

Summary
Phosphopantetheine-modified carrier domains play a central role in the template-directed, biosynthesis of several classes of primary and secondary metabolites. Fatty acids, polyketides and nonribosomal peptides are constructed on multidomain enzyme assemblies using phosphopantetheinyl thioester-linked carrier domains to traffic and activate building blocks. The carrier domain is a dynamic component of the process, shuttling pathway intermediates to sequential enzyme active sites. Here we report an approach to structurally fix carrier domain/enzyme constructs suitable for X-ray crystallographic analysis. The structure of a two-domain construct of E. coli EntF was determined with a conjugated phosphopantetheinyl-based inhibitor. The didomain structure is locked in an active orientation relevant to the chemistry of nonribosomal peptide biosynthesis. This structure provides details into the interaction of phosphopantetheine arm with the carrier domain and the active site of the thioesterase domain.
Introduction
Thiol-templated oligomerization is a central element of the biosynthetic pathways to fatty acid (FA), polyketide (PK) and nonribosomal peptide (NRP) metabolites (Meier et al., 2009). The chemistry of activation, elongation and termination occurs on large multidomain, multimodule enzyme assemblies. During the synthetic pathways, building blocks and intermediates are covalently attached to the modules via the thiol-terminated phosphopantetheine (ppant) prosthetic group. Post-translational modification of a conserved serine residue on dedicated carrier domains by reaction with coenzyme A (CoA) produces the prosthetic group (Lai et al., 2006; Mercer et al. 2007). Small molecule carboxylic acid building blocks are linked to ppant as thioesters intermediates, providing chemical activation and spatial templating, hallmarks of versatile oligomerization mechanisms.
Recent structural characterization of multidomain protein assemblies has provided significant insight into FA/PK synthase and NRP synthetase structure and function (Koglin et al., 2009; Weissman et al., 2008). The small (~90 amino acid) acyl or peptidyl carrier domain (ACP and PCP), with its attached ppant plays a central role in the overall mechanism by interacting with enzyme partners during the multiple chemical steps. Structures of the fungal and mammalian megadalton, fatty acid synthases have been determined to atomic resolution, establishing two distinct architectures for the iterative synthesis of fatty acids (Leibundgut et al., 2008; Maier et al., 2006; Lomakin et al., 2007). The carrier domain is often found to be disordered and not clearly evident in multidomain crystal structures. One exception is a structure of yeast fatty acid synthase where electron density for the ACP is observed bound near the ketosynthase domain (Leibundgut et al., 2007). In general, the large distances observed between synthase/synthetase active sites in these structures support a model where the small ACP/ppant domain is mobile and plays a key role in flux through the multidomain assembly.
Structural and functional analysis of ACP domains from fatty acid and polyketide synthases have revealed a common helical bundle structure with one helix in particular responsible for domain/domain interactions with partner proteins (Crump et al.,1997; Li et al., 2003; Findlow et al., 2003; Zornetzer et al., 2006; Sharma et al., 2006; Ploskon et al., 2008; Alekseyev et al., 2007). The PCP domain of NRP synthetases adopts a similar overall architecture as the ACPs from FA/PK synthases (Weber et al., 2000). Biochemical and combinatorial approaches have identified a surface on the PCP responsible for interaction with partner domain (thioesterase) in enterobactin synthetase (Zhou et al., 2006). NMR-based structural analysis of PCP domains demonstrated that the domain is plastic, with conformations dependent on the interacting in trans partner (Koglin et al., 2006; Koglin et al., 2008). In addition, NMR structural characterization of the excised apo-PCP-TE didomain construct of enterobactin synthetase illustrated that the carrier domain also adapts its structure in order to interact with various enzyme partners in cis (Frueh et al., 2008). This work demonstrated that the apo-didomain fragment can adopt multiple conformations including an orientation that would allow the ppant to extend to the active site of the thioesterase. A similar approach, however, applied to the NMR characterization of PK synthase ACP/TE pairs, showed that the ACP adopts a single conformation and there is minimal interaction between the two domains during relevant to catalysis (Tran et al., 2010). X-ray crystal structures of several NRP synthetase domains and multidomain constructs have been reported, however all lack structural details of an holo-PCP domain (Koglin et al., 2009). Of note, the crystal structure of an apo-four-domain NRP synthetase termination module from surfactin NRP synthetase provides valuable insight into the structure and domain organization of NRP synthetases (Tanovic et al., 2008). In this structure, among several catalytically relevant possibilities, the carrier domain is positioned to interact with the condensation domain.
The enterobactin biosynthetic pathway from E. coli is a well-studied model system for NRP synthetase chemistry (Gehring et al., 1998). The two modules (EntB/E and EntF) iteratively couple 2,3-dihydroxybenzoic acid and L-serine, producing the iron-chelating natural product. EntB activates DHB and loads the aryl acid on the ArCP domain. The four domain EntF module activates L-serine and couples the two building blocks through the condensation domain. The final two domains (PCP and thioesterase, TE) of EntF work in concert to execute the complex cyclotrimerization to form a trilactone through sequential transfer of the oligomer intermediates between the ppant of the PCP and the active site serine of the TE domain (Figure 1A) (Shaw-Reid et al., 1999).
