


Abstract
SHAPE (Selective 2′-hydroxyl acylation analysed by primer extension) technology has emerged as one of the leading methods of determining RNA secondary structure at the nucleotide level. A significant bottleneck in using SHAPE is the complex and time-consuming data processing that is required. We present here a modified data collection method and a series of algorithms, embodied in a program entitled Fast Analysis of SHAPE traces (FAST), which significantly reduces processing time. We have used this method to resolve the secondary structure of the first ∼900 nt of the hepatitis C virus (HCV) genome, including the entire core gene. We have also demonstrated the ability of SHAPE/FAST to detect the binding of a small molecule inhibitor to the HCV internal ribosomal entry site (IRES). In conclusion, FAST allows for high-throughput data processing to match the current high-throughput generation of data possible with SHAPE, reducing the barrier to determining the structure of RNAs of interest.
INTRODUCTION
An understanding of the biological role of viral RNA is facilitated by knowledge of its higher order structure. Selective 2′-hydroxyl acylation analysed by primer extension (SHAPE) has emerged as one of the most robust and well-characterized methods of mapping RNA secondary structure at single-nucleotide resolution. SHAPE has been used to map the structure of RNAs as large and diverse as the ribosome and the HIV genome (1–4).
In SHAPE, the RNA of interest is chemically interrogated by addition of an electrophile, such as N-methylisatoic anhydride (NMIA), which preferentially acylates conformationally flexible nucleotides at the ribose 2′-OH position (5). These 2′-O-adducts result in termination events during subsequent reverse transcription (RT). The result is a pool of DNA fragments whose lengths correspond to the location of flexible nucleotides. The use of fluorescent-labelled primers allows these fragments to be resolved by capillary electrophoresis (6). In SHAPE, for every experimental condition, an experimental dataset, a control dataset, and one to two sequencing ladders need to be generated. Thus, three to four electrophoretic traces are generated for each region of RNA, which must be processed in order to calculate the SHAPE reactivity of each nucleotide in the RNA of interest. This data processing is challenging and laborious because neither the y-axis scale nor the x-axis scale is the same for each trace (3).
Each trace has a different x-axis scale because each fluorophore migrates through the capillary matrix at a different rate (3). Each fluorophore also interacts with the 5′ terminal nucleotides of the DNA fragment to which it is attached; consequently, both the identity of the fluorophore and its adjacent sequence affect the rate of DNA fragment migration. In DNA sequencing, the fact that only one peak should occur at each location allows for algorithmic correction. In SHAPE, the data lacks such registry information: the control and experimental (and ladder) peaks can and often should occur at the same location (3).
Two factors predominantly govern why each trace has a different y-axis scale. First, individual fluorophores have different spectral properties (3). Thus, a DNA fragment with the same concentration but labelled with two different fluorophores will have two different fluorescent intensities. Second, human error inevitably introduces variances in RNA (and later DNA) concentrations during sample processing.
SHAPEfinder is a program that was written in order to address these scaling issues (3). This program allows the user to visually adjust traces, allowing for normalization of traces to a common x- and y-axis. Visual manipulation, however, is a time-consuming and user-specific process.
Our goal was therefore to generate a SHAPE system that would obviate or automate the need for x- and y-axis scaling by visual inspection. Here, we describe a windows program, entitled FAST, to address this goal. To enhance its accessibility, FAST has been encoded as a module within the widely used program, RNAstructure (7).
FAST builds upon the idea of using the same fluorophore-labelled primer for all arms of a study, as first introduced in the program CAFA (8). Using the same fluorophore-labelled primer holds the migration offset introduced by the fluorophore constant among all conditions. Consequently, x-axis scaling is not needed. With respect to y-axis scaling, the need to correct for spectral differences among fluorophores is also eliminated by the use of the same fluorophore-labelled primer. In this manner, we have reduced the scaling problem down to correcting for y-axis differences that result from handling error.
In order to use one fluorophore for all arms of a study, capillary to capillary comparisons must be possible. Capillary to capillary comparisons are made possible by using an internal standard—a technique common to fragment analysis (9). The use of an internal standard to normalize data among capillaries has additional advantages: the control arm of the study, and any associated sequencing ladders, need to be generated only once, rather than needing to be generated repeatedly along with each and every experimental arm (4).
The most crucial step in the SHAPE process is proper alignment between the peaks found in a trace and the RNA sequence—i.e. assigning a nucleotide position to each peak. We refer to this process as peak identification. In SHAPEfinder, this process is highly dependent on the user correctly performing x-axis calibrations. Furthermore, because the original SHAPE method does not allow for traces from different capillaries to be compared, calibration must be performed for every condition in an experiment. In contrast, FAST allows the same calibration file to be used repeatedly for all subsequent analyses of the same RNA region.
