

Abstract
Established cell transfection via nucleofection relies on nucleofection buffers with unknown and proprietary makeup due to trade secrecy, inhibiting the possibility of using this otherwise effective method for developing cell therapy. We devised a three-step method for discovering an optimal formulation for the nucleofection of any cell-line. These steps include the selection of the best nucleofection program and known buffer type, selection of the best polymer for boosting the transfection efficiency of the best buffer, and the comparison with the optimal buffer from an established commercial vendor (Amaxa). Using this 3-step selection system, competitive nucleofection formulations were discovered for multiple cell lines, which are equal to or surpass the efficiency of the Amaxa nucleofector solution in a variety of cells and cell lines, including primary adipose stem cells, muscle cells, tumor cells, and immune cells. Through the use of scanning electron microscopy, we have revealed morphological changes, which predispose for the ability of these buffers to assist in transferring plasmid DNA into the nuclear space. Our formulation may greatly reduce the cost of electroporation study in laboratory and boosts the potential of application of electroporation-based cell therapies in clinical trials.
Keywords: electroporation, cell transfection, cell therapy, adipose stem cells, formulation
INTRODUCTION
Cell-based gene therapy has a great potential for treating both incurable diseases and genetic diseases. For example, genetically modified dendritic cells can assist in the formation of vaccines to boost anti-tumor immunity (1), show promise as a treatment for graft-versus-host disease (2), and promote immunity from infectious diseases (3). More importantly, using stem cells as a cell based gene delivery therapy may have a greater potential for multiple applications. Stem cells are already tested for the treatment of autoimmune diseases and lymphomas (4) (5)and the repairing and regrowth of damaged tissue (6). Moreover, stem cells can be promoted to differentiate into dendritic cells useful for treatment (7). Recently, the adipose-derived stem cells (ASCs) have been isolated (8)and characterized (9). ASCs have shown to be another potential candidate for cell therapy (8) (10). They are present in relatively large amounts in the body, can be harvested and isolated more easily than most other stem cell lineages, and give rise to a variety of different cell lineages, including adipocytes, chondrocytes, myoblasts, and endothelial cells (9). More importantly, ASCs possess a homing ability for some cancer cell lines (11), making them excellent potential candidates for anti-tumor cell therapy.
A number of methods are used to modify the cells and “arm” them for cell therapy. Viral transduction remains an attractive method for modifying cells, due to the high efficiency of transfection (12) (13). There are a number of different viral types, including adenovirus, adeno-associated virus, and various retroviruses, including lentiviruses (14). Unfortunately, the use of viral vectors will often invoke the host’s immune response, which can lead to decreased efficacy (15). Alternatively, binding of viruses can alter cellular function beyond any desired changes from transfection (16), which has the potential to alter the “stemness” of stem cells. Some viral vector may cause oncogene activation, resulting in tumor development (17). In summary, these viral vectors present the combined challenges of immunogenicity and overall safety (18).
These drawbacks create a need for non-viral methods of gene transfection. Polymer-based, cationic lipid groups present a promising method of transfection for both dividing and non-dividing cell lines (19) (20). However, use of lipofection is effective in some cells but limited in other cells (21). Moreover, some polymer complexes, including the popular Polyethylenimine (PEI) (22), present their own issues with cytotoxicity (23). This limit is due to a number of factors, including the ability to localize genes to the nucleus (24).
Another form of non-viral transfection is the use of an electric field to increase the permeability of a cell to larger molecules, or electroporation (25). First discovered in 1982 (26), the method proved to be an effective means to move hydrophilic, nonpermanent molecules across the plasma membrane (27). The actual method by which the membrane becomes permeable is not well-known, even to this day. Polarization of the cells from the electric field only lasts a few microseconds (28), but the increased permeability of the cells can last for several minutes (29) (30). The current model states that the electric field opens small pores that allow macromolecules into the cytosol (25). The limitation of most electroporation methods for cell therapy is cell death and low transfection efficiency.
The Amaxa Nucleofector is an electroporation device combined with a set of special transfection buffers that greatly improves the transfection efficiency and increases the cell survival rate (31). This can be applied not only to cells that are generally difficult to transform, such as natural killer (NK) cells (32) (33), but also to cells that do not replicate, such as most neurons (34). The use of the nucleofector technique allows efficient transfection of stem cells with reduced risk of modifying cellular processes (35). However, the use of the Amaxa nucleofector requires the use of proprietary Nucleofector buffers, which must be purchased directly from the vendor/manufacturer. This presents a number of limitations on the use of such technology: the nucleofector solutions are very costly for a small quantity of solution; the composition of these buffers remains a trade secret by which prevents any knowledge of toxicity of these buffers and therefore clinical applications (if the compounds are used in clinical studies, it is likely that the manufacturer will have a master document on file with the FDA defining the solution’s composition); and the nucleofector solutions vary from cell line to cell line, and in many cases an optimal solution is not given. In this study, we present a method to determine an optimal in-house nucleofector formulation – one that can be made from known chemicals – for different cell lines. The formulation is more economically feasible and ideally will have the same transfection efficiency as commercial nucleofector solutions.