Figure 1. Strategy to constrain carrier domain interactions in nonribosomal peptide synthetases (NRPS).
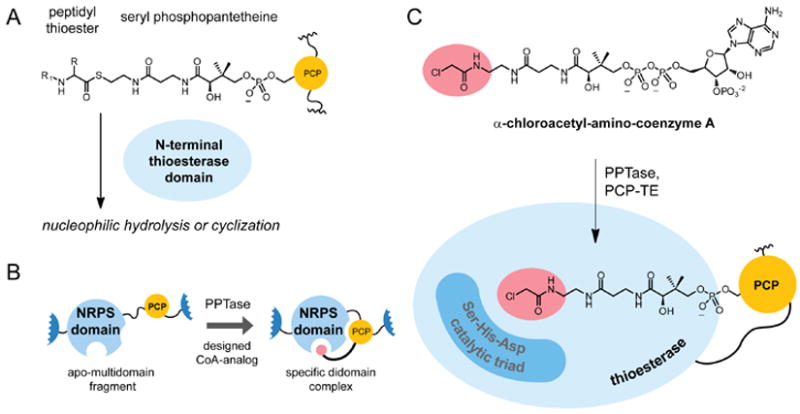
(A), Chemical structure of the phosphopantetheine substrate of terminal thioesterase (TE) domains in NRPSs, the peptide product is removed the peptide product from the NRPS assembly through nucleophilic hydrolysis or cyclization. (B), General approach to target specific enzyme active sites using a designed coenzyme A (CoA) analog loaded on to carrier domain (PCP) with a phosphopantetheinyl transferase (PPTase). (C), Targeting thioesterase domains with electrophile-based phosphopantetheine analogs. After conjugation to a PCP domain with PPTase, α-chloroacetyl-amino-coenzyme A is a stable mimic of the natural thioester substrate and the α-Cl-amide functionality targets the active site.
An approach to gain insight into the structural basis for mobile carrier domains is to chemically alter the ppant arm in order to direct assembly line complexes into a specific conformation (Figure 1B). We previously described a facile synthetic route to nonhydrolyzable amide analogs of coenzyme A thioesters that allows the production of designed carrier domain conjugates directed to different target domains of NRP synthetases (Lui et al., 2007). Similar strategies have been successfully applied to domains of NRP synthetases (Hur et al., 2009) and PK synthases (Kapur et al., 2008; Worthington et al., 2006; Worthington et al., 2010; Meier et al., 2010). Here we report the crystal structure of a designed didomain construct from enterobactin synthetase. The structure of the PCP-TE fragment was solved using a ppant analog designed to direct the PCP domain to the active site of the thioesterase. The structure provides detailed insight into the interactions between the ppant arm and the TE domain and the domain/domain interactions in the didomain complex. This general approach can be applied to gain molecular detailed insight into additional multidomain constructs in NRP, PK and fatty acid synthetase/synthase complexes.
Results
As an initial step to develop a general strategy to obtain configurationally constrained multidomain NRP synthetases, we chose the PCP-TE fragment from E. coli enterobactin synthetase. The fragment encompasses the final two domains of the four-domain EntF (domain organization: C-A-PCP-TE). As introduced, NMR structural characterization of a similar construct has been reported in the apo-form, without ppant modification (Frueh et al., 2008). A gene fragment was cloned with a start in the A-PCP linker region (residue 965 of EntF) through the end of the TE domain, overexpressed in E. coli and purified to homogeneity. The synthesis of an α-chloro-amide terminated coenzyme A (α-Cl-acetyl-amide-CoA, Figure 1C) targeting the catalytic triad of TE domains was previously reported by our group (Liu et al., 2007). α-Chloromethyl ketones are established inhibitors of serine hydrolases through covalent modification of the histidine or serine residues of the catalytic triad (Prorok et al., 1994). It was anticipated that an α-Cl-amide would interact with the TE catalytic triad through a similar electrophilic mechanism. Consistent with this hypothesis, the CoA analog was previously shown biochemically to inhibit thioesterase activity when loaded on to full length EntF with a phosphopantetheinyl transferases (PPTase) (Liu et al., 2007). The apo-PCP-TE fragment was incubated with the B. subtilis PPTase Sfp and α-Cl-acetyl-amide-CoA for two hours to allow for efficient ppant formation and subsequent reaction with the TE active site. The conjugate was purified and mass-spectral analysis indicated complete reaction to generate the PCP-TE conjugated ppant analog and loss of chlorine. This construct readily formed diffraction-quality crystals, while all efforts to crystallize either the apo- or holo-PCP-TE were not successful. The didomain protein crystallized in the P212121 space group with two molecules per asymmetric unit. The phase solution of the structure was obtained by SAD analysis of a selenomethionine-derivatized crystal and solved to 1.9 Ǻ resolution (Table S1).
