

Abstract
Although big progress has been made in sample pretreatment over the last years, there are still considerable limitations when it comes to overcoming complexity and dynamic range problems associated with peptide analyses from biological matrices. Being the little brother of proteomics, peptidomics is a relatively new field of research aiming at the direct analysis of the small proteins, called peptides, many of which are not amenable for typical trypsin-based analytics. In this paper, we present an overview of different techniques and methods currently used for reducing a sample's complexity and for concentrating low abundant compounds to enable successful peptidome analysis. We focus on techniques which can be employed prior to liquid chromatography coupled to mass spectrometry for peptide detection and identification and indicate their advantages as well as their shortcomings when it comes to the untargeted analysis of native peptides from complex biological matrices.
1. Introduction
Peptides are small (low molecular weight, LMW) proteins, built up of amino acids connected by peptide bonds. The shortest peptide is two amino acids long, and with increasing length of the amino acid chain, the name changes from peptide over polypeptide to protein, with a fuzzy border between them. Also the International Union of Pure and Applied Chemistry (IUPAC) has no clear weight or amino acid chain length limit. A somewhat arbitrary but quite generally acknowledged definition puts the boundary between a peptide and a protein at a chain length of 50 amino acids (WIKIPEDIA). In literature, often pragmatic definitions are used, such as “the small proteins typically running off a typical 2D polyacrylamide gel” or “the small proteins with zero or maximally one tryptic cleavage site”, and, therefore, different upper molecular weight limits for peptides can be found from 10 kDa and even beyond [1–6]. As proteomics is for proteins, peptidomics is the comprehensive study of all (native) peptides in a biological sample. In the last decade, proteomics has gained increasing recognition as a reliable and reproducible approach to study molecular processes in high throughput at a global level. Recently, also peptidomics is becoming more and more a “hot topic” as it is recognized that peptides play complex regulatory roles in many if not all biological processes, for example, intercellular signaling [7, 8]. As such, peptide signals secreted into the extracellular medium, reflect the state of a cell in a certain condition, and, by definition, are potential biomarkers indicative for specific physiological/pathological processes [9–11]. Currently, with a general proteomic approach, it is possible to detect and identify several hundreds to a few thousands of proteins in a single experiment [12–14]. With modern mass spectrometers excelling in sensitivity and dynamic range, also the cell's peptidomes become much more comprehensively accessible for analysis, and the discovery of novel biomarkers becomes possible, such as in innovative cancer research. For example, Ueda et al. [15] used size-exclusion-based enrichment of the peptidome in combination with label-free quantitation by nano-LC-MS/MS for peptidome profiling in lung carcinomas.
Body fluids, especially blood serum or plasma, and, in particular cases, (primary) cell culture media, serve as typical and readily available sources for a “peptidomics-driven” discovery of novel candidate disease biomarkers. However, the detection of peptide biomarkers typically present at low concentrations is hampered by the “masking” effect caused by a number of highly abundant proteins [11, 16–19]. The large dynamic concentration range, in which peptides and proteins are present in a biological system presents a major bottleneck for peptidomic discovery of new biomarkers. Figure 1 reflecting the large dynamic range of proteins and peptides in human blood plasma is very illustrative in this respect. Especially-the presence of albumin in a sample has been shown to prevent the successful identification of low-abundance biomarkers in many peptidomic studies [17, 20, 21]. Hence, different methods for capturing, partitioning, fractionating, depleting, or enriching a sample have been developed [22, 23].
Figure 1.
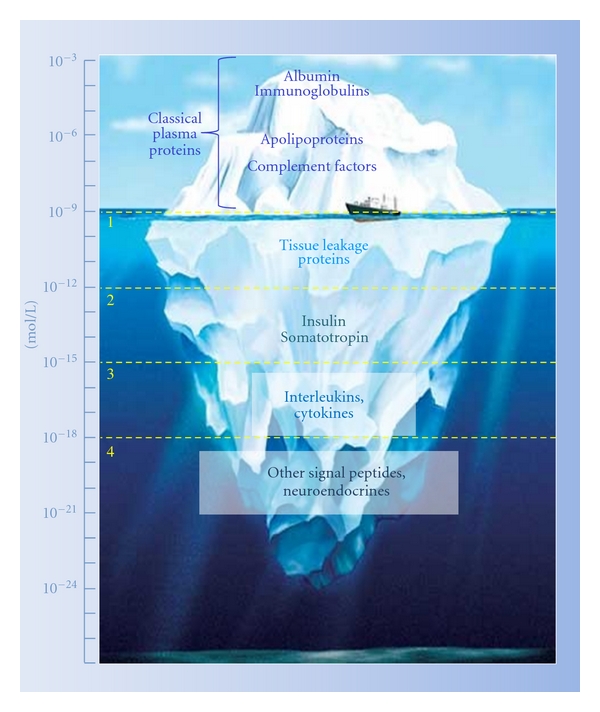
Illustrative iceberg representation of high dynamic range of proteins found in blood, showing various classes of proteins and peptides (figure composed of literature data [21] and others). Four arbitrary assemblies of proteins/peptides can be made, and representative species are indicated. Iceberg tip contains abundant classical plasma proteins detectable in 1 μL of sample or less. Tissue leakage proteins typically require, at least, 1 mL plasma volumes (typically after depletion of interfering abundant proteins), and concentration ranges of secreted signal peptides/proteins like insulin, somatotropin are yet another 3 orders of magnitude lower. Interleukins and other cytokines really push current MS systems to their very limit, whereas other neurosecretory signal peptides require extensive concentration steps to reach levels detectable by MS.
Liquid chromatography (LC) coupled with tandem mass spectrometry (MS/MS) is the analytical method of choice in today's proteomics and peptidomics research. Its major benefits include enhanced specificity (particularly over the GC-MS technologies of 25 years ago, which had very limited applicability for peptide separations), its potential for high-throughput analyses, no requirement for expensive analyte-specific reagents, high speed of assay development, and a relatively low cost per assay (the instrument itself, however, not being that cheap) [24].
In an ideal world, no sample preparation would be required for the analysis of a sample as every manipulation can lead to problems such as loss of sample and, even worse, loss of quantifiability. In particular, for the proteomic/peptidomic analysis of native peptides, which typically do not require protease (trypsin) digestion prior to LC-MS/MS analysis, a so-called “top-down” approach seems logical. However, as the complexity of samples still far exceeds the capacity of currently available analytical systems, specific sample preparation remains a crucial part of the analysis in a whole. Peptidomics sample preparation is often time consuming and laborious, involving multiple steps [25, 26]. In this paper, we present an overview of different techniques (see Figure 2) used for the simplification of complex biological samples and review advantages and possible problems related to them (see Table 1). Our focus will be on biofluids such as blood, urine, or cerebrospinal fluid (CSF), but some issues related to the extracellular peptidome (part of the so-called “secretome”) analysis of cultured cells will be considered first.
Figure 2.
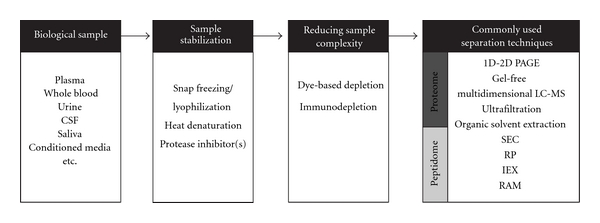
Schematic overview of methods and techniques used in proteome and peptidome analyses for sample preparation prior to LC MS/MS.
Table 1.
Summary of the strengths and weaknesses of analytical tools used in peptidome research as discussed in this review. Abbreviations: IEX: ion exchange; LC: liquid chromatography; MWCO: molecular weight cut-off; OS: organic solvent extraction; PAGE: polyacrylamide gel electrophoresis; RAM: restricted access material; RP: reversed phase; SPE: solid phase extraction; UF: ultrafiltration; SEC: size exclusion chromatography.
