

Abstract
Celastrol, an important natural product and Hsp90 inhibitor with a wide range of biological and medical activities and broad use as a biological probe, acts by an as yet undetermined mode of action. It is known to undergo Michael additions with biological sulfur nucleophiles. Here it is demonstrated that nucleophiles add to the pharmacophore of celastrol in a remarkable stereospecific manner. Extensive characterization of the addition products have been obtained using NMR spectrometry, nuclear Overhauser effects, and density functional theory to determine facial selectivity and gain insight into the orbital interactions of the reactive centers. This stereospecificity of celastrol may be important to its protein target selectivity.
The natural product celastrol (1) (Figure 1) exhibits broad biological activity, including effects on protein homeostasis1–4 neurodegeneration,5–7 inflammation,8,9 and cancer.10–12 Recently, 1 has been identified as an inhibitor of heat shock protein 90 (Hsp90),13 an essential molecular chaperone with numerous cellular functions. Hsp90 is highly expressed in cancer cells and has a critical role in conformational maturation and activation of a wide range of signaling molecules that include pro-oncogenic proteins involved in cancer.14 Therefore, small molecule inhibitors targeting Hsp90 have great potential as anticancer therapeutics.15,16 Most of the inhibitors of this protein, such as geldanamycin17 and radicicol,18 interrupt the ATPase activity of Hsp90 resulting in enhanced degradation of the client proteins via the ubiquitin-proteasome pathway.19 However, since many of the ATP site binding compounds also exhibit undesirable side effects,20 the identification of new types of inhibitors is of great importance.
Figure 1.

Structure of Celastrol
Celastrol (1) is a non-ATP-competitive inhibitor13,21 representing a new therapeutic class and offering a new strategy for development of Hsp90 inhibitors with improved properties. Although studies from our laboratory1 and others8,22,23 suggest a conjugate addition of cysteine residues to the quinone methide substructure to be the dominant mode of action, the exact mechanism is still far from understood, especially concerning the identity and nature of binding of 1 to its protein target(s). Studies confirmed that inhibition of the Hsp90 pathway is cysteine-mediated,22,24,25 but if, and how, 1 displays specificity and discriminates among the various potential targets remains elusive. Structurally, celastrol (1) belongs to a subgroup of D:A-friedo-nor-oleananes characterized by a hydroxyl group in the adjacent position to the carbonyl oxygen and a chromophore that extends over the A and B ring of the triterpenoid skeleton.26,27 These structural characteristics, along with steric effects and its conformation should influence its reactivity and stability. The hydroxyl group in the A ring also enhances the reactivity via intramolecular hydrogen bonding to the carbonyl. Extended conjugation into the B ring counteracts this reactivity. The quinone methide substructure of 1 incorporates three potentially electrophilic positions (C-2, C-4, and C-6), of which position C-4 appears to be too hindered to be susceptible to nucleophilic addition, and attack at C-6 is expected to be favored over C-2 as a result of the increased thermodynamic stability achieved by aromatization of the ring.
Here we investigate the reactivity and chemistry of the purported pharmacophore1 of 1, namely, the fused AB ring system. The reaction of celastrol with various nucleophiles was studied by 1D- and 2D-NMR spectroscopy and in silico analysis. We report studies that unequivocally establish that nucleophilic attack on celastrol is regioselective at the 1,6 electrophilic position and that this reaction possesses remarkable stereospecificity. Utilizing extensive NMR spectroscopy and theoretical calculations we establish that nucleophilic attack is favored exclusively at the β-face (as defined in Rose et al.28 with the nucleophile approach syn to the β-C9 methyl). Literature precedent29 involving the addition of acetate to the C-6 position of pristimerin, the methyl ester of celastrol, proposes α-facial addition, using the rationalization that the sterically congested β-face, having most of the angular methyl groups, especially at C-9, on that face, would preclude attack from an incoming nucleophile, although no experimental evidence was reported. Here we offer evidence that nucleophilic addition to celastrol and related quinone methide structures is to the β-face.