A short linker (residues 1044-1056) connects conserved secondary structure elements of the two domains. Common to other thioesterases involved in secondary metabolism, the mixed α,β-structure is ‘capped’ with an α-helical lid motif (Tsai et al., 2001; Bruner et al., 2002; Akey et al., 2006; Samel et al., 2006). The lid covers the catalytic triad and is postulated to play a role in binding the relatively large substrate and assist in the cyclization reaction of nonribosomal peptide products (Figure S1). The two proteins present in the asymmetric unit share an almost identical structure (RMSDall atoms=0.8 Ǻ), with the main structural difference in the lid region where residues 1173-1206 of monomer B were not evident in the electron density maps (Figure 2A). In addition, there are is a slight rotation (~2.5 Ǻ) of the PCP domain relative to the TE domain, although the positions of the catalytic residues and prosthetic group overlay (Figure S2). The lack of clear density and assumed disorder in the lid region has been previously observed in all the other NRPS TEs crystallized without substrate (Bruner et al., 2002, Samel et al., 2006). Monomer A is more complete, with only residues 1176-1180 not included in the final model and the following discussion will focus specifically on it (Figure 2B). The lid is composed of two α-helices (α5TE and α6TE) and includes an extended region with a short helix (α4TE) inserted in the α,β-fold between α5TE and β5TE. The catalytic triad (Ser1138, His1271 and Asp1165) and active site are located on loops of the α,β-fold. The PCP domain contains four α-helices along with a short (5 amino acid) helix. The configuration of the PCP domain resembles the A/H state as defined for alternate confirmations of an isolated PCP domain from B. brevis tyrocidine synthetase (Koglin et al., 2006). As expected, electron density corresponding to the ppant arm is contiguous with Ser1006 on the PCP domain (Figure 3). The modeled ppant analog extends from Ser1006 making minimal contacts with the PCP domain, while extensive interactions are evident with the TE domain. The ppant group lies in a channel in the TE domain leading to the catalytic triad. Interactions with the seryl-phosphate are minimal with only hydrogen bonding interactions with a series of ordered water molecules. A loop region between β2TE and α1TE (residues 1074-1080) is involved in a majority of the contacts between the protein and the ppant arm. A hydrophobic pocket forms around the gem-dimethyl section of the ppant (Figure 3). Among the residues in this pocket are Phe1077 and Trp1079 of the first α-helix (α1TE) of the TE domain and Ile1213 from a lid helix (α6TE). Additional interactions include hydrogen bonding interactions between Ala1074 and Ser1075 with two amide carbonyls of the ppant and van der Waals contact between Gln1080 and one of the amides.
Figure 2. The overall structure of the PCP-TE conjugate.
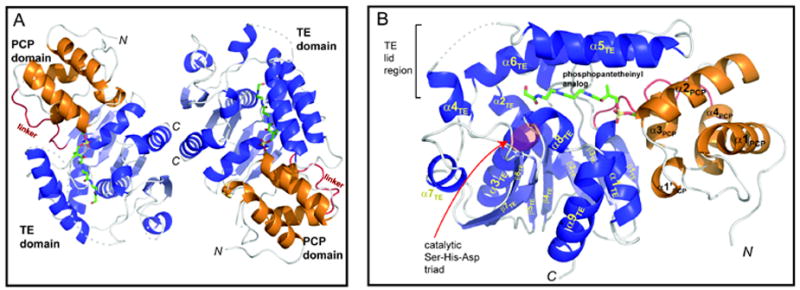
(A), Ribbon representation of the protein dimer observed in the asymmetric unit. The PCP domain (orange) is connected to the TE domain (blue) through a short linker (red). The conjugated CoA analog is shown in licorice representation. Regions not observed in the electron density maps are indicated by dashed lines. (B), Closeup view of one of the monomer didomains. See also Figures S1, and S2.
Figure 3. The structure of the ppant arm and interactions with the TE domain.
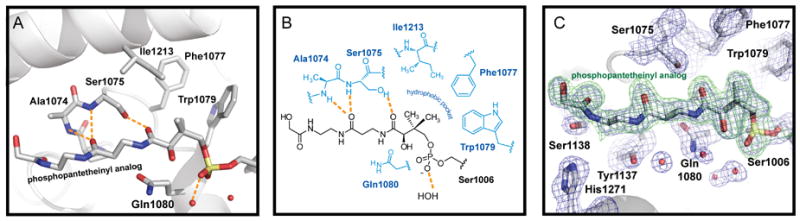
Key residues are shown in licorice format (A) and graphical representation (B). (C), A sigma-weighted composite omit electron density map (blue, contoured at 1.5σ) along with an Fo-Fc map (2.2σ) computed without the ppant arm.
The PCP domain forms extensive interactions with the TE domain encompassing ~1450 Ǻ2 of total buried surface area not including the ppant arm. The largely hydrophobic interactions between the domains primarily involve α2PCP/α3PCP and α1TE/β1TE. Additionally, the helices of the TE’s lid region (α5TE/α6TE) extend to form contacts with the carrier domain at α2PCP (Figure 4). The specific interactions largely involve hydrophobic side chains including the previously described hydrophobic pocket near the gem-dimethyl of the ppant. In addition, two leucine residues (Leu1058,1060 on β1TE) at the start of the TE domain interact with complementary hydrophobic residues in α3PCP of the PCP.