MATERIALS AND METHODS
Cell culture
Cell lines including Hela (human cervical cancer), RM1 (murine prostate cancer), C2C12 (murine muscle tissue progenitor cells), 4T1 cells (murine breast cancer), SCCVII (murine squamous cell carcinoma), B16F10 (murine melanoma), MCF7 (breast tumor), MDA-MB-231 (human breast adenocarcinoma), HT29 (human colorectal adenocarcinoma), and NCTC 1469 (mouse hepatocytes) were purchased from ATCC. K7M3 (murine osteosarcoma) were donated by Genie Kleinerman (UT MD Anderson Cancer Center). These cell lines were maintained in Dulbecco’s Modified Eagle Medium/F-12 medium containing 10% fetal bovine serum. MPC11 (mouse myeloma) cells were donated from Dr Qing Yi (MD Anderson Cancer Center, Houston, TX). MC3T3E1 (mouse osteoblast) cells were kindly donated by Dr. Marxa Figueiredo (LSU, Baton Rouge, LA) and maintained in α-MEM, without ascorbic acid, and supplanted with 10% FBS. SL12.4, Jurkat, mouse primary bone marrow, and 3LL cells were grown in RPMI supplanted with 10% FBS and 1% P/S solution. SL12.4 and Jurkat cells were purchased from ATCC. Cells were grown to 80% confluence and harvested with Trypsin/EDTA buffer at 37°C for five minutes. The murine ASC were isolated from inguinal or epididymal fat pads of male mice (C57BL/6) by collagenase digestion and culture expansion in DMEM/F12 Ham’s Medium, 10% FBS, 1% antibiotic/antimycotic according to published methods (36).
Buffers and polymers
OptiMEM (Buffer O) is a commercial buffer that was purchased from Invitrogen (Carlsbad, CA) and stored at 4°. Pulsing buffer (Buffer P), consisting of 125mM KCl, 15mM NaCl, 3mM Glucose, 25mM HEPES, and 1.2mM MgCl2 (pH7.4), is made from a 10X home-made stock solution, filtered, and stored at 4°. After preparation, the solution is filtered through a 0.2μm filter and stored at room temperature. Buffer S consists of 0.45M NaCl, and is stored at room temperature.
LMA1 stock was made to a 10% (wt/vol) of Poly-L-glutamic acid (mw 15–50 kDa, Sigma-Aldrich) in MQ-H2O. LME1 (Poly-ethylene-glycol, mw 8 kDa, Amresco), LMP8 (Poloxamer 188, mw 8kDa, Spectrum Chemicals), LMP1 (Poloxamer 181, mw 2 kDa, Spectrum), LMP7 (Poloxamer 407, mw 12 kDa, Spectrum), and LMV1 (Poly-vinylpyrrolidone, mw 40 kDa, Fisher) stocks were all made to 20% (wt/vol) in MQ-H2O. LMP3 (Pop313, donated from (Expression Genetics, Huntsville, AL) was made to a stock concentration of 0.01% (wt/vol) in MQ-H2O. LMC1 (Crown-5, donated from Expression Genetics) was provided at a stock concentration of 10%. All stock buffers were stored at 4° and were kept for 1 month.
Electroporation
Cells were pelleted and resuspended in 100μL transfection buffer at a concentration of 1.0 × 106 cells/100μL, then transferred to a sterile 3mm Amaxa nucleofection cuvette. Cells were electroporated using the appropriate nucleofection program. Nucleofected cells were then rinsed with 500μL sterile culture medium and transferred to the well of a sterile 12-well plate. Cells were incubated at 37°C for 24 hours before analysis.
Survival
Immediately after transfer to a 12-well plate, 10μL cell solution is mixed with 10μL Gibco Trypan Blue Stain (Invitrogen; Carlsbad, CA; part# 15250-061.) 10μL of the cell mixture is transferred to a hemacytometer and counted. Survival is expressed using the following equation: (number of non-blue cells)/(total number of cells) × 100%.
Flow Cytometry
Prior to luciferase and protein assays, cells are harvested by trypsinization and resuspended in 1000μL PBS. 500μL cell suspension is removed and fixed in PBS + 1.0% Formaldehyde solution prior to analysis. Cells are measured by a BD FACSCalibur (BD Biosciences; San Diego, CA)
Luciferase and Protein assays
After incubation, cells were lysed and assayed for luciferase using a commercial protein assay and cell lysis kit (Promega; Madison, WI; part# E4030). Briefly, cell pellets were rinsed with PBS and lysed with 200μL lysis reagent using the freeze/thaw method. Cell lysates were kept at 4°C during analysis and stored at −20°C when not in use. 20μL of cell lysates were transferred to a 96-well plate. Luciferase activity was measured using a Packard LumiCount (Perkin Elmer; Boston, MA.) All luciferase activity is normalized to protein assay data using the following equation: ((luciferase assay data in RLU) × 200)/(protein assay data in μg).