Table 1 summarizes some of the differences between FAST and SHAPEfinder. PeakScanner is a freely available software program by Applied Biosystems, Inc (ABI), which we use to integrate the area of peaks in a trace. As noted previously, CAFA also uses a single fluorophore for RNA probing experiments, but it lacks the automated y-axis scaling and peak identification algorithms that make up the heart of the FAST program (8). Here, we demonstrate the utility of FAST by applying it to the analysis of the 5′ end of the hepatitis C virus (HCV) RNA genome. This segment encodes a critically important region of RNA secondary structure, the internal ribosome entry site (IRES)—several stretches of which have been solved by high-resolution nuclear magnetic resonance (NMR) and crystallography that provide for ideal ‘gold standards’ against which to compare FAST-generated results—and the HCV core protein. We first examine the IRES secondary structure by FAST, and then assess how it changes in response to addition of a small molecule ligand. Finally, we use SHAPE/FAST to resolve the RNA secondary structure downstream of the IRES, the core-encoding region of the HCV genome, which has previously been suggested to contain numerous RNA structures, some of which have been implicated in packaging.
Table 1.
Comparison between FAST and SHAPEFinder
| FAST | SHAPEFinder | |
|---|---|---|
| Processing corrections | ||
| Baseline adjustments | Automateda | Automated |
| Integration of peak areas | Automateda | Automated |
| Fluorophore spectral overlap | Less important | Automated |
| Fluorophore-specific mobility shifts (x-axis) | Not needed | Partially automated |
| Peak Identification | Partially Automated | Partially automated |
| Signal Decay | Automated | Visual manipulation |
| Signal Scaling (y-axis) | Automated | Visual manipulation |
| Notable characteristics | ||
| Electrophoresis technology | Fragment Analysis | DNA sequencing |
| Time Required (expert) | 5–15 min | 1–2 h |
| Specific mobility file for each region of RNA | NO | YES |
| Cost | 1-labelled Primer/300 nt | 4-labelled Primers/600 nt |
| Read length | ∼250–350 | ∼600 |
| Sequencing ladder | Once per RNA region | Needed for every run |
| Peak identification | Once per RNA region | Needed for every run |
aBaseline adjustment and Peak Area integration are performed initially using PeakScanner
METHODS
The DNA polymerase chain reaction (PCR) template for the genotype (GN) 1b/con1 5′UTR RNA was generated from Bart79I (10). The DNA PCR template for the GN2/J6 RNA was generated from FL-J6/JFH (11). RNAs were generated by in vitro transcription, using T7 MEGAscript. RNAs for SHAPE were purified by MEGAclear, with purity and length verified by capillary electrophoresis. HCV RNA was folded [100 mM NaCl; 2.5 mM MgCl; 65°Cx1′, 5′ cooling at room temperature, 37°C for 20–30′) as previously described (12), but in 100 mM HEPES, pH = 8.
2′ acylation with NMIA (5) and reverse transcription (RT) primer extension were performed at 45°Cx1′, 52°Cx25′, 65°Cx5′, as previously described (4). 6FAM was used for all labelled primers (see Supplementary Data for a list of primers.) Exceptions to these protocols were as follows: (i) RNA purification after acylation as well as removal of micro RNA (miRNA) before RT (as appropriate), was performed using RNA C&C columns (Zymoresearch), rather than ethanol precipitation; (ii) before and after SHAPE primer buffer was added, the mixture was placed at room temperature for 2–5 min, which enhanced RT transcription yields significantly; (iii) DNA purification was performed using Sephadex G-50 size exclusion resin in 96-well format then concentrated by vacuum centrifugation, resulting in a more significant removal of primer; and (iv) 1 pmol rather than 3 pmol of RNA was used in ddGTP RNA sequencing reactions.
Compound S-1 (kindly provided by Dr. Thomas Hermann) binding experiments were carried out as follows: After folding the GN1b RNA, S-1 was added to the RNA at the following concentrations: 200 µM, 126.4 µM, 40 µM, 12.66 µM, 4 µM, 1.26 µM, 400 nM, 126.4 nM, 40 nM, and allowed to incubate for ∼10 min. Pearson's correlation coefficient (σ) was used to examine the correlation between S-1 concentration and SHAPE reactivity, for each nucleotide.
The ABI 3100 Genetic Analyzer (50 cm capillaries filled with POP6 matrix) was set to the following parameters: voltage 15 kV, T = 60°C, injection time = 15 s. The GeneScan program was used to acquire the data for each sample, which consisted of purified DNA resuspended in 9.75 ul of Hi-Di formamide, to which 0.25 ul of ROX 500 internal size standard (ABI Cat. 602912) was added.