Figure 4. Domain-domain interactions.
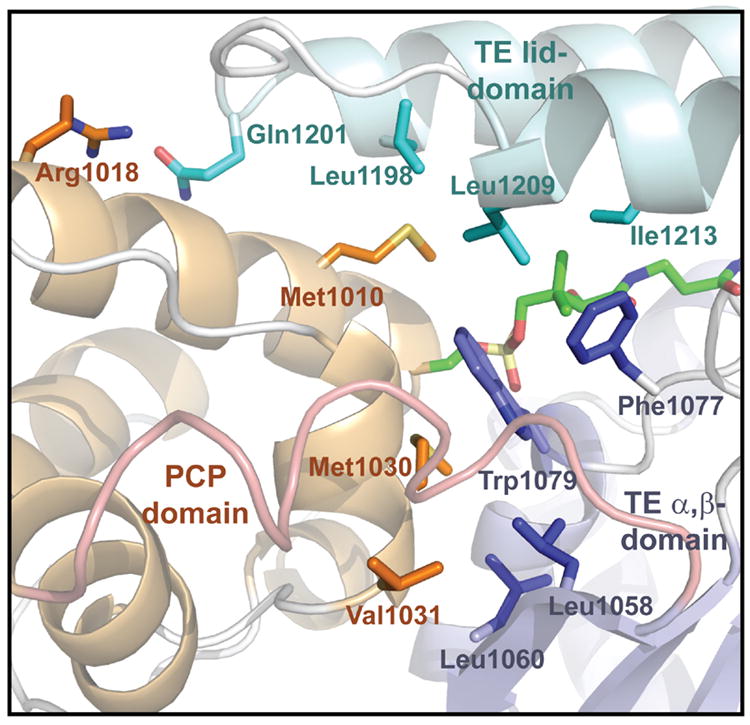
The interdomain contacts between the PCP domain (orange) and the TE domain are highlighted. The two distinct regions of the TE domain are designated; lid (light blue) and the α,β fold (dark blue). See also Figure S4.
Based on the structural model, residues of the TE predicted to be important for interaction with the ppant arm and the PCP were analyzed by biochemical characterization of site-directed mutants of the full length EntF four-domain enzyme. In order to assess the roles of mutations on the activity of the synthetase, an enterobactin reconstitution assay was used (Gehring et al., 1998). In vitro production of the natural product was monitored by HPLC (Figure S3). The results obtained are consistent with the assigned roles in the interdomain interface. Trp1079 from the TE formed substantial interactions involved in both the domain interface and in forming a hydrophobic ppant pocket. Mutation of this residue resulted in the most dramatic affect on enterobactin production with no natural product observed under the assay conditions. Gln1080 forms a van der Waals interaction with the extended ppant and Q1080A mutant retained 50% of activity. Mutation of Phe1077 had an intermediate affect on enterobactin product (2% relative activity) as expected based on its role in forming hydrophobic contacts with the gem-dimethyl of the ppant.
The residues of the catalytic triad are well-ordered in the active site of the TE domain and in an orientation common to serine hydrolases (Figure 5A). The ppant channel (approximately 8 Ǻ by 20 Ǻ) leads to the triad and the hydroxyl side chain of Ser1138 is properly positioned to attack the terminal carbonyl of the ppant analog. The resulting oxyanion is stabilized by backbone amide hydrogens of Leu1139 and Ala1074 forming a classic oxyanion hole motif. Mass spectral analysis of the protein loaded with the ppant analog was consistent with loss of chlorine. However, the main mass observed was larger (by 18 Da) than that predicted from covalent linkage to the histidine and suggested that a terminal-hydrolyzed ppant analog product is formed after loss of chlorine. Based on these observations and analysis of the electron density maps, it is predicted that α-Cl-acetyl-amide ppant arm first reacts with Ser1138 analogous to α-chloromethyl ketones (Figure 5B). The reactive intermediate can undergo nucleophilic attack by water to yield the α-hydroxy-acetyl amide. Alternatively, attack by His1271 gives a crosslinked product than can hydrolyze over the lengthy time of crystallization. It is also possible that the analog reacts directly with an activated water molecule, though this mode of reactivity is uncommon in α,β-hydrolyase enzymes.
Figure 5. Interaction of the ppant arm with the active site of the TE domain.
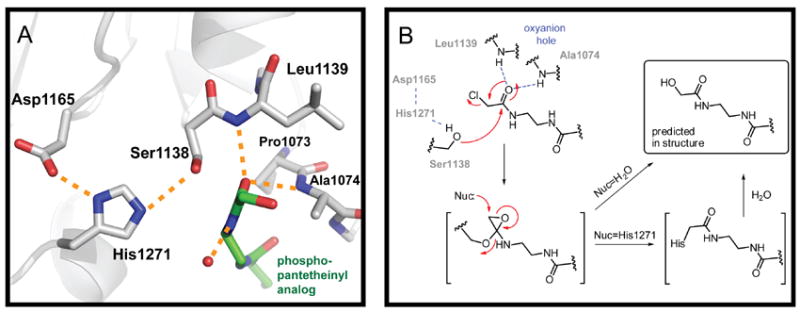
(A), Residues of the catalytic triad (Ser1138, His1271, Asp1165) and oxyanion hole (Ala1074 and Leu1139) are shown in licorice representation. (B), Proposed mechanism for fate of the α-Cl-amide moiety in the active site. See also Figures S5 and S6.