Protein assays were conducted using a commercially purchased BCA protein assay kit (Thermo Scientific; Rockford, IL; Product # 23227.) Briefly, 4 μL cell lysate prepared from luciferase assay is transferred to a clear 96-well plate. Protein levels are measured by a Packard SpectraCount (Perkin Elmer.) Protein assay measurements are calibrated to a BSA standard with a maximum of 40μg and analyzed by I-Smart 2.0 (Perkin Elmer.)
Electron Microscopy
5 × 104 murine ASC were seeded onto 13mm round Thermanox cover slips (Thermo Fisher Scientific, Rochester, NY) placed in a sterile 24-well plate. Cells are grown until they reach 40–50% confluence. All growth medium is removed from the wells, and cells are incubated at room temperature for ten minutes in one of the following solutions before processing for scanning electron microscopy: DMEM, Buffer O, Buffer O + 0.001% LMP1, and Amaxa MSC nucleofector solution. Briefly, cells are fixed in 1.25% Glutearaldehyde and 2% Formaldehyde in 0.1 M Sodium Cacodylate (CAC) buffer for 1 hour at room temperature. Cells are then washed in 0.1M CAC buffer, treated with 1% Osmium Tetroxide in 0.1M CAC, and washed again. Cells are then dehydrated in ethanol before platinum-coating.
RESULTS
First Step – Determine optimal buffer and program
To discover novel nucleofection formulation(s) for increasing cell transfection efficiency using an Amaxa Nucleofection device, we decided to use known cell transfection electroporation buffers as a starting point. We initially chose two buffers: OptiMEM and pulsing buffer (37). OptiMEM, referred in this publication as Buffer O, has been effective in the electroporation of immune cells (37). A pulsing buffer, referred in this publication as Buffer P for easy description, has shown some promise in the electroporation of CHO7 cells when used in combination with commercial devices (37). To determine which nucleofection program from Amaxa Nucleofector device has the greatest potential to boost the gene delivery to cells using these known buffers, we used seven programs suggested in Amaxa’s optimized nucleofection protocol: A-20, T-20, T-30, X-01, X-05, L-29, and D-23. The exploration of the optimal buffers and the optimal program to combine with this buffer is referred to as Step 1. Fig. 1a gives an example of the results of the first step for the murine breast cancer cell line 4T1 and the same procedure was applied to all other 20 tested cell lines listed in this study. X-01 appeared to be the most effective nucleofection program for the 4T1 cell line and for half the cell lines tested, suggesting half of cell lines in nature may share the same or similar electroporation sensitivity. Many of the cell lines favor Buffer O, though some tumor cell lines favor buffer P. Other buffers such as saline and PBS were also tested, but none stood out persistently (data not shown).
Fig. 1.

Second Step – Determine optimal polymer
Once an optimal buffer and program were determined from Step 1, we wanted to test if the addition of any polymers to the selected transfection buffer would increase the efficiency of transfection when pulsed. A number of polymers were selected based on their ability to assist other forms of transfection. Poly-L-glutamic acid (LMA1), Poly-ethylene-glycol (LME1), and Polyvinylpyrrolidone (LMV1) were selected due to their ability to enhance gene delivery to muscle cells via electroporation in vivo using another BTX electroporation device by us (unpublished data) and others (38) (39) (40). Poloxamer 188 (LMP8), a member of the family of pluronic-block co-polymers (41), has shown some promise in lipofection (42) (43). An example of result of the second step is presented in Fig. 1b, using the same 4T1 cell line displayed in the example in step 1 (Fig. 1a). We repeated the two step optimization for a total of 20 cell lines. The results of these can be seen in Fig. 2. The results show that varying degrees of effectiveness were acheived on different cell lines. Poly-L-glutamic acid (LMA1) has shown some promise in increasing transfection efficiency in many tumor cell lines. LMP8 has shown a great promise in a number of different cell lines regardless of the tissue of origin. Moreover, for many cell lines where another polymer was more effective, LMP8 proved to be a close second (not shown), indicating that LMP8 is an important component for generating universal and effective electroporation formulation for cells therapy.
Fig. 2.