| Technique | Strengths | Weaknesses |
|---|---|---|
| Depletion | (i) Removes highly abundant “household” proteins, allowing a “deeper” look into the peptidome | (i) Requires costly antibody columns |
| (ii) Each protein to be removed requires a different specific antibody | ||
| (iii) Loss of peptides by nonspecific binding | ||
| PAGE | (i) Traditional well-established method (ii) able to separate isoforms or PTMs (iii) able to remove low molecular organic and inorganic impurities |
(i) Unsuitable for highly complex samples, poor dynamic range (ii) Poor resolving power (iii) Proteins/peptides with extreme pI values cannot be separated (iv) The smallest peptides are not retained in MW separation dimension (v) Very time consuming and laborious (vi) Low dynamic range |
| OFFGel | (i) Effective prefractionation tool (ii) Medium resolution |
(i) Postconcentration is required low resolution (ii) No MWCO discrimination (iii) long separation times |
| UF | (i) Fast (ii) Inexpensive (iii) Easy to automate |
(i) Variable quality and reproducibility of commercial devices (ii) Loss of hydrophobic peptides by nonspecific binding |
| LC (SPE/RP/IEX) |
(i) High resolution (ii) Directly compatible with MS (iii) High sensitivity in nanoLC (iv) SPE flexible, easy to automate (v) Efficient preconcentration |
(i) Extensive method development for each specific matrix is required |
| SEC | (i) High resolving power (ii) Reproducible |
(i) Loading limited by small injection volume |
| RAM | (i) Effective removal of HMW compounds (ii) Relatively large injection volumes (iii) Analysis of untreated sample matrices possible (iv) Easy to automate (v) Online RAM coupled to (nano-)LC-MS/MS possible |
(i) Complicated LC setup (ii) Extensive application directed method development and optimization required |
| OS | (i) Easy to operate (ii) Inexpensive |
(i) Tedious to perform |
2. Sample Preparation Techniques Used for the Analysis of LMW Secretome/Signaling Proteins
2.1. Cell Culture Conditions
Conditioned media are cell culture media whose cells have grown in for a certain period of time. The cells “condition” the media by releasing/secreting proteins, cytokines, and other biomolecules. As such, culture supernatants or conditioned media (CM) can be considered yet another (“body”) fluid that can serve as a source for the identification of novel biomarkers, for example, in cancer research [27]. It is important to note that in order to promote a healthy growth of cells in culture, the culture medium has to be supplemented with a standard cocktail of nutrients and growth factors. In mammalian cell cultures, this is typically achieved by the addition of a substantial volume of fetal calf serum (FCS, up to 10% solution). When studying the intracellular proteome (or peptidome), cells are washed several times with FCS-free cell culture medium and/or phosphate-buffered saline (PBS) prior to lysis to reduce contamination of the sample with (bovine) serum proteins. However, this is not possible when the extracellular peptidome is under investigation. As a compromise, in most studies, the medium containing FCS is replaced by FCS-free medium or medium containing reduced amounts of FCS just before starting the secretome experiment (e.g., giving a biological/physiological stimulus). For adherent cells, it is relatively easy and noninvasive to replace the medium. However, when studying cells in suspension, several centrifugation steps are required before the medium can be replaced, which arguably is a source of unnatural stress to the cells, possibly even causing cell lysis. A main disadvantage of working with reduced amounts of FCS (or no FCS at all) is that also this is known to cause metabolic stress to the cells, not seldom inducing cell death and consequently altering the cell culture's secretome [11, 28–34].
2.2. Gel Electrophoresis (1D-2D PAGE)
A traditional well-established technique in proteomics is one- or two-dimensional (1D or 2D) polyacrylamide gel electrophoresis (PAGE). Although more a protein than a typical peptide separation technique, 1D and 2D PAGE are used in “peptidomics” workflows as well, particularly when targeting larger (poly)peptides or proteins. The latter can then be analyzed by MS after extraction from the gel as proteolytic fragments, after in-gel digestion. Denaturing agents, such as SDS, are used to unfold the macromolecules and disrupt noncovalent intra- and intermolecular protein/protein interactions. SDS PAGE is a rather simple technique and, above all, very robust. Its poor resolving power, however, often poses a problem in the analysis of complex mixtures. 1D and particularly 2D PAGE are employed to increase the depth of proteome/peptidome analysis, that is, through fractionation of the sample components and removal of LMW impurities, particularly salts, which interfere with subsequent MS analyses [35–41].
2D PAGE is very sensitive predominantly to molecular charges of a protein (by the isoelectric focusing step), making it a very effective method to reveal/separate certain posttranslational modifications like phosphorylations, sulfations, or glycosilations. Limitations are that proteins/peptides with extreme pI values cannot be separated and that the smaller peptides are typically not retained in the second (MW separation) dimension. 2D PAGE followed by in-gel digestion is relatively time consuming and laborious. The dynamic range for detection in 2D PAGE is 102–104, which is less than the protein expression range observed in biological systems. In order to achieve the detection of low abundant proteins, more protein needs to be loaded. Such higher loads, however, often further compromise the technique's resolution due to spot fusion and comigration [19, 21, 39, 42–46].
Important in PAGE is the visualization of the separated proteins, although selected areas of a gel can be processed for MS from unstained preparative gels after comparison/alligning with an analytical reference or master gel.
The most commonly used visible stains are Coomassie brilliant blue (CBB) and silver nitrate staining. CBB staining is easy, MS compatible, and linear over, at least, one order of magnitude, so it is usable for quantification to a limited extent. Silver nitrate is a more sensitive staining method—0.5 ng versus 50 ng for CBB—but the staining procedure is more labor intensive and has a more limited linear range (due to its polychrome results). Although widely regarded as the standard of rigor by which all other “ultrasensitive” staining methods are judged, silver staining remains quite a complex and variable protein-gel-staining methodology, with many dozens of published protocols, all of them requiring several steps. Silver staining quantitation is never simple, due to the complex polychromatic nature of the color development and to considerable differences in response factors between different proteins. Fluorescent dyes are also popular to visualize proteins in gels; however, they are not cheap reagents and require expensive scanners/image analyzers. They exhibit detection sensitivity rivaling that of silver staining, with workflow advantages similar to CBB staining. Fluorescence offers linear quantitation ranges 10–100-fold greater than other colorimetric methods [47–50].
2.3. Off-Gel and Gel-Free Separation
Because of the limitations associated with gel-based techniques, recently with respect to detection of the smaller proteins and peptides (see above), attention has gone to off-gel methods for peptide/protein separations, in particular in solution-pI-based peptide separations without the need for carrier ampholytes. It focuses proteins and peptides on an immobilized pH gradient (IPG) gel, which is sealed against a multichamber frame that contains both sample and focusing solutions. The sample is separated by migration through the gel, followed by diffusion into the well adjacent to the section of the IPG strip. It allows for multiple samples to run simultaneously and requires only small sample volume and no prior sample cleanup. Disadvantages are that it has a rather long separation time and requires an insulated cooling system [51–53]. Cologna et al. [52] recently described a variation of the mostly used “OFFGEL” system (Agilent Technologies, USA). Their device allows customization of the number of wells or the pH gradient. Off-gel separation has not only shown applicability for proteins but also for peptides. Hubner et al. [54] compared the technique to in-gel digestion and found that using peptide off-gel separation led to a third more protein/peptide identifications Comparing off-gel separation to reversed phase liquid chromatography (RPLC) at high pH, Manadas et al. [55] conclude that RPLC leads to the identification of more peptides and also more unique peptides.
In general, off-gel separation has clear advantages over a gel-based approach with respect to focusing and concentrating peptides, but it still requires further optimization to reach the same level of identifications as an RPLC-based separation.
2.4. Specific Depletion of Highly Abundant Sample Proteins
Many different approaches exist to separate proteins based on their biochemical and biophysical properties such as molecular weight, mass, and hydrophobicity. However, these separation methods are not protein selective. Another way to reduce a sample's complexity is to specifically remove the most abundant protein(s), by doing (immuno-) affinity capturing [17, 20, 32, 39, 56–61].
Depletion of highly abundant proteins can be done based on dyes or on antibodies. An example is the removal of albumin from serum, plasma, or cell culture samples. The most used dye for removal of albumin is Cibacron blue (often in combination with protein G for the removal of IgG). This dye however does not only show affinity for albumin but also for NAD, FAD, and ATP binding sites of proteins, which often results in the unwanted removal of proteins of interest [62–64]. Several comparisons have been made on Cibacron-blue-based depletion of highly abundant proteins from a complex sample versus immunodepletion of the same sample. The overall conclusion is that the dye method is less performant than the immuno-based affinity removal: it does not only incompletely bind all albumin from the sample (lower efficacy), but it also appears to remove a substantial portion of proteins other than the targeted albumin (lower specificity) [62, 65, 66].
Immunodepletion based on monoclonal antibodies (mAbs) is generally not preferred as, besides being very expensive, these antibodies typically remove only proteins or protein fragments with the specific targeted epitope, whereas other fragments of the protein remain untouched. Therefore, immunodepletion systems are generally based on polyclonal IgG and/or IgY antibodies, targeting multiple epitopes on the same proteins. Moreover, a mixture of polyclonal antibodies to distinct proteins are nowadays commonly used for removing multiple highly abundant proteins at once [62]. Antibody-based depletion of a sample can be performed in (low-pressure) spin cartridges or (high-pressure) liquid chromatography (LC) columns.
When using antibodies, one has to consider the number of proteins that has to be depleted from the sample. Depending on the system used, it is possible to remove between 1 and 20 abundant proteins. Roche et al. compared several systems which deplete for different amounts of proteins [60]. They observed that increasing the number of depleted proteins from 12 to 20 had only little beneficial effect and could in fact even increase the removal of peptides and proteins of interest which are associated with the abundant proteins. Also Tu et al. [67] did a comparison of 2 types of multiple affinity removal system (MARS; Agilent Technologies, Inc.) depleting, respectively, 7 (MARS-7) or 14 (MARS-14) abundant proteins. They also concluded that depleting more proteins is not by definition better. The MARS-14 column removed 7 extra proteins but showed no substantial advantage over the MARS-7 in improving peptide analysis/global protein identifications from plasma.
Recently, a creative way of depleting a sample was developed, the so-called hexapeptide library of combinatorial peptide ligands. High abundant proteins are expected to quickly saturate their specific affinity ligands leaving nonbound high abundant proteins to be washed away. In contrast, low and medium abundant proteins and peptides do not saturate their ligands and hence are concentrated on the beads. This technique has the advantage that peptides and proteins are adsorbed under native conditions and thus allow monitoring of their biological activity [68, 69], although its efficacy is still debated [70].