Discussion
The carrier domain and its attached ppant prosthetic group are integral players in secondary metabolite pathways, yet little atomic resolution information is available on holo-carrier domains interactions. Efforts to rationally engineering assembly line chemistry through approaches such as domain swapping would benefit from details of carrier domain/enzyme interactions. Presented is the structure of detailing ppant/carrier domain interactions with an active site of the natural product synthetase. A chemical approach using designed analogs of coenzyme A is exploited to produce a stable didomain conjugate suitable for X-ray crystallographic analysis. The use of stable amide-based analogs of pathway thioesters is a general strategy to target enzyme active sites and is readily applicable to other domains of NRP synthetases and polyketide/fatty acid synthases.
The presented structure provides details of protein/protein interactions between the TE and PCP. The gross structure of the two domains is similar to that observed in the NMR structure of the apo-PCP-TE construct (Frueh et al., 2008). The NMR structure showed the two domains as mobile, adopting multiple conformations depending on the context. In contrast, the X-ray structure of the holo-PCP-TE is compact and ordered with extensive domain/ppant and domain/domain interactions (see Supplementary information). As compared to the NMR structure, the helices of the PCP domain (especially α2PCP and α1PCP) move towards the TE domain and the helix α5TE in the lid region shifts closer to the PCP (Figure S4). The distance between the two conserved Ser residues in the active sites of the PCP and TE domain is shortened to ~16 Å from 19 Å of the apo structure. A helix (α3PCP) of the PCP domain and a β-sheet (β1TE) of the TE domain at the domain interface in the structure are not observed in the apo-structure. As mentioned previously, Met1030 and Val1031 are located on helix α3PCP forming hydrophobic interactions with Leu1058 and Leu1060 residues on the β1TE sheet, which may contribute to this ordered secondary structure. Met1030 is also suggested to be crucial for the in cis interaction with the TE domain by the previous combinatorial mutagenesis studies (Zhou et al., 2006). Comparison with apo-PCP-TE indicates that phosphopantetheinylation leads to enhanced interaction between the two domains. In addition, loops (between β6TE and α7TE, β5TE and α5TE, and β7TE and α9TE) are more structured than in the apo-structure by forming three short helices α4TE, α7TE, and α8TE, which may attribute to the interaction of the inhibitor arm with the substrate-binding site of the TE domain. Interaction between the lid region of the TE and the PCP was also more pronounced than in the apo-NMR structure. This contact could assist in ordering the active site of the TE in order to accommodate the relatively large substrate. The cyclotrimerization catalyzed by the EntF TE likely requires an interplay between the two domains that can translate structural changes from the carrier domain to the thioesterase active site.
The apo-structure of the four-domain surfactin synthetase, SrfA-C, provides an overall picture of the multidomain organization of NRPS assembly lines (Tanovic et al., 2008). When superimposing the TE domains of PCP-TE and SrfA-C, the TE α,β-sandwich core region is conserved between the two different TEs and the major difference is in the lid region (Figure S5). The lid in SrfA-C is very similar to the previously reported ‘open’ state of the individual SrfTE domain,(Bruner et al., 2002) while the lid of the phosphopantetheinyl amide PCP-TE conjugate is distinct from either of the ‘open’ or ‘closed’ form. Furthermore, the distance between the active sites of the PCP and TE domain in the SrfA-C structure (43Å) is much larger than that in PCP-TE complex (16Å), consistent with the previous conclusion that the PCP domain in SrfA-C is in an orientation to interact with the accepter site of the C domain.
As the ppant arm of EntF interacts with the TE domain as well as two additional domains (A and C), we were curious whether the observed contacts were shared by other NRP synthetase PCP-TE didomains. The alignment of EntF PCP-TE sequence with those of other PCP-TE didomain fragments from surfactin, fengycin, and tyrocidine NRPSs shows that most of the hydrophobic residues in the domain/domain interface discussed previously are conserved, such as M1010 and M1030 from the PCP domain, L1058, L1060 and W1079 from the TE α,β core region, and L1209 from the lid region (Figure S6). P1073 is absolutely conserved in all of the PCP-TE fragments, crucial in orienting the amide hydrogen of the backbone into the oxyanion hole. The position of F1077 in the hydrophobic pocket aligns with Phe, Tyr and Gln in other systems, though is predictable as the gem-dimethyl group interacts with the β-carbon of F1077. Based on these alignment results, the in cis interactions between the PCP and TE domain of EntF can likely be extended to other systems.