Third Step – Comparison to Amaxa buffer
Amaxa nucleofection solutions are cell-specific. Amaxa formulations are available for ten of the twenty cell lines we tested – HT29, Hela, K7M3, C2C12, Jurkat, MCF7, MC3T3E1, D1, 4T1, and B16F10. Since our purpose is to find a cell electroporation formulation that is comparable to the commercial buffers recommended by Amaxa, we tested the overall effectiveness of our optimal formulations determined in step 2 with the Amaxa recommended buffer. When using Amaxa-produced buffers, the proper corresponding program listed in the Amaxa nucleofection kit protocol was used. We determined the relative efficiency based on the amount of gfp production expressed as a relative percentage of the amount of luciferase expressed by the same cells transfected using the Amaxa formulation (Fig. 3). As figure 3 illustrates, our formulation outperforms Amaxa buffer in electroporation transfection efficiency in five out of ten cell lines.
Fig. 3.
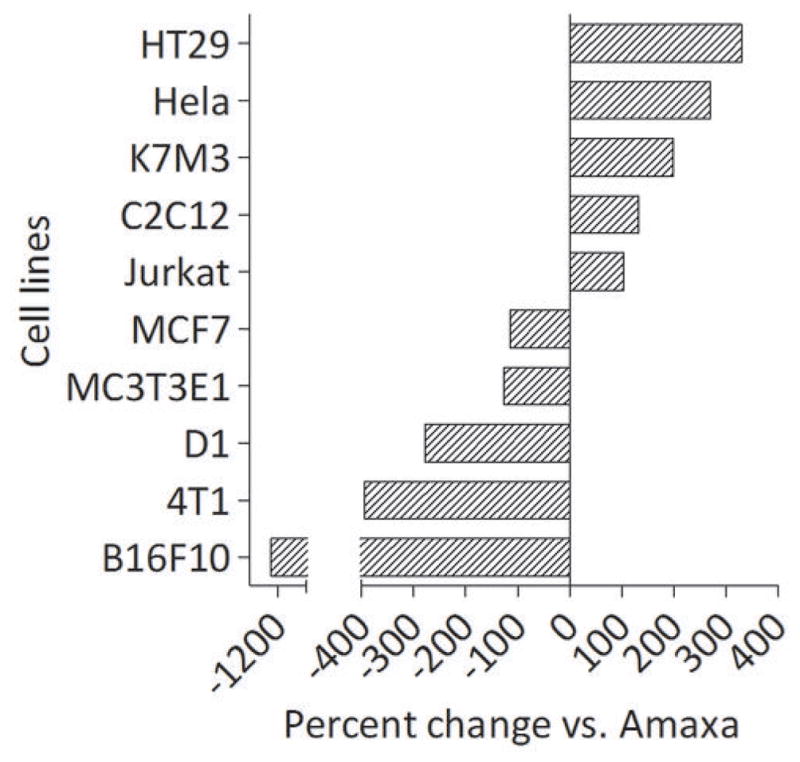
Cell transfection efficiency
While measuring the efficiency of transfection based on the production of luciferase can be a useful metric in determining transfection efficiency, we wanted to determine the total percentage of cells transfected with our formulation. For this purpose, we transfected cells with PGF1032, a plasmid coding for green florescence protein (gfp), and identified GFP positive cells using Flow cytometry. In terms of the percentage of cells positive for transfection of GFP, our formulations, match closely to Amaxa’s formulation in six out of seven tested cell lines (Fig. 4a). This results show that, while our formulation may transfect the same percentage of cells as Amaxa, but the amount of luciferase produced by the cells is much less than in cells transfected with Amaxa. The most striking example occurred when we compared luciferase production with the percentage of gfp positive B16F10 cells: our formulation easily matches Amaxa for the amount of cells transfected using GFP as the end point (Fig. 4a), though the Amaxa formulation is superior to ours in inducing a higher amount of luciferase produced by those cells (Fig. 3). This difference from GFP and luciferase expression caused by Amaxa and our formulation was not due to the transfection variation because cells were transfected with both reporter genes at the same time and split for different reporter gene analysis.
Fig. 4.

Cell Survival
Due to the heavy amount of cell death post-transfection, one major challenge is cell survival rate when electroporation is employed for in vitro transfection. Initially we postulated that Amaxa’s formulation did not improve cell transfection efficiency, but improved cell survival. To test this hypothesis, immediately after washing the transfected cells, 10μL of the cell suspension were used to measure cell viability using Trypan blue staining. We first noticed our electroporation buffer is as good as Amaxa buffer in reserving cell survival rate except for Hela cells (Fig. 4b).
