | Fold-change | p-value |
|---|---|---|---|
| Adenocarcinoma | |||
| FOS | v-fos FBJ murine osteosarcoma viral oncogene homolog | -2.02 | 9.18 × 10-4 |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B | -2.14 | 4.87 × 10-4 |
| Squamous cell carcinoma | |||
| ECT2 | Epithelial cell transforming sequence 2 oncogene | 3.59 | 2.55 × 10-8 |
| DCUN1D1 | DCN1, defective in cullin neddylation 1, domain containing 1 (S. cerevisiae) | 2.66 | 4.00 × 10-7 |
| JUN | jun oncogene | -2.30 | 7.39 × 10-9 |
| FOS | v-fos FBJ murine osteosarcoma viral oncogene homolog | -2.80 | 7.18 × 10-7 |
| ROS1 | c-ros oncogene 1 , receptor tyrosine kinase | -2.97 | 8.79 × 10-9 |
| CXCL2 | Chemokine (C-X-C motif) ligand 2 | -3.65 | 2.45 × 10-8 |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B | -4.65 | 1.60 × 10-9 |
| DLG1 | Discs, large homolog 1 (Drosophila) | 2.41 | 3.46 × 10-5 |
| DLGAP5 | Discs, large (Drosophila) homolog-associated protein 5 | 2.35 | 4.22 × 10-7 |
| TGFBR2 | Transforming growth factor, beta receptor II (70/80 kDa) | -2.09 | 1.85 × 10-11 |
Discussion
NSCLC represents a heterogeneous collection of cancer subtypes that arise as a consequence of altered gene expression and mutations acquired during cancerogenesis. Molecular signatures of NSCLC subtypes can underline mechanisms of this complex disease and more importantly can facilitate the development of novel targeted therapy for cancer patients. Here we reported major cellular pathways of human lung ADC and SCC and described similarities as well as unique differences between these two subtypes of lung cancer.
Sub-Network Enrichment Analysis (SNEA)
To build comprehensive molecular signatures of ADC and SCC, we used the SNEA algorithm, which is designed to investigate variations in gene set enrichment. Unlike previously reported approaches, such as GSEA, which uses a predefined collection of hand-curated gene sets [16], SNEA uses the global literature-extracted gene-gene expression regulation network to generate a comprehensive collection of gene sets [11]. The global expression network used for SNEA in this study is extracted and comprised over 160,000 independently reported relations [11]. The advantage of the SNEA application for this type of analysis is in the unbiased knowledge-driven nature of this approach. Sub-networks in SNEA are calculated from gene expression regulation ‘facts’ extracted across the entire Pub-Med database. Thus, each individual relation can come from specific and perhaps very narrow publications, but when combined together they provide an unbiased and comprehensive picture of cellular gene expression network. Another critically important power of SNEA is in its ability to find ‘hidden’ regulators, for example genes and proteins for which changes in cancer are not detected on the level of mRNA, but rather on a biologic activity level. This is particularly important for proteins for which activity is regulated at the post-transcriptional level, such as post-translational protein modification, protein stability, or degradation. The vast majority of cancer signaling pathways are activated or inactivated through phosphorylation of individual protein kinases, an event that is unlikely to be reflected on the level of mRNA measured in gene expression profiling. Similarly, activity of many transcription factors downstream of major signaling cascades is regulated by phosphorylation, and these changes will be overlooked in traditional gene expression profiling. SNEA can detect such regulators by looking at the changes in downstream targets, rather than the gene/protein itself. Another important advantage of SNEA is its ability to summarize the individual gene expression changes and to project them to the system-level cellular signaling map.
Molecular complexity of ADC and SCC
Our search for key transcriptional regulators involved in differential changes using the Res-Net 7 transcriptional network showed uniform involvement of E2F, CTGF, and PDGF in lung cancer pathogenesis. These observations are consistent with previous reports describing the role of CTGF and PDGF in lung cancer progression [20-22]. Our analysis showed that SCC can be uniquely characterized by the involvement of the EGF, IL1F8, and CX3CL1 pathways, while changes in Rb1, miR-200, and EMP2 targets are specific for ADC.
Consistent with the aggressively proliferative phenotype of lung cancer cells, the most significantly affected cellular processes were those involved in the cell cycle and metastasis. The biochemical ‘signature’ pinpoints changes in purine and pyrimidine biosynthesis and energy production pathways, and these changes were seen in both ADC and SCC. Up-regulation of cell-cycle-related genes was more profound in SCC than in ADC. DNA repair genes were also more profoundly up-regulated in SCC. There were no significant changes in apoptotic genes in either type of lung cancer. Surprisingly, we found that all oncogenes in ADC and most oncogenes in SCC were significantly down-regulated, including FOS and FOSB. In SCC, ECT2 and DCUN1D1 were up-regulated. Tumor suppressors were not changed significantly in ADC, whereas in SCC, the significantly changed tumor suppressors were DLG1 and DLGAP5 (elevated), and TGFBR2 (down-regulated).
In conclusion, we found that ADC and SCC subtypes of NSCLC can be characterized by unique gene signatures and distinct molecular pathways. Our data suggest that the gene expression signature of subtypes of lung cancer can be a critical tool for improved characterization of subtypes currently classified based on analysis of histology of tumor samples by light microscopy. Taken together, these data provide a better understanding of the unique molecular features of NSCLC subtypes, and may open new avenues towards the molecular-based identification of novel therapeutic strategies for NSCLC.