Celastrol (1) undergoes reduction with various hydride reagents exclusively at the 1,6-position, generating prochiral dihydrocelastrol (2)30 (Figure 2) with complete discoloration, characteristic of reduction of the quinone methide substructure. We envisioned utilizing this reaction to introduce a radio-label for subsequent biological studies without altering the parent structure. Consequently, a model reaction with sodium borodeuteride was conducted, wherein we anticipated incorporation of ~50% deuterium. Remarkably, deuterium addition was observed to occur exclusively to the β-face of the C-6 center to provide deuteriohydroxycelastrol (3). When comparison is made to dihydrocelastrol (2) (Table S1, supporting information) it can be seen that the chemical shift of C-6Hα (α-face proton) of 2 occurs at δ3.28 as a doublet of doublets with the expected splitting occurring from the C-7 hydrogen and the enantiotopic C-6H□ proton (□-face proton). Deuteriohydroxycelestrol (3) shows a slight upfield shift for the C-6Hα proton to δ3.25 because of the adjacent deuterium. What is surprising, however, is the doublet splitting pattern with the C-7 proton, suggesting stereospecific addition of the nucleophile to one face as opposed to the expected non-stereoselective addition, which would give rise to a doublet of doublets, as observed in 2. Furthermore, as expected from the principle of microscopic reversibility, aerial oxidation of 3 back to 1 resulted in complete loss of deuterium. Intrigued by this observation, we conducted extensive n.O.e studies (Figure 3) to investigate the generality of the absolute stereochemistry of the addition. No n.O.e. effect was observed for the C-6 hydrogen at δ3.25 with the C-9 methyl group occupying the β-face, suggesting that the hydrogen was facing `down' (the α-face), and, therefore, nucleophilic addition occurred from the `top' β-face. However, numerous factors, such as intraatomic distance and the steric effect of the C-9 methyl group toward top face addition, demanded further investigation.
Figure 2.

Structures of dihydroxycelastrol (2) and deuteriohydroxycelastrol (3).
Figure 3.

Nuclear Overhauser effect couplings observed for β–face addition
Further evidence that nucleophilic addition was exclusive to one face and that the face in question was the `top' β-face was sought. Hence, dihydroxycelastrol (2) was subjected to n.O.e irradiation (Figure 3) of the δ3.28 dd and δ2.99 d signals (Table S1, Supporting Information) of the two enantiotopic protons at the C-6 carbon atom to determine if they interacted with another proton. An enhancement of the C-9 methyl signal at δ1.27 was observed upon selective irradiation of the δ2.99 signal (assigned, on this basis, as the β-face C-6 proton), and no enhancement of the C-9 methyl was observed upon irradiation of the δ3.28 signal. These results provide conclusive evidence that a) the addition of hydride occurs stereospecifically to the β-face of celastrol and b) the intramolecular distance between the C-6 and C-9 centers is appropriate for analysis by n.O.e spectroscopy.
To demonstrate that the observed facial selectivity of nucleophiles is an inherent property of celastrol, we investigated a variety of nucleophilic reactions comprising hard and soft carbon, nitrogen, and sulfur nucleophiles. In all cases the experiments were conducted utilizing an excess of nucleophilic reagent to encourage the formation of both possible isomers. Initial studies with carbon nucleophiles established that 1 was inert to reactions with a large excess (~10 equiv) of methyl-magnesium bromide and methyllithium because hard organometallic nucleophiles act as bases, leading to deprotonation of the enol functionality, as confirmed by NMR spectrometry. This, in effect, renders celastrol poorly electrophilic and inert toward nucleophilic attack. Successful Michael addition of a carbon nucleophile was achieved, however, with a nitroalkane to form nitromethyl adduct 4. The nitronate anion, generated in situ from nitromethane and tetra-n-butylammonium fluoride (TBAF), was employed; it is known that fluoride ions catalyze Michael additions of nitroalkanes to electron-deficient alkenes.31 Extensive spectroscopic analysis of 4 confirmed the exclusive stereospecific addition of the nitronate anion to the C-6 carbon of 1. The signals of interest, the C-6 proton, and adjacent diastereotopic methylene protons from the newly introduced nucleophile, were observed as distinct signals devoid of overlap at δ4.27, δ4.12, and δ4.54, respectively (Table S1, Supporting Information). The use of n.O.e irradiation again provided unequivocal evidence of addition of the nucleophile to the β-face (Figure 3). As expected, no enhancement of the C-9 methyl protons was observed upon irradiation of the C-6 proton, or vice versa, confirming the α-face orientation of the C-6 proton. Furthermore, irradiation of the signal corresponding to one of the diastereotopic methylene protons (δ4.12) resulted in an enhancement of the C-9 methyl protons, confirming its position on the β-face.