Limited comparative studies are published on the different depletion and enrichment methods. In these few comparisons, most of these methods are found to be complementary to each other. Typically, the methods compared all lead to identification of a number of peptides and proteins, a part of which is generally identified by all methods under investigation and another part which has been identified uniquely in a sample that was treated with one of the methods [58, 68]. The only exception to this is the comparison of dye-based depletion to immunodepletion of albumin (see above). In this case, immunodepletion invariably resulted in the most efficient enrichment of low abundant proteins and peptides [62, 65, 66]. Recently, Polaskova et al. [71] compared 6 depletion columns. They, however, did not compare based on identification of proteins but on number of spots found by 2D PAGE and also took into account the costs of all methods. When only looking at protein spots, they concluded that one column (Seppro MIXED12-LC20, GenWay Biotech) had the best overall performance leading to the largest number of new proteins spots. A second column (multiple affinity removal column human 6, Agilent Technologies) in the same study led to almost the same quality results, while being cheaper and thence representing a more economical option which could become the preferred method when budgets are limited.
2.5. Alkylation and Digestion
In those cases where larger members of the peptidome need to be addressed, another crucial part of sample pretreatment is alkylation and digestion of the peptides/small proteins. This can be performed prior to or after the previously described techniques, that is, just before LC-MS/MS analysis.
To break the tertiary structure of peptides, disulfide bridges have to be disrupted (reduced) and blocked to prevent reoxidation. Breaking of disulfide bonds is traditionally achieved using reducing agents such as dithiothreitol (DTT) or tris(2-carboxyethyl)phophine hydrochloride (TCEP) [72, 73]. After reduction, the thiol groups are blocked, typically by alkylation, with iodoacetamide (IAM) being the mostly used alkylating agent. Another alkylating agent is 2-bromoethylamine (BrEA), which transforms cysteine into S-aminoethyl cysteine, a pseudo-lysine, hereby creating an extra cleavage site for proteolytic enzymes such as trypsin [74, 75], which can be an advantage in certain cases.
When larger (poly)peptide members of the peptidome are envisaged, a bottom-up approach, requiring proteolytic digestion prior to mass spectrometric analysis, is sometimes to be considered. Trypsin by far is the most used proteolytic enzyme for the degradation of proteins or peptides. This protease has the advantages of having a high cleavage specificity and being stable in a wide range of conditions. It cleaves C-terminal to arginine or lysine residues (except where the subsequent amino acid in the parent sequence is a proline). Thanks to the biological distribution of these amino acids among all proteins, the resulting peptide masses typically fall within the range required for analysis by mass spectrometry. Some larger peptides or even proteins have been described in literature that contain only 1 tryptic cleavage site, producing a peptide still too large to be readily detected by mass spectrometry. In those cases, a combination of 2 or more proteases and/or alkylating with BrEA i.s.o. IAM, may be used to assist in peptide identifications. Trypsin is very similar to chymotrypsin in primary structure, however chymotrypsin prefers cleaving C-terminal to amino acids with bulky aromatic residues such as phenylalanine, tyrosine, and tryptophan [48, 76–79]. Also other sequence-specific proteases have their advantages in view of subsequent MS analysis of the resulting peptides. A good example is metalloendopeptidase Lys-N, which guarantees a prominent positive charge at the aminoterminus of the cleaved product [80].
3. General Sample Preparation in Peptidomics
3.1. Ultrafiltration
Ultrafiltration is a rather easy and widely used technique to fractionate a proteomics sample into aLMW fraction (the “peptidome”) and aHMW fraction (the rest of the “proteome”) by centrifugation [81]. For example, in a recent study on CSF, Zougman et al. [81] demonstrated the power of ultrafiltration to separate neuropeptides from the set of larger proteins present in CSF. The ultrafiltrate low molecular weight fraction of the CSF was directly separated by nano-LC-MS/MS. Ultimately, this led to the identification of 563 peptides derived from 91 protein precursors. Several of the identified peptides were found to carry features typical of regulatory peptides. For example, they exhibited evidence of being produced by proconvertase cleavage, having a high cysteine content, bearing an aminoterminal pyroglutamate and/or carboxyterminal amidation. Interestingly, in the same study, a parallel proteome profiling by 1D PAGE was performed, and this yielded 798 proteins. For many of the identified neuropeptides in the ultrafiltrate, the corresponding precursor protein was not identified in the proteome screen, which evidently demonstrates the necessity of separating peptides from larger protein species. Many different devices are available, each with specific molecular weight cut-off (MWCO) and type of membrane. Depending on the sample and the experiment, different devices are preferred. Ultrafiltration is not seldom reported as a poorly reproducible technique, with possible removal of proteins and peptides below and above the stated MWCO [82–84].
To disrupt potential protein-protein/peptide interactions, acetonitrile (ACN) is added to the sample before ultrafiltration. ACN this way improves the recovery of LMW peptides [20, 85]. Harper et al. [86] investigated several factors which might influence the ultrafiltration such as centrifugation time and speed. In their study, slower speed and longer time than advised by the manufacturer lead to improved recovery of LMW proteins and peptides. However, at the same time, also more HMW proteins appeared to pass through the filters. Greening and Simpson [87] compared 4 different ultrafiltration devices. They concluded that devices with vertical or angular membrane configuration reduce membrane fouling, allowing continuous flow rates, even with high protein concentrations.
Ultrafiltration units can also be used as “reactors” for digestion of proteins and chemical modifications. This approach is known as filter-aided sample preparation (FASP) and it can be used to combine the advantages of in-gel and in-solution digestion [88, 89].
3.2. Organic Solvent Precipitation
The addition of organic solvents to serum causes high molecular weight (HMW) proteins to precipitate, leaving the LMW protein fraction—including the peptides—in solution. By also adding ion-pairing reagents such as trifluoroacetic acid, peptides, and smaller proteins can be dissociated from high abundant proteins, thereby facilitating their extraction [90–92]. In general, proteins are precipitated by adding ice-cold acetone or ACN. However, also other organic solvents can be used. Tucholska et al. [82] compared several solvents for organic precipitation and concluded that acetone precipitated the serum proteins most completely. They found that methanol was actually the least favorable solvent for precipitation as it still contained the largest amount of albumin in solution. Chertov et al. [90] report that the use of methanol also leads to a much larger and less dense precipitate which was more difficult to separate from the supernatant. Polson et al. [93] likewise compared several precipitation methods, concluding that ACN is the most efficient precipitant for protein removal.
As an alternative to organic solvents, ammonium sulfate (AS) can be used for precipitation of proteins. Although AS is a very efficient precipitant, it can cause interface contamination when combining with LC-MS [93].
Kawashima et al. [94] recently described a method for differential solubilization. This method consists of 2 steps, the first of which denatures proteins and peptides and by dropping the denatured sample in ice-cold acetone causes all proteins and peptides to precipitate. In the second step, the LMW proteins and peptides which dissolve in 70% ACN containing 12 mM HCl are separated from most of the other proteins. When compared to normal organic precipitation (with ACN) and ultrafiltration, they conclude this method gives a much better yield for low molecular weight peptide extraction from human serum (see Figure 3).
Figure 3.
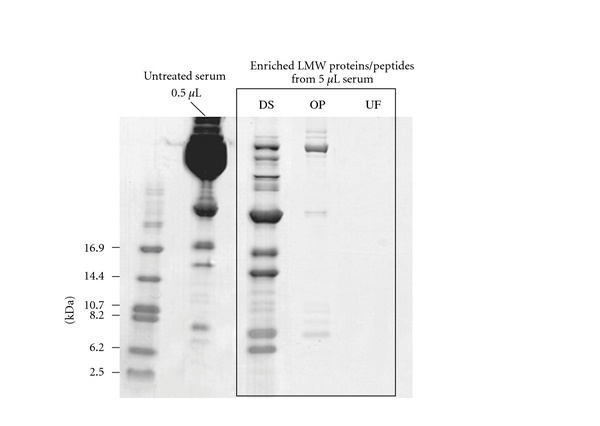
Comparison of the yield of low molecular weight protein/peptide extraction from serum by means of differential solubilization (DS), organic precipitation (OP), and ultrafiltration (UF). All techniques effectively remove the high molecular weight serum protein, whereas recovery of LMW proteins/peptides is highest with DS. Reprinted with permission from Kawashima et al. [94]. Copyright 2011 American Chemical Society.
3.3. Liquid Chromatography
3.3.1. Size Exclusion Chromatography
Another analytical tool to separate LMW compounds from HMW compounds is size exclusion chromatography (SEC). This is a widely used technique for the purification and analysis of synthetic and biological polymers based on their size, which is not by definition the same as their molecular weight. It separates polysaccharides, nucleic acids but also proteins and peptides. The material used for SEC consists of porous beads, which either exclude the peptide/protein analytes from the internal space or allow them to enter based on their size. Peptides and smaller proteins, which can enter the beads, will move at a slower rate through the column than bigger proteins which cannot penetrate the beads, thus migrating faster. A disadvantage of this technique is its low resolving power, which can be improved by using it in combination with other separations, such as in multidimensional separation approaches (see below). Other drawbacks are the high elution volumes which cause dilution of the sample, increased costs when having to use multiple columns, and the need for high sample loads [44]. In literature, some reports have appeared on the use of a one-step peptidome enrichment method based on SEC [15, 95, 96]. Ueda et al. [15] mention the separation of over 12,000 molecules by using SEC followed by C18 nano-LC-MS. They show that their procedure allows a precise separation of serum proteins and peptides based on their molecular weights. Tucholska et al. [82] showed that SEC can be used to remove substantial amounts of albumin from serum (although a small amount could still be detected in the depleted fraction). Hu et al. [97] performed a comprehensive peptidome analysis of mouse liver by combining SEC prefractionation with nano-LC-MSMS. From the low molecular weight fractions (<3 kDa) that eluted from the SEC column, 1181 unique peptides originating from 371 proteins were identified [97].