Significance
Presented is the 1.9 Å resolution structure of a ‘holo’-PCP/TE didomain from the termination module of a NRP synthetase assembly line. This work represents the first structure of a holo-NRPS multidomain fragment in a catalytically-relevant orientation. The crystallization and structure determination were feasible by employing a designed coenzyme A analog with an electrophilic group attached to the end and subsequently a ppant analog loaded on to a truncated didomain construct. This allowed the construct to specifically form a compact conformation, mimicking the catalytic state of the enzyme bound to the natural substrate. The conformationally restricted didomain structure, for the first time, provides detailed insight into how the phosphopantetheinyl arm interacts with its partner enzyme and stabilizes domain/domain interactions. This methodology can be extended towards the crystallographic and mechanistic study of other multidomain NRP synthetases and FA/PK synthases.
Experimental Procedures
Cloning and expression of EntF PCP-TE didomain construct
A gene encoding EntF PCP-TE didomain was cloned from an expression plasmid for full length EntF(Gehring et al., 1998) into the BamHI and XhoI sites of pET30a. The vector was transformed into BL21 (DE3) cells and grown in 1L of LB medium at 37 °C to an O.D. of 0.6. IPTG (50 μM) was then added and incubation continued at 18 °C for 18 h. Cells were harvested and resuspended in lysis buffer (20 mM Tris·HCl pH 7.5, 500 mM NaCl) and lysed with a French Press homogenizer. The lysate was clarified by centrifugation at 10,000 rpm and was subjected to Ni-NTA affinity chromatography (Qiagen). The protein was washed with lysis buffer four times, then elution buffer (lysis buffer with 250 mM imidazole). Elution fractions were dialyzed against 100 mM NaCl, 20 mM Tris·HCl pH 7.5, 1 mM βME and concentrated to ~1mL. 1 μL of enterokinase (New England Biolabs) added and incubated at 25 °C for 1.75 h. Cleaved protein was separated from the uncleaved by Ni-NTA affinity chromatography then purified using HiTrap-Q (GE Biosciences) ion exchange column using a gradient of 0-80% buffer B over 30 min at a flow rate of 2 ml/min (buffer A: 50 mM Tris·HCl pH 7.5, 1 mM βME; buffer B: 1 M NaCl, 20 mM Tris·HCl pH 7.5, 1 mM βME). For the production of SeMet-labeled EntF PCP-TE, the gene was cloned into the NdeI and EcoRI sites of pET28a vector as the SeMet-protein was not stable under the enterokinase cleavage condition as described above. The vector was transformed into B834 (DE3) methionine auxotroph cells (Novagen). 1L of M9 minimal medium was inoculated with 1mL of an LB-preculture pre-washed with M9 medium. To the 1L culture was added 50 mg each of 19 amino acids (w/o Met), 10 mL 20% glucose, 2 mL 1M MgSO4, 0.05 mL 2M CaCl2, 0.1 mL of 0.5% (w/v) thiamine, DL-seleno-methionine (60 mg) and FeSO4 (15 μM). When an OD of 0.6 was reached, the temperature was reduced to 15 °C and additional DL-seleno-methionine (30 mg) was added to the culture. After 15 min, IPTG (50 μM) was added and incubation continued for 18 h. Purification of SeMet-labeled protein followed the protocol described above except: purification buffers contatined 5 mM DTT and were degassed prior to usage. The His-tag was cleaved by incubating with thrombin (7.5 U per 1 mL concentrated protein solution in 100 mM NaCl, 20 mM Tris·HCl 7.5, 2 mM CaCl2) at 4 °C overnight.
Preparation of an α-chloroacetyl amide conjugated EntF PCP-TE construct
In a 5 mL reaction mixture, purified EntF PCP-TE (30 μM) was incubated with 75 mM Tris·HCl (pH 8.0), 1 mM TCEP, 10 mM MgCl2, 150 μM α-chloroacetyl amide CoA (Liu et al., 2007), 3 μM PPTase Sfp from B. subtilis (Quadri et al., 1998). The reaction was incubated for 2 h at 20 °C and immediately purified by Superdex 200 (GE Biosciences) gel filtration chromatography in 100 mM NaCl, 20 mM Tris·HCl pH 7.5, 1 mM βME (or 5 mM DTT for SeMet protein) to give the α-chloroacetyl amide EntF PCP-TE construct. Loading of the α-chloroacetyl amide phosphopantetheinyl arm was verified by LC-MS.