Optimize buffers for adipose-derived stem cells
One of the intended uses of our method is to determine an optimized buffer for cells intended for therapeutic uses. One such primary cell target would be adipose-derived stem cells (ASCs). Their abundance, ease of harvesting, and multipotency make ASCs a very attractive target to transform (9), and engineering these cells would prove to be a valuable tool in cell therapy. We performed the three-step optimization on murine ASCs. The use of Buffer O and program X-01 yielded the most effective transfection rate. As with other cells, addition of LMP8 improved transfection efficiency by 3-fold, yielding 13.65% but failed to match the efficiency of the Amaxa buffer (29.1%). The consistent improvement in transfection efficiency by using LMP8 (Fig. 2) suggested that we should look into the other family members of this polymer (43). In that regard, we have tested two of them: LMP8 (poloxamer 188, see the structure in Fig. 5a) and LMP1 (poloxamer 181). LMP8 and LMP1 are both similar in structure (Fig. 5a). The internal, hydrophobic polyoxypropylene chain is the same size in both copolymers. However the outer, more hydrophilic polyoxyelthylene chains are much shorter in LMP1 than in LMP8. Apart from making the copolymer smaller, it also serves to decrease the solubility of LMP1. At concentrations of 1.0% or greater, we found LMP1 to be very toxic, with no living cells remaining in culture. However, at very low concentrations (<0.01%), LMP1 proved to be even more effective at transfecting cells than LMP8. Transfecting murine ASC using a low concentration of LMP1 in place of 0.5% LMP8 results in a larger population of cells positive for gfp (Fig. 5b). FACS sorting of the cells shows that 0.005% LMP1 is equally as efficient as the recommended Amaxa MSC nucleofection buffer (Fig. 5c), both formulations achieved about 29% transfection rate. This rate may be adequate for cell therapy as a non-viral delivery approach.
Fig. 5.

Nuclei Projection Under Scanning Electron Microscopy (SEM)
DNA or cell formulations are generally considered to change the electric, chemical, and biological properties of cells, increasing cell transfection efficiency. In this study, a different angle was used to analyze the possible underlying mechanism by which formulation increases the cell transfection efficiency. In this regard, ultra-morphology change in the presence of different cell formulations was explored using SEM. We incubated the seeded mASCs in DMEM, OptiMEM, OptiMEM + 0.001% LMP1, and Amaxa MSC Nucleofector Solution. The SEM images show that, in the presence of either Amaxa buffer (Fig. 6b) or our transfection buffers (Fig. 6c), the majority of nuclei (>80%) are projected to cell membrane, pushing the cell membrane to a noticeable bump (Figs. 6b,c). This is in contrast to the control mASC, where most of the nuclei (>80%) are masked by the plasma membrane with a flat plateau morphology, making the nuclei more difficult to observe (Fig. 6a).
Fig. 6.

DISCUSSION
Our primary goal is to develop an effective but public known cell formulation for boosting cell transfection via Nucleofection or other electroporation devices at a low cost. Our current result suggests that both LMP8 and LMA have a great potential to surpass or match Amaxa buffer in some cell lines (Fig. 3). Independently, one study (44) used a new assembled electroporation device with ability of varying pulses during the cell transfection and found that sequential delivery of high and low pulses achieved the same transfection efficiency as Amaxa buffer/device. It is our expectation that this new device in combination with our formulation may further boost the transfection efficiency and allowing gene expression in most, if not all, cells, surpassing currently available commercial systems. This study should be initiated.
In some tested cell lines, such as the transfection of 4T1 cells, the Amaxa nucleofector buffer still produced greater transfection efficiency than our formulations. It should be noted that, in either case, the use of nucleofection yields a moderate level of cell transfection. We observed, in most cases, 10–30% cells transfected by nucleofection, whether by our own formulation or the recommended Amaxa nucleofection buffer. This number is much less than the number advertised by Amaxa. Other studies using the reagents and guidelines published by Amaxa showed results similar to ours (45). These results suggest that further improvement is needed, regardless if using Amaxa or our buffer.
No single polymer exhibited the ability to increase the transfection rates of all cell lines. We had the most consistent success with Poloxamer-188 (LMP8). We have had comparable positive results with the use of pluronic block copolymers.
The exact mechanism of electroporation remains poorly understood, though current thought suggests a very brief opening of cellular pores that allow macromolecules to flow into the cell (46) (25). Originally, we thought that the use of certain polymers would surround the DNA, allowing it to more easily enter the cell in a manner similar to lipofection (20). However, no difference in transfection efficiency between mixing the nucleofection buffer with cells first and mixing the buffer with DNA first (data not shown). This result suggests that such a model may not be true.