3.3.2. Reversed Phase Chromatography
Reversed phase liquid chromatography (RPLC) separates molecules based on differences in their hydrophobicity. The mobile phase is a water and nonpolar organic solvent mixture, whereas the stationary phase is hydrophobic. Factors influencing the selectivity and resolution of separation include the hydrophobic ligand, the particle size, sample volume, column length, and pH. The most commonly used hydrophobic ligand for peptide analysis is C18, but C4 and C8 are preferred for the larger peptides and proteins. In terms of peptides extraction, the solid phase material can be packed in syringe-shaped cartridges, or even in 96-well plate formats, which allows for high throughput extractions. Another advantage of this technique is its capability to desalt samples, which is desirable prior to mass spectrometric analysis [43, 44, 98, 99]. In proteomics/peptidomics analysis, nowadays, capillary reversed phase columns with an internal diameter of 75 to 100 μm are commonly used, mainly because of the increased sensitivity this offers. Because of these small columns, low flow rates have to be used, without decreasing the sensitivity, and (often environmentally unfriendly) solvents are saved. Reducing the particle size to 2 μm or less allows the use of a wider variety of linear velocities while maintaining acceptable chromatographic resolution in substantially reduced analysis times. This, however, requires new hardware since typical HPLC systems generally are not able to handle the high pressures required to pump the mobile phase through a column packed with such minute particles. Ultrahigh-pressure LC (UPLC) systems have been developed for this purpose, allowing pressures of 400 bar and higher. Thanks to the greater analytical power of UPLC, downscaling of sample volumes and increasing the throughput of sample handling have become possible [98, 100–103].
3.3.3. Ion Exchange Chromatography
Ion exchange chromatography (IEX) separates peptides (and proteins) based on their charge in a specific salt environment and pH of the mobile phase. Two types of IEX exist, namely, cation or anion exchange (respectively CX and AX). IEX has a disadvantage, being the use of salts which makes the eluate incompatible with MS. The use of salts can also lead to irreversible peptide or protein absorption to the resin, resulting in sample loss. The principles of IEX are well understood, and other advantages include the high resolution that can be achieved, high capacity, selectivity, and robust operation.
Usually, simple salt buffers are sufficient and concentrations are used in a defined range, in which the so-called salting-in effect on proteins is observed. This is the range where a protein becomes more soluble with increasing salt concentration. Cation exchangers are negatively charged, and anion exchangers; are positively charged. Above its pI, a protein is negatively charged and binds to an anion exchanger; below its pI, it is positively charged and binds to cation exchangers. The ion exchanger itself behaves like an acid or base, and the disproportionation of the charges depends on the pH. Strong ion exchangers behave like a strong acid or base and do not change the charge within a wide range of pH, whereas weak ion exchangers do. This property can also be exploited to gain selectivity or by applying pH gradients for elution [44, 105, 106].
3.3.4. Restricted Access Material
Restricted access material (RAM) has been described for use in the separation of large biomolecules and the extraction of LMW analytes. RAM could be considered to be an “upgrade” of normal size exclusion material. The outer surface of the RAM particles is coated with a protective, nonadsorptive hydrophilic packing, while the surface of the pores can be coated with a variety of different affinity matrices [107]. In principle, it prevents access of macromolecules (e.g., proteins) to the bonded phase in the pores, which can be compared to the size exclusion process. Simultaneously, LMW target compounds (such as peptides) are selectively retained on the affinity matrix in the pores [108, 109].
The most used type of internal selection material is strong cation exchange (SCX) as this type of material is highly suitable for the Online extraction of target peptides from complex biological samples such as plasma [110, 111]. The method allows repetitive, direct injections of untreated sample matrices and can also handle higher injection volumes in comparison to SEC [95, 98]. Also, this method allows for automation of process. A few setups have been described, in which RAM is even used in an Online LC-MS/MS setup [108, 110–113]. This possibility is an important advantage of the RAM technology. Rieux et al. [113] even described the use of a macroscopic RAM cartridge Online with nano-LC-MS for the quantification of a peptide analyte in samples containing high amounts of bovine serum albumin (BSA). Although the separation process can be automated, important parameters in RAM chromatography, such as loading flow rate, pH, and amount of dilution and injected volume should be optimized as they have a large effect on the amount of adsorbed peptides and proteins as well as on their recovery from the column. The use of RAM has been demonstrated for the analysis of peptides in complex biological matrices [114]. For example, Machtejevras et al. [108] isolated and detected more than 1500 peptides and protein fragments from only 3 mL of urine using an online RAM-SCX system.
3.3.5. Multidimensional
Because of the high complexity of biological samples, often a single fractionation or separation step is insufficient. Therefore, several techniques can be combined to what is referred to as a multidimensional separation. Combining 2 (different) separation techniques leads to an increased number of peptides measurable, an enlarged overall dynamic range and thus an improved peptidome coverage. A multidimensional approach can be achieved both offline and online. For the offline separation, fractions are collected after the first dimension, which are later reinjected into the second dimension separation. In between both steps, the fractions can be manipulated if necessary. A disadvantage of this method is the potential sample loss when transferring between both dimensions. When doing Online multidimensional separation, the samples are automatically transferred from the first to the second dimension. A drawback of this approach is that both dimensions have to be compatible. Also the solvents should not cause salt precipitation or immediate elution of the compounds in the second dimension. Most often, RPLC is the second dimension for its high speed, desalting capability, and compatibility with mass spectrometry [19, 43, 115–118].
4. Sample Quality: Preserving the Integrity of the Peptidome
It is clear that, besides selecting the best methodology to comprehensively separate peptides from a biological mixture, an essential part of peptidomics sample preparation is the preservation of the integrity of the in vivo peptidome. It is capital to prevent all types of peptide degradation throughout the procedure. Various protease inhibitor cocktails can be introduced in the sample as early as viable, to try and inhibit ubiquitous peptide degrading enzymes [118, 119]. In addition, peptidomics samples are traditionally snap-frozen, at a very low temperature as soon as possible, and subsequently freeze dried. Yet, such strategies donot always appear adequate (rapid enough) to prevent bioactive peptide degradation (several peptidases “survive” repeated freeze-thaw cycles). This has clearly been demonstrated for the (neuro)peptidome, which appears particularly sensitive to rapid ex vivo (postsampling) degradation (Figure 4). Microwaves were found to be effective to rapidly and permanently inactivate enzymes [104, 120], although poorly controllable. An elegant automated instant thermal denaturation system, based on conductive heating, at controlled pressure under vacuum, was developed for successful and reproducible blocking of ex vivo sample degradation, thereby stabilizing the peptidome (Denator, Gothenburg, Sweden). Depending on the sample-origin-specific “indicator peptides” can be used to monitor/assess the sample quality, prior to analysis. One example is the post mortem appearance in mammalian neuronal tissue of the peptide stathmin 2–20, which indicates proteolytic degradation [104].
Figure 4.
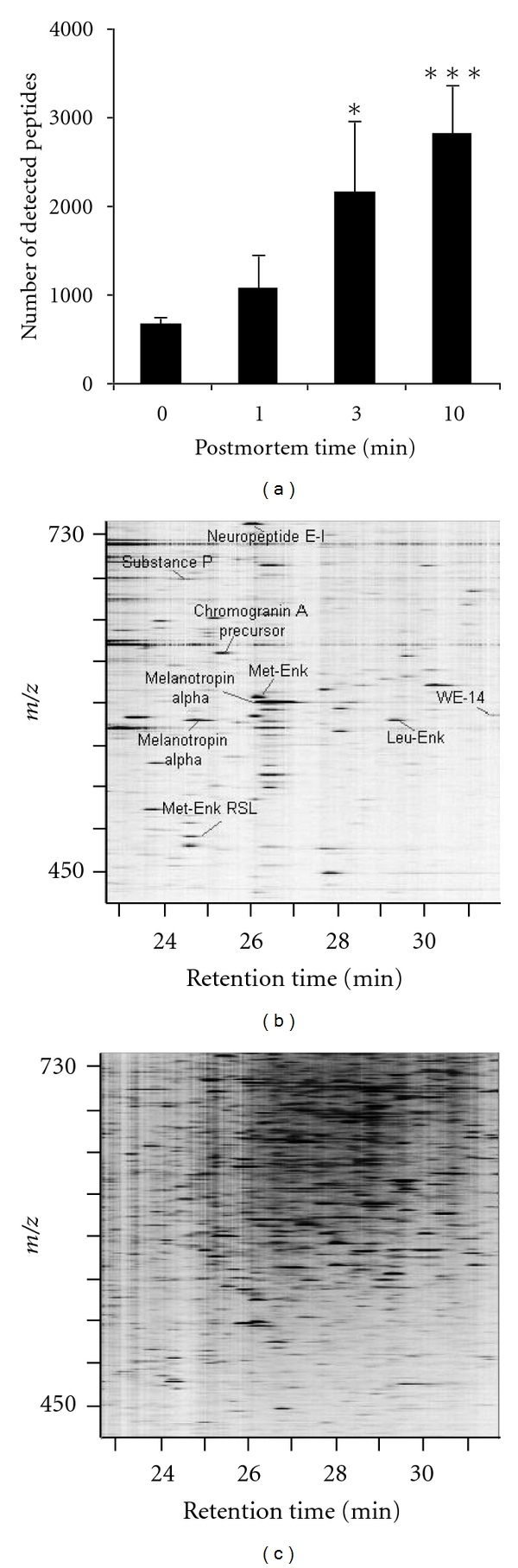
Effect on the number of detected (neuro)peptides (from mouse hypothalamus) by postmortem time (time between tissue collection and heat denaturation/stabilization) [104]. (a) Number of detected peptides by nano-LC ESI MS rapidly increases with postmortem time. (b, c) Two-dimensional peptide displays of control group and 10 min postmortem group, respectively. Picture adapted from Sköld et al. [104].