Acknowledgments
We thank Ann Contijoch, Sanofi, for reviewing this paper. Editorial assistance was provided by ArticulateScience Ltd., supported by Sanofi.
Conflict of interest
Valeria Ossovskaya is employee of BiPar Sciences Inc. (subsidiary of Sanofi). Yipeng Wang, Adam Budoff, Gordon Vansant, and Joseph Monforte are employees of AltheaDx Inc. Qiang Xu is former employee of AltheaDx Inc. Alexander Lituev and Olga Potapova are employees of Cureline Inc. Nikolai Daraselia is employee of Ariadne Inc.
Supplementary material
Figure S1.

An overview of Sub-Network Enrichment Analysis (SNEA) algorithms
References
- 1.Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol. 2007;18:581–592. doi: 10.1093/annonc/mdl498. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 4.NCCN Clinical practice guidelines in oncology: non-small cell lung cancer. NCCN Clinical Practice Guidelines in Oncology, v2. 2009 [Google Scholar]
- 5.Borczuk AC, Toonkel RL, Powell CA. Genomics of lung cancer. Proc Am Thorac Soc. 2009;6:152–158. doi: 10.1513/pats.200807-076LC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borczuk AC, Powell CA. Expression profiling and lung cancer development. Proc Am Thorac Soc. 2007;4:127–132. doi: 10.1513/pats.200607-143JG. [DOI] [PubMed] [Google Scholar]
- 7.Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A, Spitznagel EL, Piccirillo J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol. 2006;24:4539–4543. doi: 10.1200/JCO.2005.04.4859. [DOI] [PubMed] [Google Scholar]
- 8.Langer CJ, Besse B, Gulaberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small cell lung cancer. J Clin Oncol. 2010;28:5311–5320. doi: 10.1200/JCO.2010.28.8126. [DOI] [PubMed] [Google Scholar]
- 9.Daraselia N, Yuryev A, Egorov S, Mazo I, Ispolatov I. Automatic extraction of gene ontology annotation and its correlation with clusters in protein network. BMC Bioinformatics. 2007;8:243. doi: 10.1186/1471-2105-8-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Novichkova S, Egorov S, Daraselia N. Med-Scan, a natural language processing engine for MEDLINE abstracts. Bioinformatics. 2011;19:1699–1706. doi: 10.1093/bioinformatics/btg207. [DOI] [PubMed] [Google Scholar]
- 11.Sivachenko AY, Yuryev A, Daraselia N, Mazo I. Molecular networks in microarray analysis. J Bioinform Comput Biol. 2007;5:429–456. doi: 10.1142/s0219720007002795. [DOI] [PubMed] [Google Scholar]
- 12.Sivachenko AY, Yuryev A, Daraselia N, Mazo I. Identifying local gene expression patterns in biomolecular networks. 2005 IEE Computational Systems Bioinformatics Conference – Workshops (CSBW'O5); 2005. pp. 180–184. [Google Scholar]
- 13.Sivachenko AY, Kalinin A, Yuryev A. Pathway analysis for design of promiscuous drugs and selective drug mixtures. Curr Drug Discov Technol. 2006;3:269–277. doi: 10.2174/157016306780368117. [DOI] [PubMed] [Google Scholar]
- 14.Yuryev A, Mulyukov Z, Kotelnikova E, Maslov S, Egorov S, Nikitin A, Daraselia N, Mazo I. Automatic pathway building in biological association networks. BMC Bioinformatics. 2006;7:171. doi: 10.1186/1471-2105-7-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuryev A. In silico pathway analysis: the final frontier towards completely rational drug design. Expert Opinion Drug Discov. 2008;3:867–876. doi: 10.1517/17460441.3.8.867. [DOI] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis; a knowledge-based approach for interpreting genomewide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–7854. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 18.Wadehra M, Goodglick L, Braun J. The tetraspan protein EMP2 modulates the surface expression of caveolins and glycosylphosphatidyl inositol-linked proteins. Mol Biol Cell. 2004;15:2073–2083. doi: 10.1091/mbc.E03-07-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood RD, Mitchell M, Lindhal T. Human DNA repair genes, 2005. Mutat Res. 2005;577:275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Chen PP, Li WJ, Wang Y, Zhao S, Li DY, Feng LY, Shi XL, Koeffler HP, Tong XJ, Xie D. Expression of Cyr61, CTGF and WISP-1 correlates with clinical features of lung cancer. PLoS ONE. 2007;2:e534. doi: 10.1371/journal.pone.0000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donnem T, Al Saad S, Al Shibli K, Busund LT, Bremnes RM. Co-expression of PDGF-B and VEGFR-3 strongly correlates with lymph node metastasis and poor survival in nonsmall-cell lung cancer. Ann Oncol. 2010;21:223–231. doi: 10.1093/annonc/mdp296. [DOI] [PubMed] [Google Scholar]
- 22.Kinoshita K, Nakagawa K, Hamada J, Hida Y, Tada M, Kondo S, Moriuchi T. Imatinib mesylate inhibits the proliferation-stimulating effect of human lung cancer-associated stromal fibroblasts on lung cancer cells. Int J Oncol. 2010;37:869–877. doi: 10.3892/ijo_00000738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.