Nitrogen nucleophiles were found to be inert to reaction with celastrol (1). A range of amines (primary and secondary, alkyl and aryl substituted) and reaction conditions (excess amine reagent, base strength) all failed to furnish an addition product. Other studies from our laboratory to convert the carboxylic acid functionality of 1 to various amides (including the nucleophilic amino acids lysine and histidine) also supported this observation.
Of critical interest to the potential biological mode of action of 1 is the stereoselectivity of sulfur nucleophiles, such as cysteine residues in biological milieu, which is the reaction purported1 to be essential for its biological activity. To the best of our knowledge, previous studies have not reported on stereo-selectivity of this addition. The N-acetylcysteine adduct of 1 (compound 5) was synthesized and subjected to n.O.e irradiation (Figure 3). Irradiation of the δ4.70 doublet of the C-6 proton failed to cause enhancement of the C-9 methyl protons, indicating an α-facial orientation for the C-6 proton. This was confirmed by irradiation of the δ3.37 diastereotopic methylene proton, which clearly displayed an n.O.e. to the C-9 methyl protons signal. These results provide conclusive evidence for β-face addition of N-acetylcysteine to celastrol, which is a model for the addition of cysteine nucleophiles contained in proteins that add to 1. To achieve a closer approximation to intracellular conditions, we also investigated the reaction of 1 with glutathione in buffered conditions (pH 7.5). Although mass spectrometric analysis (Supporting Information) showed the generation of the expected C-6 adduct, NMR spectrometry proved futile because of a combination of poor solubility and diminished and complex signals.
We postulate that soft nucleophiles are more reactive in nucleophilic additions to celastrol because of favorable orbital interactions. These nucleophiles possess large diffuse orbitals for interaction with the accepting orbitals of celastrol (1). In comparison, the highly polarized nature of hard, basic nucleophiles results instead in deprotonation of the celastrol hydroxyl functionality.
In an attempt to rationalize these observations and suppositions regarding a high degree of orbital involvement, which result in facial selectivity, comprehensive theoretical calculations were performed. Density functional theory (DFT) calculations were performed on 1 and one of the nucleophiles (the nitronate anion) using Jaguar 7.8 implemented in the Schrodinger 9.1 package.32 The B3LYP functional with 6–31G** basis set was utilized for optimizing the compounds along with the computation of Fukui functions. The Fukui indices are known to be reactivity indices, and they give the information about which atoms in a molecule have a larger tendency to either lose or accept an electron or, in other words, which atoms are more prone to undergo a nucleophilic or an electrophilic attack. The molecule undergoing nucleophilic or an electrophilic attack has the tendency of becoming polarized in the presence of an external field or upon change in electron density. The idea behind Fukui's indices calculations lies in the realm of conceptual DFT. Mineva et.al.33 carried out selectivity studies of compounds used as reagents in organic synthesis by using density functional local reactivity indices. They computed orbital Fukui indices from Kohn-Sham orbitals and also from the atomic resolved hardness matrix to study the reaction mechanism along with regioselectivity.33 The analysis of the Fukui indices is primarily done for the HOMO and LUMO of the reactants. Jaguar computes the atomic Fukui indices according to the method developed by Perez et.al.34,35 These indices are based on Fukui functions, which are partial derivatives of the electron and spin density with respect to a change in either the electron count or the unpaired spin count. Atomic Fukui indices are an attempt to quantify the anticipated reactive center in a molecule. Each integrated index has two subscripts, N or S, which represent the electron density and spin density, respectively. Usually f_NN indices are of interest, because the other indices require a change in either the spin density or the spin multiplicity. The f_NN of HOMO indices are related to f (−) Fukui function, and f_NN LUMO indices are related to f (+) Fukui function.
We have computed all the Fukui functions for structure 1 and the nucleophilic nitronate anion. It was observed that the f_NN LUMO of 1 at position C-6 (#10 in DFT number, see SI) has the highest index (0.2116), depicting that nucleophilic attack has to be at this center. For the nucleophile, the f_NN HOMO index of the carbanion center is highest and found to be 0.6124 (see SI). This clearly indicates that the carbanionic center of the nucleophile is expected to attack carbon C-6 of 1, consistent with the hard-soft-acid-base (HSAB) principle.36 After identifying the reactive centers in 1 and the nucleophile, we analyzed the HOMO contour of the nucleophile and LUMO contour of 1. The orbital analysis clearly indicates the stereo-specific conjugate addition of 1 (Figure 4A). We then computed the final product obtained in the above reaction. To identify the stable product, we carried out DFT calculations for both geometries of 4 (Figure 4B). The DFT optimized geometries reveal a 11.6 kcal/mol (see SI) energy difference between the two structures in favor of 4β. This clearly indicates that the reaction site at C-6 produces a much more stable product (4β) by β-facial attack, which is consistent with the experimental observations detailed above.