What makes nucleofection such a desirable form of transfection is the ability to transfer the desired material directly into the nucleus. Our observation of mASC using scanning electron microscopy reveals a possible explanation for how Buffer O promotes cell transfection via electroporation. In the presence of transfection Buffer O and the Amaxa nucleofector solution, the majority of mASC nuclei are juxtaposed to the cell membrane, giving the nuclei the appearance of a projection above the plane of the cell (Fig. 6b, c). Theoretically, the nuclei projection to cell membrane would reduce the physical distance from the extracellular space to the nucleus. The same study was performed with other optimum buffer and polymer combinations without any success because cells fallen off the Thermanox cover slips in presence of LMP1 solution, inhibiting to determine the cell morphology under SEM. Many pluronic-block copolymers exhibit surfactant qualities (41), and LMP1 tends to be more hydrophobic (43) which may be impeding the ability of the mASC to remain firmly attached to the Thermanox cover slip. This may explain our inability to permanently attach cells to the cover slip for SEM study. Other materials such as glass fiber disks were used to grow cells for SEM study because this material is tolerant to the hot temperature under SEM and could prevent from cells stripped off in the presence of LMP1 solution prior to loading on SEM but the noising background from glass fibers prevented from observing any cell morphology. Other materials need to be identified for testing the model that some of the buffers and polymers may simply boosts the projects of nuclei to cell membrane and cut the distance between cell membrane to nuclei.
Adipose stem cells are a very attractive option for cell therapy, due to their ease of acquisition and isolation (8). Moreover, ASCs can be manipulated to differentiate into a number of different cell types (9). Since ASCs have shown some promise in homing to certain tumors in vivo (47), it is feasible to envision their potential use for cell-driven anti-tumor treatment, capable of promoting anti-tumor immunity or altering the tumor microenvironment. The ease of acquiring and isolating ASCs, combined with the ease and effectiveness of nucleofection along with the clinical safety and cost-effectiveness of an in-house nucleofection buffer, makes for a very attractive method of cell therapy.
Acknowledgments
This study was supported by National Institutes of Health grant RO1CA120895 and NIH/NIBIB grant R21EB007208. Authors also are thankful to the assistance in cell culture from Jiemiao Hu and in plasmid DNA preparation by Xueqing Xia.
Footnotes
Place of work completed: Baton Rouge, LA, USA
Conflict of Interest
The author has stated that there is no conflict of interest in this manuscript.
References
- 1.Pulendran B, Palucka K, Banchereau J. Sensing pathogens and tuning immune responses. Science. 2001;293(5528):253–6. doi: 10.1126/science.1062060. [DOI] [PubMed] [Google Scholar]
- 2.Paczesny S, Choi SW, Ferrara JL. Acute graft-versus-host disease: new treatment strategies. Curr Opin Hematol. 2009;16(6):427–36. doi: 10.1097/MOH.0b013e3283319a6f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rappa G, Anzanello F, Alexeyev M, Fodstad O, Lorico A. Gamma-glutamylcysteine synthetase-based selection strategy for gene therapy of chronic granulomatous disease and graft-vs-host disease. Eur J Haematol. 2007;78(5):440–8. doi: 10.1111/j.1600-0609.2007.00833.x. [DOI] [PubMed] [Google Scholar]
- 4.Lee ST, Jang JH, Cheong JW, Kim JS, Maemg HY, Hahn JS, et al. Treatment of high-risk acute myelogenous leukaemia by myeloablative chemoradiotherapy followed by co-infusion of T cell-depleted haematopoietic stem cells and culture-expanded marrow mesenchymal stem cells from a related donor with one fully mismatched human leucocyte antigen haplotype. Br J Haematol. 2002;118(4):1128–31. doi: 10.1046/j.1365-2141.2002.03767.x. [DOI] [PubMed] [Google Scholar]
- 5.Prata K, de L, Orellana MD, De Santis GC, Kashima S, Fontes AM, Carrara R, de C, et al. Effects of high-dose chemotherapy on bone marrow multipotent mesenchymal stromal cells isolated from lymphoma patients. Exp Hematol. 38(4):292–300 e4. doi: 10.1016/j.exphem.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Dupont KM, Sharma K, Stevens HY, Boerckel JD, Garcia AJ, Guldberg RE. Human stem cell delivery for treatment of large segmental bone defects. Proc Natl Acad Sci U S A. 107(8):3305–10. doi: 10.1073/pnas.0905444107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senju S, Hirata S, Motomura Y, Fukuma D, Matsunaga Y, Fukushima S, et al. Pluripotent stem cells as source of dendritic cells for immune therapy. Int J Hematol. doi: 10.1007/s12185-010-0520-1. [DOI] [PubMed] [Google Scholar]