5. Conclusion and Future Perspective
In general, it can be concluded that sample preparation for the purpose of capturing the peptidome from biofluids still has room for improvement, and a single generic methodology is not (yet) available. Many methods exist for the analysis of proteins which can, sometimes with minor adjustments, be used for peptides as well. Ideally, sample preparation/handling should be minimal, but current mass spectrometers are not quite able to handle complex biological matrices. Manufacturers of mass spectrometry instrumentation are continually improving the performance of their systems. For example, over the last decades, the sensitivity of mass spectrometers has tremendously increased, where at the moment they have reached the low attomole level. Also, improvements in mass accuracy and advances in peptide fragmentation techniques permit us now to obtain highly confident identification and even fully de novo sequence novel peptides. Despite these ongoing improvements, it should be noted that at the moment, a peptidomics analysis MS system that can operate without sample preparation is still far way, which is mainly due to the high complexity of the biological matrices. The biggest bottleneck in biofluid research is the high dynamic range at which the proteins/peptides occur. For example, albumin concentration is ~40 mg/mL in serum, whereas the concentration of biologically active compounds such as cytokines is ~1 pg/mL. The removal of abundant plasma/serum proteins by dye- or immuno-based depletion is at moment the most popular strategy to “mine” deeper into proteome/peptidome. However, one should also realize that albumin and other major proteins in blood plasma carry some number of other “adsorbed” peptides and even small proteins on their surface, and, therefore, these will risk to get lost during this depletion step. The precipitation of the abundant protein fraction in human biofluids by organic solvents has probably the longest history among all the methods used for abundant protein removal. This procedure is still one of the most effective methods as it permits fast and, although not always highly reproducible, cheap obtainment of the peptide fraction for their analysis by mass spectrometry.
Several other analytical strategies are at hand that aid in the reduction of the complexity of samples, and some specifically focus on capturing the LMW fraction, that is, the smaller proteins and peptides from the bulk of larger proteins, for example, ultrafiltration as method to split a sample into a proteome and a peptidome fraction. However, one should be aware that several studies have shown dramatic differences in the performance (both in terms of reproducibility, recovery, and separation) of commercially available ultrafiltration devices.
A more preferred analytical tool to separate biomolecules on basis of size is liquid chromatography. Size exclusion chromatography and more specifically restricted access material have high potential in peptidome research, due to their high resolution and selectivity. Reversed phase and ion exchange chromatography are highly suitable for peptide fractionation and separation. These last two chromatography techniques also can be miniaturized, making them even more favorable in term of sensitivity.
In general, as sample preparation is often time consuming and laborious, high-throughput and automated approaches are highly desirable. Whereas up to now, when doing multidimensional LC, the first dimension led to the collection of fractions in an offline setup, online multidimensional LC setups gain popularity. Also the column material used in liquid chromatography is continually being improved, and the possibility of combining separation based on different physicochemical properties, such as RAM, is a big step forward. Even though it usually takes some effort to completely set up and optimize an LC-based method, once done, it usually results in a very robust and reliable technique, which can be fully automated. Also miniaturization is an important aspect of the evolution, as a miniature system (e.g., lab-on-a-chip) will allow for lower amounts of solvents and less complicated equipment to be used, decreasing the cost of an analysis. With the development of new sample pretreatment methods and lower detection limits on mass spectrometers, future peptidome analysis will aid substantially in biological and medical research, for example, by discovering new biomarkers and uncovering novel signaling pathways.
Acknowledgments
The authors wish to thank the European Marie Curie Training Network (Grant number MCRTN-CT-2006-035854) and The Netherlands Proteomics Centre for their financial support.
References
- 1.Schrader M, Schulz-Knappe P. Peptidomics technologies for human body fluids. Trends in Biotechnology. 2001;19(supplement 10):S55–S60. doi: 10.1016/S0167-7799(01)01800-5. [DOI] [PubMed] [Google Scholar]
- 2.Hu L, Ye M, Zou H. Recent advances in mass spectrometry-based peptidome analysis. Expert Review of Proteomics. 2009;6(4):433–447. doi: 10.1586/epr.09.55. [DOI] [PubMed] [Google Scholar]
- 3.Jimenez CR, Piersma S, Pham TV. High-throughput and targeted in-depth mass spectrometry-based approaches for biofluid profiling and biomarker discovery. Biomarkers in Medicine. 2007;1(4):541–565. doi: 10.2217/17520363.1.4.541. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X, Ye M, Zou H. Technologies and methods for sample pretreatment in efficient proteome and peptidome analysis. Proteomics. 2008;8(4):686–705. doi: 10.1002/pmic.200700617. [DOI] [PubMed] [Google Scholar]
- 5.Schulz-Knappe P, Zucht HD, Heine G, Jürgens M, Hess R, Schrader M. Peptidomics: the comprehensive analysis of peptides in complex biological mixtures. Combinatorial Chemistry and High Throughput Screening. 2001;4(2):207–217. doi: 10.2174/1386207013331246. [DOI] [PubMed] [Google Scholar]
- 6.McNaught AD, Wilkinson A, editors. Compendium of Chemical Terminology—The Gold Book, Second Edition. 2nd edition. Blackwell Science; 1997. [Google Scholar]
- 7.Gelman JS, Sironi J, Castro LM, Ferro ES, Fricker LD. Peptidomic analysis of human cell lines. Journal of Proteome Research. 2011;10(4):1583–1592. doi: 10.1021/pr100952f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soloviev M, Finch P. Peptidomics: bridging the gap between proteome and metabolome. Proteomics. 2006;6(3):744–747. doi: 10.1002/pmic.200500878. [DOI] [PubMed] [Google Scholar]
- 9.Hathout Y. Approaches to the study of the cell secretome. Expert Review of Proteomics. 2007;4(2):239–248. doi: 10.1586/14789450.4.2.239. [DOI] [PubMed] [Google Scholar]
- 10.Liang X, Huuskonen J, Hajivandi M, et al. Identification and quantification of proteins differentially secreted by a pair of normal and malignant breast-cancer cell lines. Proteomics. 2009;9(1):182–193. doi: 10.1002/pmic.200700957. [DOI] [PubMed] [Google Scholar]
- 11.Makridakis M, Vlahou A. Secretome proteomics for discovery of cancer biomarkers. Journal of Proteomics. 2010;73(12):2291–2305. doi: 10.1016/j.jprot.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nature Biotechnology. 2010;28(7):710–721. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- 13.Kim MS, Kandasamy K, Chaerkady R, Pandey A. Assessment of resolution parameters for CID-based shotgun proteomic experiments on the LTQ-orbitrap mass spectrometer. Journal of the American Society for Mass Spectrometry. 2010;21(9):1606–1611. doi: 10.1016/j.jasms.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilsson T, Mann M, Aebersold R, Yates JR, Bairoch A, Bergeron JJM. Mass spectrometry in high-throughput proteomics: ready for the big time. Nature Methods. 2010;7(9):681–685. doi: 10.1038/nmeth0910-681. [DOI] [PubMed] [Google Scholar]
- 15.Ueda K, Saichi N, Takami S, et al. A comprehensive peptidome profiling technology for the identification of early detection biomarkers for lung adenocarcinoma. PLoS One. 2011;6(4) doi: 10.1371/journal.pone.0018567. Article ID e18567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu T, Qian WJ, Mottaz HM, et al. Evaluation of multiprotein immunoaffnity subtraction for plasma proteomics and candidate biomaker discovery using mass spectrometry. Molecular and Cellular Proteomics. 2006;5(11):2167–2174. doi: 10.1074/mcp.T600039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richens JL, Lunt EAM, Sanger D, McKenzie G, O’Shea P. Avoiding nonspecific interactions in studies of the plasma proteome: practical solutions to prevention of nonspecific interactions for label-free detection of low-abundance plasma proteins. Journal of Proteome Research. 2009;8(11):5103–5110. doi: 10.1021/pr900487y. [DOI] [PubMed] [Google Scholar]