PeakScanner parameters were set to the following parameters: smoothing = none; window size = 25; size calling = local southern; baseline window = 51; peak threshold = 15. Fragments 250 and 340 were computationally excluded from the ROX500 standard (13). RNAstructure parameters: slope and intercept parameters of 2.6 and −0.8 kcal/mol, were initially tried, as suggested (14); however, we found that smaller intercepts closer to 0.0 kcal/mol (e.g. ∼−0.3) produced fewer less optimal structures (within a maximum energy difference of 10%). We speculate that this minor parameter difference may be due to the precise fitting achieved between experimental and control data sets by the automated FAST algorithm. FAST was written in ANSI C/C++ and the code is available upon request; in its current implementation, FAST is integrated into RNAstructure, which requires MFC (Microsoft Foundation Classes). RNA structures were drawn and coloured using RNAViz2 (15)
RESULTS AND DISCUSSION
Automated y-axis scaling
The various processing steps inherent to the SHAPE chemical probing method inevitably result in undesired differences among samples. In FAST, these differences are automatically corrected for in an algorithm referred to as load error correction.
The goal of load correction is to normalize two traces to one another, by scaling the traces such that the low-reactive nucleotides in the two traces overlap. This requires distinguishing low-reactive nucleotides from high-reactive nucleotides, which result in peaks of interest. In SHAPEfinder, the user attempts to visually overlap the least reactive 5–10% of peaks in the control arm to those in the experimental arm.
The challenge of visually manipulating one trace such that its least-reactive peaks overlap with the least-reactive peaks in a second trace is exemplified in experiment 1 (Figure 1A) and experiment 2 (Figure 1B). In experiment 1, the control arm has been deliberately overloaded with respect to the experimental arm, resulting in the area of each peak in the control data being, on average, higher than the peak area in the experimental data. Visually, because the control arm data are so prominent, a user might conclude that the data are too noisy to be meaningful. In experiment 2, the control arm has been diluted down with respect to the experimental arm. Visually, because the control arm looks virtually flat, the user may be tempted to assume that no gain adjustment is necessary. [In CAFA, there does not appear to be a normalization step: instead, an arbitrary cutoff of a 3-fold change over the mean background indicates positions of concern (8).]
Figure 1.

Load error correction. (A) Experiment 1, in which the control sample concentration is greater than the experimental sample concentration (∼2×). (B) Experiment 2, in which the control sample concentration has been decreased relative to the experimental sample concentration (∼1/2×). (C, D) Peak area traces are transformed into box-plot distributions, which are then normalized by their median values. After normalization, the resultant SHAPE reactivity traces (E) demonstrate that the two experiments have captured the same experimental data, despite their differing visual appearance.
In FAST, y-axis scaling is conceptualized not as a process of overlapping two traces, but instead as a process of normalizing the distribution of peak areas in the control data to the distribution of peak areas in the experimental data (Figure 1C and D). This makes scaling relatively trivial. A scaling factor is computed that increases or decreases the median of the experimental data such that it matches the median of the control data. The power of this method is illustrated in Figure 1E. Despite the notable difference in absolute and relative peak areas for the control and experimental data sets in experiment 1 compared to the data in experiment 2, each experiment has captured the same shape reactivity data, as evidenced by the overlapping traces. No user visualization or manipulation is required. If more than one experimental arm exists, all arms can be normalized to the control arm using this method—this results in the same scale for all experimental conditions tested. Practically, high load correction factors >2 indicate that the data are of limited quality; conversely, overfitting of high quality data can be avoided by excluding highly reactive nucleotides in the experimental trace from the process by which the scaling factor is determined (see Supplementary Data for a detailed description of this process).
Thus, this algorithm enables correction along the y-axis for any differences that may result from electrokinetic injection, loss of sample or loading error.
Signal decay correction
An overall trend is sometimes observed in electrophoretic traces: shorter fragments have higher signals, on average, than longer fragments. This phenomenon is called ‘signal decay’ (6). One reason for signal decay is that, while the probing reaction with the electrophile is carried out such that any given RNA molecule is modified only once, multiple hits still do occur. As a reverse transcriptase will pause at any first modification, this biases the distribution of fragments towards shorter ones. Another reason is the imperfect processivity of the superscript III reverse transcriptase (3). Additionally, we have observed that salt and primer concentration appear to effect signal decay. Desalting and removal of the primer by size exclusion chromatography appear to significantly reduce the decay rate. We suspect that one explanation for this observation is that DNA samples are taken up into the capillary by electrokinetic injection, a process that is dependent on solvent conductivity and solute electrophoretic mobility (9).