Crystallization and structure determination
Crystals of native and SeMet-labeled α-chloroacetyl amide EntF PCP-TE were obtained by the hanging drop method at 20 °C. Native protein was mixed with 1.5 μL of reservoir solution containing 24.5% (w/v) PEG 6K, 0.1 M Tris·HCl pH8.5 and 70 mM MgCl2. The crystallization reservoir solution for SeMet-labeled protein contained 21% (w/v) PEG 8K, 0.1 M Tris·HCl pH 8.5 and 72.5 mM MgCl2. Crystals were observed after two days and continued growth for several additional days. Crystals were soaked in cryoprotectant solution (reservoir solution with 20% glycerol) for 10 min and then flash frozen in liquid nitrogen. X-ray diffraction data of native α-chloroacetyl amide EntF PCP-TE were collected to 1.9 Å resolution on the X25A beamline at the National Synchrotron Light Source at Brookhaven National Labs. A 2.2 Ǻ SAD dataset a SeMet derivativized crystal was collected on the same beamline. All diffraction data were indexed, integrated and scaled using the HKL2000 program (Otwinowski et al., 1997) in the space group P212121 (a=69.40 Ǻ, b=90.36 Ǻ, 97.53 Ǻ, α=β=γ=90°). Two monomers are present in the asymmetric unit (Matthews coefficient of 2.12 and 42% solvent content). 10 selenium sites were found using hkl2map software (Pape et al., 2004). Heavy atom phasing was carried out with the PHENIX software package (Adams et al., 2004). Initial electron density maps were calculated and a first model of EntF PCP-TE was obtained using PHENIX. The initial model contained a significant part of TE domains and a limited portion of the PCP domains. The initial model was further built with the assistance of non-crystallographic symmetry operations on the existing fragments using the program COOT (Emsley et al., 2004). The structure was refined until Rworking/Rfree converged to 20.9%/24.2% using CNS (Brünger et al., 1998) (Table S1). Sigma-weighted simulated composite omit maps (CNS) were used to judge and verify structures throughout refinement. Water molecules were added manually and evaluated based on inspection of the maps. Figures in the manuscript were made using the program PYMOL.
Supplementary Material
Highlights.
Formation and purification of a phosphopantethenyl conjugate of a synthetase didomain with a mechanism-based inhibitor.
Crystallization and structure solution of the didomain in a catalytically relevant conformation.
Use of site-directed mutagenesis to probe the didomain interface using a natural product reconstitution assay
Acknowledgments
This work was supported by grants from the NIH (GM086570) and NSF(CAREER-0645653). We are grateful to C. T. Walsh for plasmids containing genes in the enterobactin pathway and we thank members of the Bruner laboratory for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Akey DL, et al. Structural basis for macrolactonization by the pikromycin thioesterase. Nat Chem Biol. 2006;2:537–542. doi: 10.1038/nchembio824. [DOI] [PubMed] [Google Scholar]
- Alekseyev VY, Liu CW, Cane DE, Puglisi JD, Khosla C. Solution structure and proposed domain domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase. Protein Sci. 2007;16:2093–2107. doi: 10.1110/ps.073011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner SD, et al. Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure. 2002;10:301–310. doi: 10.1016/s0969-2126(02)00716-5. [DOI] [PubMed] [Google Scholar]
- Brünger AT, et al. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Crump MP, et al. Solution structure of the actinorhodin polyketide synthase acyl carrier protein from Streptomyces coelicolor A3(2) Biochemistry. 1997;36:6000–6008. doi: 10.1021/bi970006+. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Findlow SC, Winsor C, Simpson TJ, Crosby J, Crump MP. Solution structure and dynamics of oxytetracycline polyketide synthase acyl carrier protein from Streptomyces rimosus. Biochemistry. 2003;42:8423–8433. doi: 10.1021/bi0342259. [DOI] [PubMed] [Google Scholar]
- Frueh DP, et al. Dynamic thiolation-thioesterase structure of a non-ribosomal peptide synthetase. Nature. 2008;454:903–906. doi: 10.1038/nature07162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring AM, Mori I, Walsh CT. Reconstitution and characterization of the Escherichia coli enterobactin synthetase from EntB, EntE, and EntF. Biochemistry. 1998;37:2648–2659. doi: 10.1021/bi9726584. [DOI] [PubMed] [Google Scholar]
- Hur GH, et al. Crosslinking studies of protein-protein interactions in nonribosomal peptide biosynthesis. Chem Biol. 2009;16:372–381. doi: 10.1016/j.chembiol.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S, et al. Mechanism based protein crosslinking of domains from the 6-deoxyerythronolide B synthase. Bioorg Med Chem Lett. 2008;18:3034–3038. doi: 10.1016/j.bmcl.2008.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koglin A, et al. Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science. 2006;312:273–276. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- Koglin A, et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–911. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koglin A, Walsh CT. Structural insights into nonribosomal peptide enzymatic assembly lines. Nat Prod Rep. 2009;26:987–1000. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JR, Koglin A, Walsh CT. Carrier protein structure and recognition in polyketide and nonribosomal peptide biosynthesis. Biochemistry. 2006;45:14869–14879. doi: 10.1021/bi061979p. [DOI] [PubMed] [Google Scholar]