- 8.Katz AJ, Llull R, Hedrick MH, Futrell JW. Emerging approaches to the tissue engineering of fat. Clin Plast Surg. 1999;26(4):587–603. viii. [PubMed] [Google Scholar]
- 9.Gimble J, Guilak F. Adipose-derived adult stem cells: isolation, characterization, and differentiation potential. Cytotherapy. 2003;5(5):362–9. doi: 10.1080/14653240310003026. [DOI] [PubMed] [Google Scholar]
- 10.Morizono K, De Ugarte DA, Zhu M, Zuk P, Elbarbary A, Ashjian P, et al. Multilineage cells from adipose tissue as gene delivery vehicles. Hum Gene Ther. 2003;14(1):59–66. doi: 10.1089/10430340360464714. [DOI] [PubMed] [Google Scholar]
- 11.Josiah DT, Zhu D, Dreher F, Olson J, McFadden G, Caldas H. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol Ther. 18(2):377–85. doi: 10.1038/mt.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Godbey WT. Viral vectors for gene delivery in tissue engineering. Adv Drug Deliv Rev. 2006;58(4):515–34. doi: 10.1016/j.addr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Hendrie PC, Russell DW. Gene targeting with viral vectors. Mol Ther. 2005;12(1):9–17. doi: 10.1016/j.ymthe.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Schaffer DV, Koerber JT, Lim KI. Molecular engineering of viral gene delivery vehicles. Annu Rev Biomed Eng. 2008;10:169–94. doi: 10.1146/annurev.bioeng.10.061807.160514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–7. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 16.Howitt J, Anderson CW, Freimuth P. Adenovirus interaction with its cellular receptor CAR. Curr Top Microbiol Immunol. 2003;272:331–64. doi: 10.1007/978-3-662-05597-7_11. [DOI] [PubMed] [Google Scholar]
- 17.Strauss BE, Costanzi-Strauss E. Combating oncogene activation associated with retrovirus-mediated gene therapy of X-linked severe combined immunodeficiency. Braz J Med Biol Res. 2007;40(5):601–13. doi: 10.1590/s0100-879x2007000500002. [DOI] [PubMed] [Google Scholar]
- 18.Check E. Gene therapy put on hold as third child develops cancer. Nature. 2005;433(7026):561. doi: 10.1038/433561a. [DOI] [PubMed] [Google Scholar]
- 19.Halama A, Kulinski M, Librowski T, Lochynski S. Polymer-based non-viral gene delivery as a concept for the treatment of cancer. Pharmacol Rep. 2009;61(6):993–9. doi: 10.1016/s1734-1140(09)70160-4. [DOI] [PubMed] [Google Scholar]
- 20.Narang AS, Thoma L, Miller DD, Mahato RI. Cationic lipids with increased DNA binding affinity for nonviral gene transfer in dividing and nondividing cells. Bioconjug Chem. 2005;16(1):156–68. doi: 10.1021/bc049818q. [DOI] [PubMed] [Google Scholar]
- 21.Bally MB, Harvie P, Wong FM, Kong S, Wasan EK, Reimer DL. Biological barriers to cellular delivery of lipid-based DNA carriers. Adv Drug Deliv Rev. 1999;38(3):291–315. doi: 10.1016/s0169-409x(99)00034-4. [DOI] [PubMed] [Google Scholar]
- 22.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A. 1995;92(16):7297–301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine)-mediated gene delivery affects endothelial cell function and viability. Biomaterials. 2001;22(5):471–80. doi: 10.1016/s0142-9612(00)00203-9. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A, Ranganathan P, Diamond SL. Nuclear targeting peptide scaffolds for lipofection of nondividing mammalian cells. Nat Biotechnol. 1999;17(9):873–7. doi: 10.1038/12860. [DOI] [PubMed] [Google Scholar]
- 25.Mir LM. Nucleic acids electrotransfer-based gene therapy (electrogenetherapy): past, current, and future. Mol Biotechnol. 2009;43(2):167–76. doi: 10.1007/s12033-009-9192-6. [DOI] [PubMed] [Google Scholar]
- 26.Neumann E, Schaefer-Ridder M, Wang Y, Hofschneider PH. Gene transfer into mouse lyoma cells by electroporation in high electric fields. Embo J. 1982;1(7):841–5. doi: 10.1002/j.1460-2075.1982.tb01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mir LM, Banoun H, Paoletti C. Introduction of definite amounts of nonpermeant molecules into living cells after electropermeabilization: direct access to the cytosol. Exp Cell Res. 1988;175(1):15–25. doi: 10.1016/0014-4827(88)90251-0. [DOI] [PubMed] [Google Scholar]
- 28.Cukjati D, Batiuskaite D, Andre F, Miklavcic D, Mir LM. Real time electroporation control for accurate and safe in vivo non-viral gene therapy. Bioelectrochemistry. 2007;70(2):501–7. doi: 10.1016/j.bioelechem.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Kotnik T, Mir LM, Flisar K, Puc M, Miklavcic D. Cell membrane electropermeabilization by symmetrical bipolar rectangular pulses. Part I. Increased efficiency of permeabilization. Bioelectrochemistry. 2001;54(1):83–90. doi: 10.1016/s1567-5394(01)00114-1. [DOI] [PubMed] [Google Scholar]