Weeks and colleagues observed that signal decay can be modelled as (6)
| (1) |
In this equation, D is the correction factor by which each peak area is divided; A and C are scaling factors for the initial and final trace intensities, respectively; and P is the probability of extension. Since our fragment analysis technique assigns a standardized fragment length to each peak, elution time in this equation is replaced by the fragment length of each peak.
In SHAPEfinder, the user can rapidly try out different values for P, such that after the peak area is divided by D (corrected peak area), the corrected peaks at the start, middle and end of the trace have similar heights by visual inspection. In FAST, we automate this procedure by (i) performing a linear fit of corrected peak areas versus their location and (ii) empirically determining the value of P that minimizes the magnitude of the slope of this linear fit. Because a slope of zero is a horizontal line, then when the magnitude of the slope of the linear fit is minimized, the P-value that results in the most even peak intensities at the beginning, middle and end of the trace has been identified. This empirical process is illustrated graphically in Figure 2A. As discrete values of P are tested (increments of 0.001), the absolute value of the slope reveals the inflection point where the slope changes from negative to positive. The slope changes sign because over-correction inverts the trace, resulting in corrected peaks at the start of the trace being lower than peaks at the end of the trace. In the example in Figure 2A, P was calculated to be 0.964.
Figure 2.

(A) Signal decay correction. Graphical depiction of the empirical determination of P in equation (1). Signal decay in an electrophoretic trace—in which smaller fragments have larger peak areas compared with longer fragments—can be described by an exponential decay function. Parameter P (probability of extension) can be determined empirically by testing values for P such that a line fit to the corrected peak area (peak area/D) versus DNA fragment length, has a slope, s, that is minimized. (B) Fragment length is an imperfect reflection of nucleotide position. Fragment length does not perfectly correlate with nucleotide position; their difference is depicted (grey line). This changing offset is the result of band compression. FAST algorithmically calculates this changing offset (grey line), using the observation that the offset can only change (delta–delta, black filled circles) in an incremental fashion. This allows for proper peak-to-nucleotide position correlation (peak identification). NP = nucleotide position; FL = fragment length. See text for further discussion.
Peak identification
Nucleotide positions are integer values. Thus, in an ideal trace, the spacing between successive fragments, as denoted by the width of valleys between peaks in the electrophoretogram, should all be uniform. In such a scenario, fragment lengths, as assigned using an internal size standard, would be identical to nucleotide position.
Band compression describes the irregularities in spacing that occur in an actual trace, which are thought to result from secondary structure. Band compression results in a changing offset between fragment length and nucleotide position (Figure 2B). A critical and novel component of the FAST algorithm is its ability to algorithmically adjust for band compression.
The input for this algorithm is an RNA sequence and the table of peak areas and their fragment lengths generated from a ddGTP sequencing ladder. FAST begins by assigning the nearest G in the RNA sequence to the peak whose fragment length is closest to the nucleotide number for that G (but where such assignments resulting in offsets larger than 5.5 nt are not allowed). This results in a first gross alignment between peak fragment length and nucleotide position, as illustrated in the Figure 3A. Notably, we have observed, and the algorithm assumes, that fragment length is always smaller than its corresponding nucleotide position.
Figure 3.
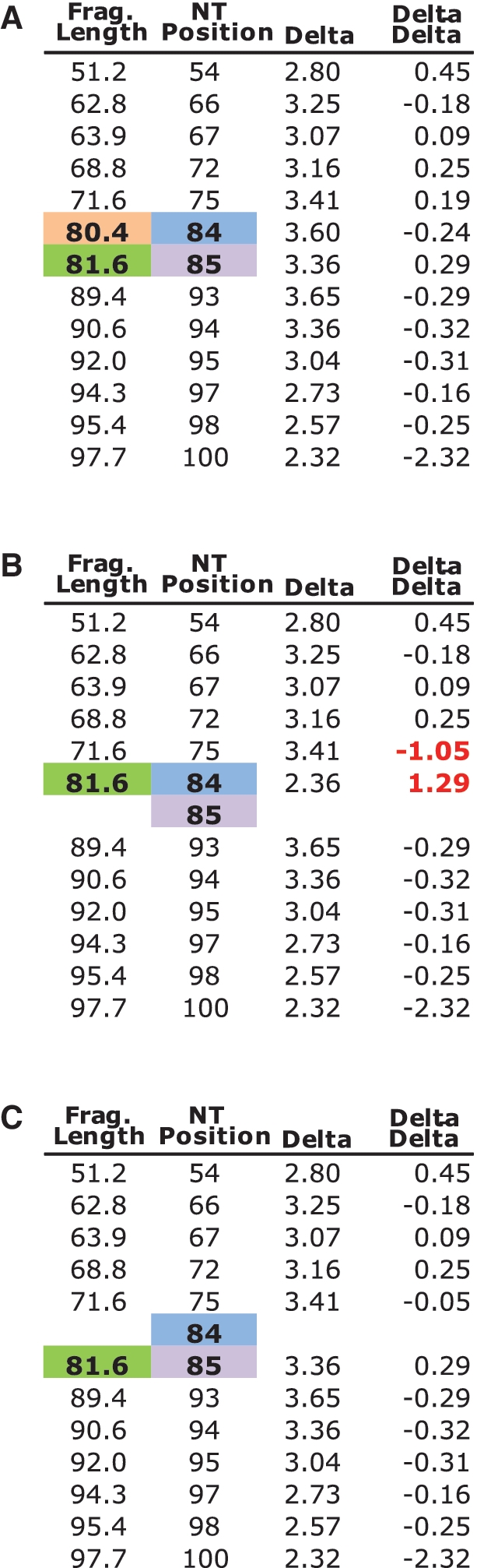
Simulation of peak identification algorithm. (A) Example calibration table of fragment lengths with their true nucleotide positions, assigned based on a gross alignment (see text). (B) The peak found at 80.4 is deleted, to simulate a missing peak or oversaturated peak at this location. This results in the mistaken assignment of NT position 84 to the peak at 81.6. At the same time, this causes the delta–delta in column 4 to become large, signifying to the algorithm that a mistake in the calibration table has been made. (C) The algorithm attempts local reassignments, in order to minimize the delta–delta, resulting in the correct re-assignment of nucleotide 85 to the peak with fragment length 81.6.