Figure 4.
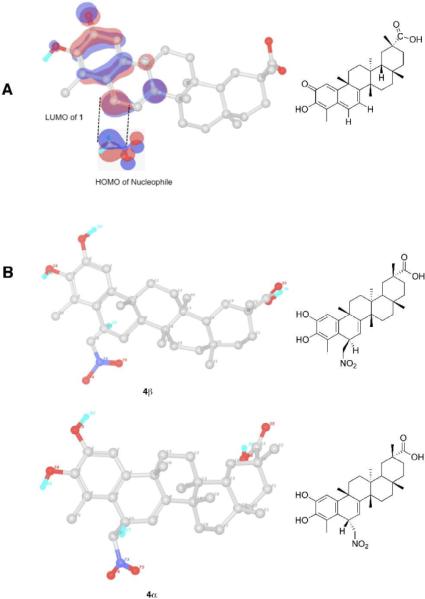
A; Orbital analysis of celastrol (1) clearly shows the LUMO (red) positioned on the β-face. The nitronate anion HOMO (blue) attacks at the upper face. Blue - highest electron density; red – lowest electron density. B; Density functional theory (DFT) minimized structures of 4β and 4α.
It is conceivable that this stereospecific addition may be an important factor in selectivity of reaction of protein targets with celastrol; binding to some proteins containing cysteine residues might not lead to reaction because the cysteine residue is oriented on the wrong face of bound celastrol. The notion of facial selectivity is well known in biology; an analogy is with reactions catalyzed by pyridine nucleotide (NAD(P)+ and NAD(P)H)-dependent enzymes. These cofactors are prochiral and bind to their respective enzymes in a specific orientation that favors stereospecific addition of hydride to either the substrate from NAD(P)H or to NAD+ by the substrate, the stereochemistry of which is determined by the orientation of bound substrate.37
In conclusion, celastrol (1) is believed to undergo formation of a Michael adduct with its intracellular target(s),17–20 and this interaction appears to be crucial for a number of biological properties that this natural product exhibits. We have demonstrated unprecedented stereospecificity in the regioselective Michael reaction of celastrol with a range of nucleophiles, especially soft nucleophiles such as biologically-relevant cysteine and glutathione. This, heretofore, unknown chemistry of celastrol may help to unlock its selectivity for protein targets and mode of biological action.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to the National Institutes of Health (R43NS057849) for financial support of this research, to Dr. Yuyang Wu in the Integrated Molecular Structure, Education and Research Center at Northwestern University for carrying out n.O.e experiments, and to Prof. Karl A. Scheidt for helpful suggestions. The Center for Molecular Innovation and Drug Discovery is funded by the Chicago Biomedical Consortium with support from The Searle Fund at The Chicago Community Trust.
Footnotes
Author Contributions The manuscript was written through contributions of all authors.
Supporting Information. Synthetic procedures, 1H-, 13C-, and n.O.e NMR spectra, detailed DFT calculations, and mass spectrum of glutathione adduct. This material is available free of charge via the Internet at http://pubs.acs.org.Instructions for Authors at (http://pubs.acs.org/page/jacsat/submission/authors.html).