- 18.Makridakis M, Roubelakis MG, Bitsika V, et al. Analysis of secreted proteins for the study of bladder cancer cell aggressiveness. Journal of Proteome Research. 2010;9(6):3243–3259. doi: 10.1021/pr100189d. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Fang A, Riley CP, Wang M, Regnier FE, Buck C. Multi-dimensional liquid chromatography in proteomics-A review. Analytica Chimica Acta. 2010;664(2):101–113. doi: 10.1016/j.aca.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Characterization of the low molecular weight human serum proteome. Molecular & Cellular Proteomics. 2003;2(10):1096–1103. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Molecular & Cellular Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Fang X, Zhang WW. Affinity separation and enrichment methods in proteomic analysis. Journal of Proteomics. 2008;71(3):284–303. doi: 10.1016/j.jprot.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Hernandez P, Müller M, Appel RD. Automated protein identification by tandem mass spectrumetry: issues and strategies. Mass Spectrometry Reviews. 2006;25(2):235–254. doi: 10.1002/mas.20068. [DOI] [PubMed] [Google Scholar]
- 24.Shushan B. A review of clinical diagnostic applications of liquid chromatography- tandem mass spectrometry. Mass Spectrometry Reviews. 2010;29(6):930–944. doi: 10.1002/mas.20295. [DOI] [PubMed] [Google Scholar]
- 25.Mirzaei H, Regnier F. Structure specific chromatographic selection in targeted proteomics. Journal of Chromatography B. 2005;817(1):23–34. doi: 10.1016/j.jchromb.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 26.Hernández-Borges J, Borges-Miquel TM, Rodríguez-Delgado MA, Cifuentes A. Sample treatments prior to capillary electrophoresis-mass spectrometry. Journal of Chromatography A. 2007;1153(1-2):214–226. doi: 10.1016/j.chroma.2006.10.070. [DOI] [PubMed] [Google Scholar]
- 27.Planque C, Kulasingam V, Smith CR, Reckamp K, Goodglick L, Diamandis EP. Identification of five candidate lung cancer biomarkers by proteomics analysis of conditioned media of four lung cancer cell lines. Molecular and Cellular Proteomics. 2009;8(12):2746–2758. doi: 10.1074/mcp.M900134-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chevallet M, Diemer H, Van Dorssealer A, Villiers C, Rabilloud T. Toward a better analysis of secreted proteins: the example of the myeloid cells secretome. Proteomics. 2007;7(11):1757–1770. doi: 10.1002/pmic.200601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colzani M, Waridel P, Laurent J, Faes E, Rüegg C, Quadroni M. Metabolic labeling and protein linearization technology allow the study of proteins secreted by cultured cells in serum-containing media. Journal of Proteome Research. 2009;8(10):4779–4788. doi: 10.1021/pr900476b. [DOI] [PubMed] [Google Scholar]
- 30.Dowling P, Clynes M. Conditioned media from cell lines: a complementary model to clinical specimens for the discovery of disease-specific biomarkers. Proteomics. 2011;11(4):794–804. doi: 10.1002/pmic.201000530. [DOI] [PubMed] [Google Scholar]
- 31.Greco TM, Seeholzer SH, Mak A, Spruce L, Ischiropoulos H. Quantitative mass spectrometry-based proteomics reveals the dynamic range of primary mouse astrocyte protein secretion. Journal of Proteome Research. 2010;9(5):2764–2774. doi: 10.1021/pr100134n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peter JF, Otto AM, Wolf B. Enrichment and detection of molecules secreted by tumor cells using magnetic reversed-phase particles and LC-MALDI-TOF-MS. Journal of Biomolecular Techniques. 2007;18(5):287–297. [PMC free article] [PubMed] [Google Scholar]
- 33.Sintiprungrat K, Singhto N, Sinchaikul S, Chen ST, Thongboonkerd V. Alterations in cellular proteome and secretome upon differentiation from monocyte to macrophage by treatment with phorbol myristate acetate: insights into biological processes. Journal of Proteomics. 2010;73(3):602–618. doi: 10.1016/j.jprot.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Xu BJ, Yan W, Jovanovic B, et al. Quantitative analysis of the secretome of TGF-β signaling-deficient mammary fibroblasts. Proteomics. 2010;10(13):2458–2470. doi: 10.1002/pmic.200900701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alldridge L, Metodieva G, Greenwood C, et al. Proteome profiling of breast tumors by gel electrophoresis and nanoscale electrospray ionization mass spectrometry. Journal of Proteome Research. 2008;7(4):1458–1469. doi: 10.1021/pr7007829. [DOI] [PubMed] [Google Scholar]
- 36.Garfin DE. One-dimensional gel electrophoresis. Methods in Enzymology. 2009;463:497–513. doi: 10.1016/S0076-6879(09)63029-9. [DOI] [PubMed] [Google Scholar]
- 37.Moebiust J, Zahedi RP, Lewandrowski U, Berger C, Walter U, Sickmann A. The human platelet membrane proteome reveals several new potential membrane proteins. Molecular and Cellular Proteomics. 2005;4(11):1754–1761. doi: 10.1074/mcp.M500209-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Shevchenko A, Tomas H, Havliš J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols. 2006;1(6):2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 39.Wu L, Han DK. Overcoming the dynamic range problem in mass spectrometry-based shotgun proteomics. Expert Review of Proteomics. 2006;3(6):611–619. doi: 10.1586/14789450.3.6.611. [DOI] [PubMed] [Google Scholar]
- 40.Zhang G, Fenyö D, Neubert TA. Use of DNA ladders for reproducible protein fractionation by Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) for quantitative proteomics. Journal of Proteome Research. 2008;7(2):678–686. doi: 10.1021/pr700601y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walsh GM, Rogalski JC, Klockenbusch C, Kast J. Mass spectrometry-based proteomics in biomedical research: emerging technologies and future strategies. Expert Reviews in Molecular Medicine. 2010;12:p. e30. doi: 10.1017/S1462399410001614. [DOI] [PubMed] [Google Scholar]
- 42.Friedman DB, Hoving S, Westermeier R. Isoelectric focusing and two-dimensional gel electrophoresis. Methods in Enzymology. 2009;463:515–540. doi: 10.1016/S0076-6879(09)63030-5. [DOI] [PubMed] [Google Scholar]
- 43.Liu H, Lin D, Yates JR., III Multidimensional separations for protein/peptide analysis in the post-genomic era. BioTechniques. 2002;32(4):898–902. doi: 10.2144/02324pt01. [DOI] [PubMed] [Google Scholar]
- 44.Ly L, Wasinger VC. Protein and peptide fractionation, enrichment and depletion: tools for the complex proteome. Proteomics. 2011;11(4):513–534. doi: 10.1002/pmic.201000394. [DOI] [PubMed] [Google Scholar]
- 45.Rabilloud T. Two-dimensional gel electrophoresis in proteomics: old, old fashioned, but it still climbs up the mountains. Proteomics. 2002;2(1):3–10. [PubMed] [Google Scholar]
- 46.Schirle M, Heurtier MA, Kuster B. Profiling core proteomes of human cell lines by one-dimensional PAGE and liquid chromatography-tandem mass spectrometry. Molecular & Cellular Proteomics. 2003;2(12):1297–1305. doi: 10.1074/mcp.M300087-MCP200. [DOI] [PubMed] [Google Scholar]
- 47.Chevalier F. Highlights on the capacities of ’Gel-based’ proteomics. Proteome Science. 2010;8, article no. 23 doi: 10.1186/1477-5956-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Granvogl B, Plöscher M, Eichacker LA. Sample preparation by in-gel digestion for mass spectrometry-based proteomics. Analytical and Bioanalytical Chemistry. 2007;389(4):991–1002. doi: 10.1007/s00216-007-1451-4. [DOI] [PubMed] [Google Scholar]
- 49.Miller I, Crawford J, Gianazza E. Protein stains for proteomic applications: which, when, why? Proteomics. 2006;6(20):5385–5408. doi: 10.1002/pmic.200600323. [DOI] [PubMed] [Google Scholar]
- 50.Steinberg TH. Protein gel staining methods. An introduction and overview. Methods in Enzymology. 2009;463:541–563. doi: 10.1016/S0076-6879(09)63031-7. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed FE. Sample preparation and fractionation for proteome analysis and cancer biomarker discovery by mass spectrometry. Journal of Separation Science. 2009;32(5-6):771–798. doi: 10.1002/jssc.200800622. [DOI] [PubMed] [Google Scholar]
- 52.Cologna SM, Russell WK, Lim PJ, Vigh G, Russell DH. Combining isoelectric point-based fractionation, liquid chromatography and mass spectrometry to improve peptide detection and protein identification. Journal of the American Society for Mass Spectrometry. 2010;21(9):1612–1619. doi: 10.1016/j.jasms.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ros A, Faupel M, Mees H, et al. Protein purification by off-gel electrophoresis. Proteomics. 2002;2(2):151–156. doi: 10.1002/1615-9861(200202)2:2<151::aid-prot151>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 54.Hubner NC, Ren S, Mann M. Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics. 2008;8(23-24):4862–4872. doi: 10.1002/pmic.200800351. [DOI] [PubMed] [Google Scholar]