Further refinement is then necessary because the ladder trace generated by performing RT in the presence of ddGTP is imperfect, due to missing or aberrant peaks. In order to automatically correct for these phenomena, FAST takes advantage of the observation that the offset (delta) between fragment length (FL) and true nucleotide position (NP), Δ(NP–FL), varies in an incremental fashion. Successive deltas are always quite similar, such that delta–delta [where ΔΔi = (NPi–FLi) – (NP(i-1) – FL(i-1)), and i is the specific nucleotide being assigned] is small (<∼1.0) (Figure 2B). It also follows that each individual Δ(NP–FL) does not deviate significantly from the average of all Δ(NP–FL) values, <Δ(NP–FL)>.
These observations allow FAST to analyse the gross alignment previously generated, and make adjustments that minimize the delta–delta value for assignments. This type of local correction is illustrated in Figure 3B. If we imagine that the peak highlighted in light orange (80.4) in Figure 3A is missed—because of high background or being missed due to a low level of termination at that location—this causes the peak at 81.6 (highlighted in green) to be incorrectly assigned during the gross alignment to nucleotide 84. This also causes the absolute value of the ΔΔ in this local area to become high (bolded red text). FAST corrects this by attempting to find the peak most likely to correspond to unassigned nucleotide 85 (light purple), using the same heuristics as for the gross alignment. FAST then checks to see if this reassignment (nucleotide 85 to peak 81.6) minimizes the ΔΔs observed as compared to the original alignment (Figure 3C). This process is then repeated for nucleotide 84, which is now newly unassigned. In this case, however, because the nearest peak with a smaller fragment length is 71.6, and the difference between the fragment length and its assigned nucleotide position (84–71.6 > 5.5) is too high to be a reasonable assignment; this terminates the local re-assignment process for this region.
FAST also performs a linear regression fit of fragment length to nucleotide position, and determines the residual for each peak. When this residual is high, the calibration file is flagged for inspection by the words ‘REVIEW THIS REGION’. These assignments are beyond the ability of the algorithm, and user inspection is requested. Usually, assignments are correct, but can be manually adjusted within the program as needed.
FAST then uses the Local Southern method (16) along with this calibration table to convert fragment length to nucleotide numbers for each peak in the experimental and control data of interest. We have observed the Local Southern method to be quite robust. We have not found it necessary for the calibration table to capture all ddGTP termination events (peaks), which allows for specificity to be emphasized over sensitivity. For this reason, assignments are attempted by default for only the top 25% (by area) of peaks in a ddGTP trace, although this number can be varied by the user. Furthermore, because the fit is local, locally incorrect assignments do not affect assignments elsewhere.
The final output of the FAST module is the same as for SHAPEfinder: a SHAPE file that lists nucleotide positions and their SHAPE reactivities. This file is read into RNAstructure as a forced pseudo-free energy constraint, resulting in a proposed RNA secondary structure.
FAST has been optimized to work specifically with SHAPE data. For example, FAST interprets values that are higher than control values as meaningful and values that are lower than control values as meaningless/noise. Thus, while the algorithms described here are, in theory, agnostic to the probing method used, recoding would likely be necessary for its use with other techniques. For example, FAST cannot readily be used to analyse hydroxyl radical footprinting data, as control values (unfolded state) for such experiments are higher in value than experimental values (folded state) (17,18). Conversely, other chemical probing techniques, such as those that use DMS or CMCT (17,19) and result in data more similar to that of SHAPE, may be compatible with FAST analysis without much modification, if any.