REFERENCES
- (1).Trott A, West JD, Klaic L, Westerheide SD, Silverman RB, Morimoto RI, Morano KA. Mol Biol Cell. 2008;19:1104. doi: 10.1091/mbc.E07-10-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. J Biol Chem. 2004;279:56053. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- (3).Cleren C, Calingasan NY, Chen J, Beal MF. J Neurochem. 2005;94:995. doi: 10.1111/j.1471-4159.2005.03253.x. [DOI] [PubMed] [Google Scholar]
- (4).Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L, Kelly JW. Cell. 2008;134:769. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kiaei M, Kipiani K, Petri S, Chen J, Calingasan NY, Beal MF. Neurodegener Dis. 2005;2:246. doi: 10.1159/000090364. [DOI] [PubMed] [Google Scholar]
- (6).Wang J, Gines S, MacDonald ME, Gusella JF. BMC Neurosci. 2005;6:1. doi: 10.1186/1471-2202-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang YQ, Sarge KD. J Mol Med. 2007;85:1421. doi: 10.1007/s00109-007-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Huang FC, Chan WK, Moriarty KJ, Zhang DC, Chang MN, He W, Yu KT, Zilberstein A. Bioorg Med Chem Lett. 1998;8:1883. doi: 10.1016/s0960-894x(98)00331-x. [DOI] [PubMed] [Google Scholar]
- (9).Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:1341. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- (10).Ngassapa O, Soejarto DD, Pezzuto JM, Farnsworth NR. J Nat Prod. 1994;57:1. doi: 10.1021/np50103a001. [DOI] [PubMed] [Google Scholar]
- (11).Chang FR, Hayashi K, Chen IH, Liaw CC, Bastow KF, Nakanishi Y, Nozaki H, Cragg GM, Wu YC, Lee KH. J Nat Prod. 2003;66:1416. doi: 10.1021/np030241v. [DOI] [PubMed] [Google Scholar]
- (12).Abbas S, Bhoumik A, Dahl R, Vasile S, Krajewski S, Cosford ND, Ronai ZA. Clin Cancer Res. 2007;13:6769. doi: 10.1158/1078-0432.CCR-07-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR. Cancer Cell. 2006;10:321. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- (14).Kamal A, Boehm MF, Burrows FJ. Trends Mol Med. 2004;10:283. doi: 10.1016/j.molmed.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Solit DB, Chiosis G. Drug Discov Today. 2008;13:38. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]
- (16).Trepel J, Mollapour M, Giaccone G, Neckers L. Nat Rev Cancer. 2010;10:537. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Proc Natl Acad Sci U S A. 1994;91:8324. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM. Cell Stress Chaperones. 1998;3:100. doi: 10.1379/1466-1268(1998)003<0100:arbttn>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Whitesell L, Lindquist SL. Nat Rev Cancer. 2005;5:761. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- (20).Sharp S, Workman P. Adv Cancer Res. 2006;95:323. doi: 10.1016/S0065-230X(06)95009-X. [DOI] [PubMed] [Google Scholar]
- (21).Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, Sun D. Mol Cancer Ther. 2008;7:162. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- (22).Sreeramulu S, Gande SL, Gobel M, Schwalbe H. Angew Chem Int Ed Engl. 2009;48:5853. doi: 10.1002/anie.200900929. [DOI] [PubMed] [Google Scholar]
- (23).Yang H, Chen D, Cui QC, Yuan X, Dou QP. Cancer Res. 2006;66:4758. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- (24).Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. J Biol Chem. 2009;284:35381. doi: 10.1074/jbc.M109.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chadli A, Felts SJ, Wang Q, Sullivan WP, Botuyan MV, Fauq A, Ramirez-Alvarado M, Mer G. J Biol Chem. 2010;285:4224. doi: 10.1074/jbc.M109.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gunatilaka AA. Triterpenoid Quinonemethides and Realted Compounds (Celastroloids) Vol. 67. Springer Wien; New York: 1996. [Google Scholar]
- (27).Zhou Q. Natural Diterpene and Triterpene Quinone Methides: Strucutres, Synthesis, and Biological Potentials. Vol. one. New Jersey; John Wiley & Sons: 2009. [Google Scholar]
- (28).Rose IA, Hanson KR, Wilkinson KD, Wimmer MJ. Proc Natl Acad Sci U S A. 1980;77:2439. doi: 10.1073/pnas.77.5.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gunatilaka AAW, W.R. J. Chem. Research. 1992:31. [Google Scholar]
- (30).Nakanishi K, Takahashi Y, Budzikiewicz H. J. Org. Chem. 1965;30:1729. [Google Scholar]
- (31).Clark JH, Miller JM, So K-H. J. Chem. Soc., Perkin Trans. 1978;1:941. [Google Scholar]
- (32).Jaguar . version 7.8 Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- (33).(a) Mineva T. Theochem. 2006;762:79. [Google Scholar]; (b) Mineva T, Parvanov V, Petrov I, Neshev N, Russo N. J Phys Chem A. 2001;105:1959. [Google Scholar]
- (34).Contreras PR, Fuentealba P, Galvan M, Perez O. Chem Phys Lett. 1999;304:405. [Google Scholar]
- (35).Chamorro E, Perez P. J Chem Phys. 2005;123:114107. doi: 10.1063/1.2033689. [DOI] [PubMed] [Google Scholar]
- (36).Pearson RG. J Am Chem Soc. 1963;85:3533. [Google Scholar]
- (37).Silverman RB. The Organic Chemistry of Enzyme-Catalyzed Reactions; Chapter 3. Academic Press; San Diego: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.