- Leibundgut M, Jenni S, Frick C, Ban N. Structural basis for substrate delivery by acyl carrier protein in the yeast fatty acid synthase. Science. 2007;316:288–290. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- Leibundgut M, Maier T, Jenni S, Ban N. The multienzyme architecture of eukaryotic fatty acid synthases. Curr Opin Struct Biol. 2008;18:714–725. doi: 10.1016/j.sbi.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Li Q, Khosla C, Puglisi JD, Liu CW. Solution structure and backbone dynamics of the holo form of the frenolicin acyl carrier protein. Biochemistry. 2003;42:4648–4657. doi: 10.1021/bi0274120. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bruner SD. Rational manipulation of carrier-domain geometry in nonribosomal peptide synthetases. Chembiochem. 2007;8:617–621. doi: 10.1002/cbic.200700010. [DOI] [PubMed] [Google Scholar]
- Lomakin IB, Xiong Y, Steitz TA. The crystal structure of yeast fatty acid synthase, a cellular machine with eight active sites working together. Cell. 2007;129:319–332. doi: 10.1016/j.cell.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Maier T, Jenni S, Ban N. Architecture of mammalian fatty acid synthase at 4.5 A resolution. Science. 2006;311:1258–1262. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- Meier JL, Burkart MD. The chemical biology of modular biosynthetic enzymes. Chemical Society Reviews. 2009;38:2012–2045. doi: 10.1039/b805115c. [DOI] [PubMed] [Google Scholar]
- Meier JL, Haushalter RW, Burkart MD. A mechanism based protein crosslinker for acyl carrier protein dehydratases. Bioorg Med Chem Lett. 2010;20:4936–4939. doi: 10.1016/j.bmcl.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer AC, Burkart MD. The ubiquitous carrier protein-a window to metabolite biosynthesis. Nat Prod Rep. 2007;24:750–773. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: C CW, S JRM, editors. Methods in Enzymology: Macromolecular Crystallography, part A. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Pape T, Schneider TR. HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J App Crystallography. 2004;37:843–844. [Google Scholar]
- Ploskon E, et al. A mammalian type I fatty acid synthase acyl carrier protein domain does not sequester acyl chains. J Biol Chem. 2008;283:518–528. doi: 10.1074/jbc.M703454200. [DOI] [PubMed] [Google Scholar]
- Prorok M, Albeck A, Foxman BM, Abeles RH. Chloroketone hydrolysis by chymotrypsin and N-methylhistidyl-57-chymotrypsin: implications for the mechanism of chymotrypsin inactivation by chloroketones. Biochemistry. 1994;33:9784–9790. doi: 10.1021/bi00198a050. [DOI] [PubMed] [Google Scholar]
- Quadri LE, et al. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- Samel SA, Wagner B, Marahiel MA, Essen LO. The thioesterase domain of the fengycin biosynthesis cluster: a structural base for the macrocyclization of a non-ribosomal lipopeptide. J Mol Biol. 2006;359:876–889. doi: 10.1016/j.jmb.2006.03.062. [DOI] [PubMed] [Google Scholar]
- Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure. 2007;15:781–792. doi: 10.1016/j.str.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Sharma SK, Surolia A, Surolia N, Sarma SP. Solution structures of conformationally equilibrium forms of holo-acyl carrier protein (PfACP) from Plasmodium falciparum provides insight into the mechanism of activation of ACPs. Biochemistry. 2006;45:6904–6916. doi: 10.1021/bi060368u. [DOI] [PubMed] [Google Scholar]
- Shaw-Reid CA, et al. Assembly line enzymology by multimodular nonribosomal peptide synthetases: the thioesterase domain of E. coli EntF catalyzes both elongation and cyclolactonization. Chem Biol. 1999;6:385–400. doi: 10.1016/S1074-5521(99)80050-7. [DOI] [PubMed] [Google Scholar]
- Tanovic A, Samel SA, Essen LO, Marahiel MA. Crystal structure of the termination module of a nonribosomal peptide synthetase. Science. 2008;321:659–663. doi: 10.1126/science.1159850. [DOI] [PubMed] [Google Scholar]
- Tran L, Broadhurst RW, Tosin M, Cavalli A, Weissman KJ. Insights into protein-protein and enzyme-substrate interactions in modular polyketide synthases. Chem Biol. 2010;17:705–716. doi: 10.1016/j.chembiol.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Tsai SC, et al. Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: versatility from a unique substrate channel. Proc Natl Acad Sci USA. 2001;98:14808–14813. doi: 10.1073/pnas.011399198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Baumgartner R, Renner C, Marahiel MA, Holak TA. Solution structure of PCP, a prototype for the peptidyl carrier domains of modular peptide synthetases. Structure Fold Des. 2000;8:407–418. doi: 10.1016/s0969-2126(00)00120-9. [DOI] [PubMed] [Google Scholar]
- Weissman KJ, Muller R. Protein-protein interactions in multienzyme megasynthetases. Chembiochem. 2008;9:826–848. doi: 10.1002/cbic.200700751. [DOI] [PubMed] [Google Scholar]
- Worthington AS, Rivera H, Torpey JW, Alexander MD, Burkart MD. Mechanism-based protein cross-linking probes to investigate carrier protein-mediated biosynthesis. ACS Chem Biol. 2006;1:687–691. doi: 10.1021/cb6003965. [DOI] [PubMed] [Google Scholar]
- Worthington AS, Porter DF, Burkart MD. Mechanism-based crosslinking as a gauge for functional interaction of modular synthases. Org Biomol Chem. 2010;8:1769–1772. doi: 10.1039/b925966j. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Lai JR, Walsh CT. Interdomain communication between the thiolation and thioesterase domains of EntF explored by combinatorial mutagenesis and selection. Chem Biol. 2006;13:869–879. doi: 10.1016/j.chembiol.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Zornetzer GA, Fox BG, Markley JL. Solution structures of spinach acyl carrier protein with decanoate and stearate. Biochemistry. 2006;45:5217–5227. doi: 10.1021/bi052062d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.