Reproducibility
To assess the precision of FAST, we performed a series of analyses. Run-to-run variability in calculated shape reactivity (inter-experimental error) is plotted in Figure 4A. The same primer was used to initiate RT on the same region of RNA, from two different NMIA-probed RNA samples. A high correlation (Pearson's coefficient = 0.97) was observed. Run–to-run variability was also evaluated for an entirely different RNA, the hepatitis delta virus, and found to be similarly small. Next, we assessed the accuracy of peak-to-nucleotide assignment by using two different primers, located near each other, and determined the shape reactivity of the same HCV RNA region (Figure 4B). The SHAPE reactivity patterns for this region are in registry, suggesting that FAST algorithms have not been tuned only to one specific region of a given RNA or one specific primer.
Figure 4.

Reproducibility of FAST. (A) Two separate SHAPE experiments analyzed by FAST, in which the same primer was used during RT. The resultant SHAPE reactivities are plotted against one another and demonstrate the strong reproducibility of the FAST method to accurately assign NT position, correct for signal decay, and adjust for loading differences. See also Figure 1. (B) Two separate SHAPE experiments, in which two different but nearby primers were used during RT. The resultant SHAPE reactivities are plotted against one another. The observed correlation suggests against the possibility that the peak identification algorithm is specific to one primer, and that instead, it is a general means of correlating peaks with NT positions.
Additionally, we tested the reproducibility of FAST by determining the SHAPE reactivity of the same HCV region (nucleotide 29 through 112) in six independent experiments. Using these data, we calculated the standard deviation in SHAPE values for every position in this region. The average of these standard deviations was determined to be 0.04 SHAPE units (scale 0–1.5). The range (high and low) for every position was also calculated. The average of these ranges was determined to be 0.11 SHAPE units (scale 0–1.5). Thus, in the worst-case scenario of the highest versus lowest SHAPE reactivity determined for a given position, the measurement varies, on average, by 0.11 SHAPE units (see Supplementary Data Figure S1).
Examples
We have used this SHAPE/FAST system in a number of contexts. We have recently determined the structure of the genotype 1b/con1 5′UTR and found it to be consistent with all available NMR and crystallographic information. We have also used it to determine the structure of the complex formed between miRNAs-122 and its second target site in HCV (Pang et al., submitted for publication).
To further demonstrate the utility of this approach, we have also used it to (i) identify the binding site of a small molecule to domain II of the HCV IRES (Figure 5) and (ii) propose a structure for the core gene of HCV (Figure 6).
Figure 5.

Effect of S-1 compound on SHAPE reactivity of nucleotides in the HCV 5′UTR. (A) Varying concentrations of S-1, an ISIS-11 analogue, were incubated with folded HCV RNA. The resultant SHAPE reactivities versus S-1 concentration were examined using Pearson's correlation coefficient (σ). An adjusted Pearson's score (σ4sqrt(RANGE)), for each NT position, which emphasizes strong correlations and incorporates the magnitude of the change in SHAPE reactivity versus nucleotide position, is plotted. Five nucleotides in the HCV 5′UTR were identified as affected by the presence of compound S-1. (B) These 5 nt map to the domain IIa bulge of the HCV 5′UTR, where it has previously been shown that analogue compound ISIS-11 binds, distorting the IIa bulge (22).
Figure 6.

Postulated RNA secondary structure of the first ∼900 nt of the HCV genome, based on a SHAPE/FAST analysis. RNA structures in the core gene are labelled A–J. The HCV IRES (domains II–IV) are accurately determined, along with previously postulated RNA structures within the core gene labelled A, B, G and I. Six novel RNA helical/stem–loop structures were also determined, labelled C, D, E, F, H and J. Structure G (SL248) contains codon 91, where another internal initiation of translation has been proposed to occur. The region between domain IV and V (Structure A), is unstructured and proposed to encode an alternative reading frame protein.
Small ligand binding
Domain II of the HCV IRES is important for viral translation (20). A small molecule, known as ISIS-11 (21), has been found that binds to the first bulge in domain II (22), known as IIa, altering its conformation and inhibiting viral translation (23,24). Given the long history of using chemical probing methods to detect the binding of small molecules to ribosomal RNA (25), we evaluated whether or not SHAPE/FAST could be used to detect the binding of a small molecule to domain II. We performed SHAPE experiments in the presence of 0.5 nM to 200 µM of an ISIS-11 derivative, S-1. We then calculated the Pearson correlation coefficient for each nucleotide in domain II as a function of the log concentration of S-1. In this manner, we identified 5 nt whose conformation was significantly altered by the presence of S-1 (Figure 5A). Each of these nucleotides is found near the binding pocket for the S-1 compound (Figure 5B), suggesting that SHAPE can be used to characterize and potentially identify the binding site of small molecules to viral RNA.