- 30.Satkauskas S, Bureau MF, Puc M, Mahfoudi A, Scherman D, Miklavcic D, et al. Mechanisms of in vivo DNA electrotransfer: respective contributions of cell electropermeabilization and DNA electrophoresis. Mol Ther. 2002;5(2):133–40. doi: 10.1006/mthe.2002.0526. [DOI] [PubMed] [Google Scholar]
- 31.Jacobsen F, Mertens-Rill J, Beller J, Hirsch T, Daigeler A, Langer S, et al. Nucleofection: a new method for cutaneous gene transfer? J Biomed Biotechnol. 2006;2006(5):26060. doi: 10.1155/JBB/2006/26060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maasho K, Marusina A, Reynolds NM, Coligan JE, Borrego F. Efficient gene transfer into the human natural killer cell line, NKL, using the Amaxa nucleofection system. J Immunol Methods. 2004;284(1–2):133–40. doi: 10.1016/j.jim.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 33.Trompeter HI, Weinhold S, Thiel C, Wernet P, Uhrberg M. Rapid and highly efficient gene transfer into natural killer cells by nucleofection. J Immunol Methods. 2003;274(1–2):245–56. doi: 10.1016/s0022-1759(02)00431-3. [DOI] [PubMed] [Google Scholar]
- 34.Leclere PG, Panjwani A, Docherty R, Berry M, Pizzey J, Tonge DA. Effective gene delivery to adult neurons by a modified form of electroporation. J Neurosci Methods. 2005;142(1):137–43. doi: 10.1016/j.jneumeth.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Aluigi M, Fogli M, Curti A, Isidori A, Gruppioni E, Chiodoni C, et al. Nucleofection is an efficient nonviral transfection technique for human bone marrow-derived mesenchymal stem cells. Stem Cells. 2006;24(2):454–61. doi: 10.1634/stemcells.2005-0198. [DOI] [PubMed] [Google Scholar]
- 36.Dubois SG, Floyd EZ, Zvonic S, Kilroy G, Wu X, Carling S, et al. Isolation of human adipose-derived stem cells from biopsies and liposuction specimens. Methods Mol Biol. 2008;449:69–79. doi: 10.1007/978-1-60327-169-1_5. [DOI] [PubMed] [Google Scholar]
- 37.Li S. Optimizing Electrotransfection of Mammalian Cells In Vitro. In: Freidmann T, editor. Gene Transfer: Delivery and Expression of DNA and RNA, A Laboratory Manual. Cold Spring Harbor Laboratory Press; 2006. pp. 419–25. [Google Scholar]
- 38.Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244(1–2):105–15. doi: 10.1016/s0378-5173(02)00315-0. [DOI] [PubMed] [Google Scholar]
- 39.Moore NM, Barbour TR, Sakiyama-Elbert SE. Synthesis and characterization of four-arm poly(ethylene glycol)-based gene delivery vehicles coupled to integrin and DNA-binding peptides. Mol Pharm. 2008;5(1):140–50. doi: 10.1021/mp700072n. [DOI] [PubMed] [Google Scholar]
- 40.Saxena A, Mozumdar S, Johri AK. Ultra-low sized cross-linked polyvinylpyrrolidone nanoparticles as non-viral vectors for in vivo gene delivery. Biomaterials. 2006;27(32):5596–602. doi: 10.1016/j.biomaterials.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 41.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82(2–3):189–212. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 42.Hartikka J, Sukhu L, Buchner C, Hazard D, Bozoukova V, Margalith M, et al. Electroporation-facilitated delivery of plasmid DNA in skeletal muscle: plasmid dependence of muscle damage and effect of poloxamer 188. Mol Ther. 2001;4(5):407–15. doi: 10.1006/mthe.2001.0483. [DOI] [PubMed] [Google Scholar]
- 43.Kabanov AV, Lemieux P, Vinogradov S, Alakhov V. Pluronic block copolymers: novel functional molecules for gene therapy. Adv Drug Deliv Rev. 2002;54(2):223–33. doi: 10.1016/s0169-409x(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 44.Stroh T, Erben U, Kuhl AA, Zeitz M, Siegmund B. Combined pulse electroporation - a novel strategy for highly efficient transfection of human and mouse cells. PLoS One. 5(3):e9488. doi: 10.1371/journal.pone.0009488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gresch O, Engel FB, Nesic D, Tran TT, England HM, Hickman ES, et al. New non-viral method for gene transfer into primary cells. Methods. 2004;33(2):151–63. doi: 10.1016/j.ymeth.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 46.Zaharoff DA, Henshaw JW, Mossop B, Yuan F. Mechanistic analysis of electroporation-induced cellular uptake of macromolecules. Exp Biol Med (Maywood) 2008;233(1):94–105. doi: 10.3181/0704-RM-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kucerova L, Altanerova V, Matuskova M, Tyciakova S, Altaner C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67(13):6304–13. doi: 10.1158/0008-5472.CAN-06-4024. [DOI] [PubMed] [Google Scholar]