- 55.Manadas B, English JA, Wynne KJ, Cotter DR, Dunn MJ. Comparative analysis of OFFGel, strong cation exchange with pH gradient, and RP at high pH for first-dimensional separation of peptides from a membrane-enriched protein fraction. Proteomics. 2009;9(22):5194–5198. doi: 10.1002/pmic.200900349. [DOI] [PubMed] [Google Scholar]
- 56.Adkins JN, Varnum SM, Auberry KJ, et al. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Molecular & Cellular Proteomics. 2002;1(12):947–955. doi: 10.1074/mcp.m200066-mcp200. [DOI] [PubMed] [Google Scholar]
- 57.Baussant T, Bougueleret L, Johnson A, et al. Effective depletion of albumin using a new peptide-based affinity medium. Proteomics. 2005;5(4):973–977. doi: 10.1002/pmic.200401065. [DOI] [PubMed] [Google Scholar]
- 58.Gong Y, Li X, Yang B, et al. Different immunoaffinity fractionation strategies to characterize the human plasma proteome. Journal of Proteome Research. 2006;5(6):1379–1387. doi: 10.1021/pr0600024. [DOI] [PubMed] [Google Scholar]
- 59.Granger J, Siddiqui J, Copeland S, Remick D. Albumin depletion of human plasma also removes low abundance proteins including the cytokines. Proteomics. 2005;5(18):4713–4718. doi: 10.1002/pmic.200401331. [DOI] [PubMed] [Google Scholar]
- 60.Roche S, Tiers L, Provansal M, et al. Depletion of one, six, twelve or twenty major blood proteins before proteomic analysis: the more the better? Journal of Proteomics. 2009;72(6):945–951. doi: 10.1016/j.jprot.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 61.Urh M, Simpson D, Zhao K. Affinity chromatography. General methods. Methods in Enzymology. 2009;463:417–438. doi: 10.1016/S0076-6879(09)63026-3. [DOI] [PubMed] [Google Scholar]
- 62.Zolotarjova N, Martosella J, Nicol G, Bailey J, Boyes BE, Barrett WC. Differences among techniques for high-abundant protein depletion. Proteomics. 2005;5(13):3304–3313. doi: 10.1002/pmic.200402021. [DOI] [PubMed] [Google Scholar]
- 63.Zhou M, Lucas DA, Chan KC, et al. An investigation into the human serum "interactome". Electrophoresis. 2004;25(9):1289–1298. doi: 10.1002/elps.200405866. [DOI] [PubMed] [Google Scholar]
- 64.Gallant SR, Koppaka V, Zecherle N. Dye ligand chromatography. Methods in Molecular Biology. 2008;421:61–69. doi: 10.1007/978-1-59745-582-4_5. [DOI] [PubMed] [Google Scholar]
- 65.Echan LA, Tang HY, Ali-Khan N, Lee K, Speicher DW. Depletion of multiple high-abundance proteins improves protein profiling capacities of human serum and plasma. Proteomics. 2005;5(13):3292–3303. doi: 10.1002/pmic.200401228. [DOI] [PubMed] [Google Scholar]
- 66.Maccarrone G, Milfay D, Birg I, et al. Mining the human cerebrospinal fluid proteome by immunodepletion and shotgun mass spectrometry. Electrophoresis. 2004;25(14):2402–2412. doi: 10.1002/elps.200305909. [DOI] [PubMed] [Google Scholar]
- 67.Tu C, Rudnick PA, Martinez MY, et al. Depletion of abundant plasma proteins and limitations of plasma proteomics. Journal of Proteome Research. 2010;9(10):4982–4991. doi: 10.1021/pr100646w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye H, Sun L, Huang X, Zhang P, Zhao X. A proteomic approach for plasma biomarker discovery with 8-plex iTRAQ labeling and SCX-LC-MS/MS. Molecular and Cellular Biochemistry. 2010;343(1-2):91–99. doi: 10.1007/s11010-010-0502-x. [DOI] [PubMed] [Google Scholar]
- 69.Righetti PG, Boschetti E, Lomas L, Citterio A. Protein Equalizer Technology : the quest for a "democratic proteome". Proteomics. 2006;6(14):3980–3992. doi: 10.1002/pmic.200500904. [DOI] [PubMed] [Google Scholar]
- 70.Keidel EM, Ribitsch D, Lottspeich F. Equalizer technology—equal rights for disparate beads. Proteomics. 2010;10(11):2089–2098. doi: 10.1002/pmic.200900767. [DOI] [PubMed] [Google Scholar]
- 71.Polaskova V, Kapur A, Khan A, Molloy MP, Baker MS. High-abundance protein depletion: comparison of methods for human plasma biomarker discovery. Electrophoresis. 2010;31(3):471–482. doi: 10.1002/elps.200900286. [DOI] [PubMed] [Google Scholar]
- 72.Hansen RE, Winther JR. An introduction to methods for analyzing thiols and disulfides: reactions, reagents, and practical considerations. Analytical Biochemistry. 2009;394(2):147–158. doi: 10.1016/j.ab.2009.07.051. [DOI] [PubMed] [Google Scholar]
- 73.Grabski AC. Advances in preparation of biological extracts for protein purification. Methods in Enzymology. 2009;463:285–303. doi: 10.1016/S0076-6879(09)63018-4. [DOI] [PubMed] [Google Scholar]
- 74.Řehulková H, Marchetti-Deschmann M, Pittenauer E, Allmaier G, Řehulka P. Improved identification of hordeins by cysteine alkylation with 2-bromoethylamine, SDS-PAGE and subsequent in-gel tryptic digestion. Journal of Mass Spectrometry. 2009;44(11):1613–1621. doi: 10.1002/jms.1675. [DOI] [PubMed] [Google Scholar]
- 75.Thevis M, Ogorzalek Loo RR, Loo JA. In-gel derivatization of proteins for cysteine-specific cleavages and their analysis by mass spectrometry. Journal of Proteome Research. 2003;2(2):163–172. doi: 10.1021/pr025568g. [DOI] [PubMed] [Google Scholar]
- 76.Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Molecular and Cellular Proteomics. 2004;3(6):608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 77.Baldwin MA. Protein identification by mass spectrometry: issues to be considered. Molecular and Cellular Proteomics. 2004;3(1):1–9. doi: 10.1074/mcp.R300012-MCP200. [DOI] [PubMed] [Google Scholar]
- 78.Olsen JV, de Godoy LMF, Li G, et al. Parts per million mass accuracy on an orbitrap mass spectrometer via lock mass injection into a C-trap. Molecular and Cellular Proteomics. 2005;4(12):2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- 79.de Godoy LM, Olsen JV, de Souza GA, Li G, Mortensen P, Mann M. Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. Genome Biology. 2006;7(6, article R50) doi: 10.1186/gb-2006-7-6-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scholten A, Mohammed S, Low TY, et al. In-depth quantitative cardiac proteomics combining electron transfer dissociation and the metalloendopeptidase Lys-N with the SILAC mouse. doi: 10.1074/mcp.O111.008474. Molecular & Cellular Proteomics. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zougman A, Pilch B, Podtelejnikov A, et al. Integrated analysis of the cerebrospinal fluid peptidome and proteome. Journal of Proteome Research. 2008;7(1):386–399. doi: 10.1021/pr070501k. [DOI] [PubMed] [Google Scholar]
- 82.Tucholska M, Scozzaro S, Williams D, et al. Endogenous peptides from biophysical and biochemical fractionation of serum analyzed by matrix-assisted laser desorption/ionization and electrospray ionization hybrid quadrupole time-of-flight. Analytical Biochemistry. 2007;370(2):228–245. doi: 10.1016/j.ab.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 83.Zheng X, Baker H, Hancock WS. Analysis of the low molecular weight serum peptidome using ultrafiltration and a hybrid ion trap-Fourier transform mass spectrometer. Journal of Chromatography A. 2006;1120(1-2):173–184. doi: 10.1016/j.chroma.2006.01.098. [DOI] [PubMed] [Google Scholar]
- 84.Orvisky E, Drake SK, Martin BM, et al. Enrichment of low molecular weight fraction of serum for MS analysis of peptides associated with hepatocellular carcinoma. Proteomics. 2006;6(9):2895–2902. doi: 10.1002/pmic.200500443. [DOI] [PubMed] [Google Scholar]
- 85.Merrell K, Southwick K, Graves SW, Esplin MS, Lewis NE, Thulin CD. Analysis of low-abundance, low-molecular-weight serum proteins using mass spectrometry. Journal of Biomolecular Techniques. 2004;15(4):238–248. [PMC free article] [PubMed] [Google Scholar]
- 86.Harper RG, Workman SR, Schuetzner S, Timperman AT, Sutton JN. Low-molecular-weight human serum proteome using ultrafiltration, isoelectric focusing, and mass spectrometry. Electrophoresis. 2004;25(9):1299–1306. doi: 10.1002/elps.200405864. [DOI] [PubMed] [Google Scholar]
- 87.Greening DW, Simpson RJ. A centrifugal ultrafiltration strategy for isolating the low-molecular weight (<or = 25 K) component of human plasma proteome. Journal of Proteomics. 2010;73(3):637–648. doi: 10.1016/j.jprot.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 88.Wiśniewski JR, Zielinska DF, Mann M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Analytical Biochemistry. 2011;410(2):307–309. doi: 10.1016/j.ab.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 89.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 90.Chertov O, Biragyn A, Kwak LW, et al. Organic solvent extraction of proteins and peptides from serum as an effective sample preparation for detection and identification of biomarkers by mass spectrometry. Proteomics. 2004;4(4):1195–1203. doi: 10.1002/pmic.200300677. [DOI] [PubMed] [Google Scholar]