RNA secondary structure of HCV core gene
The core gene of HCV has been postulated to contain a number of RNA structures by both computational and enzymatic probing (26–28). How these structures fit together in the context of the entire core gene, and whether other structures are present in this gene, is unknown. Additionally, it was shown that the core gene contains elements that allow for translation to initiate downstream of the classic HCV IRES start site (29). The core gene is also controversially postulated to contain an alternate reading frame, producing a protein product named F (30–32). Using the SHAPE/FAST system described here, we have derived a proposed RNA secondary structure of the entire core gene of HCV (Figure 6). The RNA stem–loop structures in the core gene have been labelled A–J. This structure reveals a number of new putative structures (C–F, H and J), and is notably consistent with previous determined RNA structures A, B and I. The nucleotides in the region of structure G have been previously postulated to form a structure known as SL248, which was enzymatically probed in isolation (28). Our structure is largely consistent with this structure, but varies in its bottom half. We postulate that this difference is due to the presence of other RNA core elements which did not exist in the construct originally used to probe SL248. Stem–loop G also contains the postulated initiation codon for mini-core, an alternate site of in-frame translation initiation.
As has been previously observed, the core gene contains a number of codons whose third positions are absolutely conserved (26,30) (Figure 7, bottom right), suggesting either an RNA structure of interest or as previously mentioned, an alternate reading frame protein. It is intriguing that the region between domain IV of the HCV IRES and Stem–Loop (SL)-A/domain V/SL47, contains such a high density of codons whose third positions are absolutely conserved, given that this region has high SHAPE reactivity and is expected to be unstructured. The absence of an RNA structure in this region favours the possibility of an alternate reading frame protein.
Figure 7.

Average SHAPE reactivity (window size = 100 nt) as a function of nucleotide position. The HCV IRES has the lowest SHAPE reactivity. The region after the AUG start site is unstructured yet possesses a high density of codons whose third position is conserved. Codon 91, the putative site of internal core translation initiation is indicated, and represents the region with the second lowest SHAPE reactivity.
An obvious difference in average SHAPE reactivity was observed for the non-coding versus the coding region of HIV, with the latter having significantly higher overall reactivity (6). In contrast, we observe that while the HCV IRES region has the lowest average SHAPE reactivity, the core region has some areas of high reactivity, but some areas of notably low reactivity as well. This is consistent with the observations by Simmonds et al. (33) that HCV contains significant RNA structure throughout its coding region. Intriguingly, the region near codon 91, where internal initiation is thought to also occur (29), defines a second region of high RNA structure density/low shape reactivity. Whether these structures form a second IRES can only be speculated (34). Mutagenesis studies will be needed to first confirm the RNA secondary structure shown here, and any relevance it may or may not have to internal initiation at codon 91.
In summary, we present here a novel, rapid method for determining RNA secondary structure, based on SHAPE technology. We have then used this technology to map the binding of a small molecule to the HCV IRES, as well as to postulate for the first time the structure of the first ∼900 nt of HCV, including the core gene. The software for RNAstructure with FAST can be found at http://glennlab.stanford.edu. The source code for this software is available upon request.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A Burroughs Wellcome Fund Clinical Scientist Award in Translational Research (to J.S.G.), RO1AI087917 (to J.S.G.); and an NRSA F32AI082930 (to P.S.P.) and HHMI Research Training Fellowships for Medical Students (to E.A.P.). Funding for open access charge: NIH-5R01AI087917-02.
Conflict of interest statement: Certain aspects of the technique outlined here are part of a patent filed by Stanford University.
ACKNOWLEDGEMENTS
We thank David Mathews for critical manuscript feedback, Peter De Rijk for a custom version of RNAViz2 and Thomas Hermann for the ISIS-11 derivative, S-1.
REFERENCES
- 1.Low JT, Weeks KM. SHAPE-directed RNA secondary structure prediction. Methods. 2010;52:150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–716. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasa SM, Guex N, Wilkinson KA, Weeks KM, Giddings MC. ShapeFinder: a software system for high-throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA. 2008;14:1979–1990. doi: 10.1261/rna.1166808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mortimer SA, Weeks KM. Time-resolved RNA SHAPE chemistry: quantitative RNA structure analysis in one-second snapshots and at single-nucleotide resolution. Nat. Protoc. 2009;4:1413–1421. doi: 10.1038/nprot.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008;6:e96. doi: 10.1371/journal.pbio.0060096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathews DH. RNA secondary structure analysis using RNAstructure. Curr Protoc. Bioinform. 2006 doi: 10.1002/0471250953.bi1206s13. Chapter 12, Unit 12 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitra S, Shcherbakova IV, Altman RB, Brenowitz M, Laederach A. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res. 2008;36:e63. doi: 10.1093/nar/gkn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mitchelson,K.R. and Cheng,J. (2001) Capillary electrophoresis of nucleic acids. Vol. I: Introduction to the capillary electorphoresis of nucleic acids. Springer, Heidelberg.