- 91.Kay R, Barton C, Ratcliffe L, et al. Enrichment of low molecular weight serum proteins using acetonitrile precipitation for mass spectrometry based proteomic analysis. Rapid Communications in Mass Spectrometry. 2008;22(20):3255–3260. doi: 10.1002/rcm.3729. [DOI] [PubMed] [Google Scholar]
- 92.Williams D, Ackloo S, Zhu P, et al. Precipitation and selective extraction of human serum endogenous peptides with analysis by quadrupole time-of-flight mass spectrometry reveals posttranslational modifications and low-abundance peptides. Analytical and Bioanalytical Chemistry. 2010;396(3):1223–1247. doi: 10.1007/s00216-009-3345-0. [DOI] [PubMed] [Google Scholar]
- 93.Polson C, Sarkar P, Incledon B, Raguvaran V, Grant R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography-tandem mass spectrometry. Journal of Chromatography B. 2003;785(2):263–275. doi: 10.1016/s1570-0232(02)00914-5. [DOI] [PubMed] [Google Scholar]
- 94.Kawashima Y, Fukutomi T, Tomonaga T, et al. High-yield peptide-extraction method for the discovery of subnanomolar biomarkers from small serum samples. Journal of Proteome Research. 2010;9(4):1694–1705. doi: 10.1021/pr9008018. [DOI] [PubMed] [Google Scholar]
- 95.Albrethsen J, Bøgebo R, Gammeltoft S, Olsen J, Winther B, Raskov H. Upregulated expression of human neutrophil peptides 1,2 and 3 (HNP 1–3) in colon cancer serum and tumours: a biomarker study. BMC Cancer. 2005;5, article 8 doi: 10.1186/1471-2407-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sasaki K, Takahashi N, Satoh M, Yamasaki M, Minamino N. A peptidomics strategy for discovering endogenous bioactive peptides. Journal of Proteome Research. 2010;9(10):5047–5052. doi: 10.1021/pr1003455. [DOI] [PubMed] [Google Scholar]
- 97.Hu L, Li X, Jiang X, et al. Comprehensive peptidome analysis of mouse livers by size exclusion chromatography prefractionation and nanoLC-MS/MS identification. Journal of Proteome Research. 2007;6(2):801–808. doi: 10.1021/pr060469e. [DOI] [PubMed] [Google Scholar]
- 98.Makarov A, Scigelova M. Coupling liquid chromatography to Orbitrap mass spectrometry. Journal of Chromatography A. 2010;1217(25):3938–3945. doi: 10.1016/j.chroma.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 99.Ebert MP, Niemeyer D, Deininger SO, et al. Identification and confirmation of increased fibrinopeptide A serum protein levels in gastric cancer sera by magnet bead assisted MALDI-TOF mass spectrometry. Journal of Proteome Research. 2006;5(9):2152–2158. doi: 10.1021/pr060011c. [DOI] [PubMed] [Google Scholar]
- 100.Guillarme D, Ruta J, Rudaz S, Veuthey JL. New trends in fast and high-resolution liquid chromatography: a critical comparison of existing approaches. Analytical and Bioanalytical Chemistry. 2010;397(3):1069–1082. doi: 10.1007/s00216-009-3305-8. [DOI] [PubMed] [Google Scholar]
- 101.Sandra K, Moshir M, D'hondt F, Verleysen K, Kas K, Sandra P. Highly efficient peptide separations in proteomics. Part 1. Unidimensional high performance liquid chromatography. Journal of Chromatography B. 2008;866(1-2):48–63. doi: 10.1016/j.jchromb.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 102.Waterval WA, Scheijen JLJM, Ortmans-Ploemen MMJC, Habets-van der Poel CD, Bierau J. Quantitative UPLC-MS/MS analysis of underivatised amino acids in body fluids is a reliable tool for the diagnosis and follow-up of patients with inborn errors of metabolism. Clinica Chimica Acta. 2009;407(1-2):36–42. doi: 10.1016/j.cca.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 103.Peters FT. Recent advances of liquid chromatography-(tandem) mass spectrometry in clinical and forensic toxicology. Clinical Biochemistry. 2011;44(1):54–65. doi: 10.1016/j.clinbiochem.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 104.Sköld K, Svensson M, Norrman M, Sjögren B, Svenningsson P, Andrén PE. The significance of biochemical and molecular sample integrity in brain proteomics and peptidomics: stathmin 2-20 and peptides as sample quality indicators. Proteomics. 2007;7(24):4445–4456. doi: 10.1002/pmic.200700142. [DOI] [PubMed] [Google Scholar]
- 105.Jungbauer A, Hahn R. Ion-exchange chromatography. Methods in Enzymology. 2009;463:349–371. doi: 10.1016/S0076-6879(09)63022-6. [DOI] [PubMed] [Google Scholar]
- 106.Kelley BD, Switzer M, Bastek P, et al. High-throughput screening of chromatographic separations: IV. Ion-exchange. Biotechnology and Bioengineering. 2008;100(5):950–963. doi: 10.1002/bit.21905. [DOI] [PubMed] [Google Scholar]
- 107.Boos KS, Grimm CH. High-performance liquid chromatography integrated solid-phase extraction in bioanalysis using restricted access precolumn packings. Trends in Analytical Chemistry. 1999;18(3):175–180. [Google Scholar]
- 108.Machtejevras E, Marko-Varga G, Lindberg C, Lubda D, Hendriks R, Unger KK. Profiling of endogenous peptides by multidimensional liquid chromatography: on-line automated sample cleanup for biomarker discovery in human urine. Journal of Separation Science. 2009;32(13):2223–2232. doi: 10.1002/jssc.200900058. [DOI] [PubMed] [Google Scholar]
- 109.Willemsen O, Machtejevas E, Unger KK. Enrichment of proteinaceous materials on a strong cation-exchange diol silica restricted access material: protein-protein displacement and interaction effects. Journal of Chromatography A. 2004;1025(2):209–216. doi: 10.1016/j.chroma.2003.10.072. [DOI] [PubMed] [Google Scholar]
- 110.Račaityte K, Lutz ESM, Unger KK, Lubda D, Boos KS. Analysis of neuropeptide Y and its metabolites by high-performance liquid chromatography-electrospray ionization mass spectrometry and integrated sample clean-up with a novel restricted-access sulphonic acid cation exchanger. Journal of Chromatography A. 2000;890(1):135–144. doi: 10.1016/s0021-9673(00)00639-7. [DOI] [PubMed] [Google Scholar]
- 111.Wagner K, Miliotis T, Marko-Varga G, Bischoff R, Unger KK. An automated on-line multidimensional HPLC system for protein and peptide mapping with integrated sample preparation. Analytical Chemistry. 2002;74(4):809–820. doi: 10.1021/ac010627f. [DOI] [PubMed] [Google Scholar]
- 112.Hu L, Boos KS, Ye M, Wu R, Zou H. Selective on-line serum peptide extraction and multidimensional separation by coupling a restricted-access material-based capillary trap column with nanoliquid chromatography-tandem mass spectrometry. Journal of Chromatography A. 2009;1216(28):5377–5384. doi: 10.1016/j.chroma.2009.05.030. [DOI] [PubMed] [Google Scholar]
- 113.Rieux L, Bischoff R, Verpoorte E, Niederländer HAG. Restricted-access material-based high-molecular-weight protein depletion coupled on-line with nano-liquid chromatography-mass spectrometry for proteomics applications. Journal of Chromatography A. 2007;1149(2):169–177. doi: 10.1016/j.chroma.2007.02.118. [DOI] [PubMed] [Google Scholar]
- 114.Souverain S, Rudaz S, Veuthey JL. Restricted access materials and large particle supports for on-line sample preparation: an attractive approach for biological fluids analysis. Journal of Chromatography B. 2004;801(2):141–156. doi: 10.1016/j.jchromb.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 115.Sandra K, Moshir M, D'hondt F, et al. Highly efficient peptide separations in proteomics. Part 2: Bi- and multidimensional liquid-based separation techniques. Journal of Chromatography B. 2009;877(11-12):1019–1039. doi: 10.1016/j.jchromb.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 116.Tang J, Gao M, Deng C, Zhang X. Recent development of multi-dimensional chromatography strategies in proteome research. Journal of Chromatography B. 2008;866(1-2):123–132. doi: 10.1016/j.jchromb.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.François I, Sandra K, Sandra P. Comprehensive liquid chromatography: fundamental aspects and practical considerations—a review. Analytica Chimica Acta. 2009;641(1-2):14–31. doi: 10.1016/j.aca.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 118.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 119.Yuan X, Desiderio DM. Human cerebrospinal fluid peptidomics. Journal of Mass Spectrometry. 2005;40(2):176–181. doi: 10.1002/jms.737. [DOI] [PubMed] [Google Scholar]
- 120.Dowell JA, Heyden WV, Li L. Rat neuropeptidomics by LC-MS/MS and MALDI-FTMS: enhanced dissection and extraction techniques coupled with 2D RP-RP HPLC. Journal of Proteome Research. 2006;5(12):3368–3375. doi: 10.1021/pr0603452. [DOI] [PubMed] [Google Scholar]