- 10.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 11.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 12.Kieft JS, Zhou K, Jubin R, Murray MG, Lau JY, Doudna JA. The hepatitis C virus internal ribosome entry site adopts an ion-dependent tertiary fold. J. Mol. Biol. 1999;292:513–529. doi: 10.1006/jmbi.1999.3095. [DOI] [PubMed] [Google Scholar]
- 13.Akbari A, Marthinsen G, Lifjeld JT, Albregtsen F, Wennerberg L, Stenseth NC, Jakobsen KS. Improved DNA fragment length estimation in capillary electrophoresis. Electrophoresis. 2008;29:1273–1285. doi: 10.1002/elps.200700523. [DOI] [PubMed] [Google Scholar]
- 14.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc. Natl Acad. Sci. USA. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Rijk P, Wuyts J, De Wachter R. RnaViz 2: an improved representation of RNA secondary structure. Bioinformatics. 2003;19:299–300. doi: 10.1093/bioinformatics/19.2.299. [DOI] [PubMed] [Google Scholar]
- 16.Southern EM. Measurement of DNA length by gel electrophoresis. Anal. Biochem. 1979;100:319–323. doi: 10.1016/0003-2697(79)90235-5. [DOI] [PubMed] [Google Scholar]
- 17.Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powers T, Noller HF. Hydroxyl radical footprinting of ribosomal proteins on 16S rRNA. RNA. 1995;1:194–209. [PMC free article] [PubMed] [Google Scholar]
- 19.Tijerina P, Mohr S, Russell R. DMS footprinting of structured RNAs and RNA–protein complexes. Nat. Protoc. 2007;2:2608–2623. doi: 10.1038/nprot.2007.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraser CS, Doudna JA. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 2007;5:29–38. doi: 10.1038/nrmicro1558. [DOI] [PubMed] [Google Scholar]
- 21.Seth PP, Miyaji A, Jefferson EA, Sannes-Lowery KA, Osgood SA, Propp SS, Ranken R, Massire C, Sampath R, Ecker DJ, et al. SAR by MS: discovery of a new class of RNA-binding small molecules for the hepatitis C virus: internal ribosome entry site IIA subdomain. J. Med. Chem. 2005;48:7099–7102. doi: 10.1021/jm050815o. [DOI] [PubMed] [Google Scholar]
- 22.Paulsen RB, Seth PP, Swayze EE, Griffey RH, Skalicky JJ, Cheatham TE, III, Davis DR. Inhibitor-induced structural change in the HCV IRES domain IIa RNA. Proc. Natl Acad. Sci. USA. 2010;107:7263–7268. doi: 10.1073/pnas.0911896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons J, Castaldi MP, Dutta S, Dibrov SM, Wyles DL, Hermann T. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat. Chem. Biol. 2009;5:823–825. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Q, Han Q, Kissinger CR, Hermann T, Thompson PA. Structure of hepatitis C virus IRES subdomain IIa. Acta Crystallogr. 2008;64:436–443. doi: 10.1107/S0907444908002011. [DOI] [PubMed] [Google Scholar]
- 25.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 26.Walewski JL, Gutierrez JA, Branch-Elliman W, Stump DD, Keller TR, Rodriguez A, Benson G, Branch AD. Mutation Master: profiles of substitutions in hepatitis C virus RNA of the core, alternate reading frame, and NS2 coding regions. RNA. 2002;8:557–571. doi: 10.1017/s1355838202029023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuplin A, Wood J, Evans DJ, Patel AH, Simmonds P. Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA. 2002;8:824–841. doi: 10.1017/s1355838202554066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuplin A, Evans DJ, Simmonds P. Detailed mapping of RNA secondary structures in core and NS5B-encoding region sequences of hepatitis C virus by RNase cleavage and novel bioinformatic prediction methods. J. Gen. Virol. 2004;85:3037–3047. doi: 10.1099/vir.0.80141-0. [DOI] [PubMed] [Google Scholar]
- 29.Eng FJ, Walewski JL, Klepper AL, Fishman SL, Desai SM, McMullan LK, Evans MJ, Rice CM, Branch AD. Internal initiation stimulates production of p8 minicore, a member of a newly discovered family of hepatitis C virus core protein isoforms. J. Virol. 2009;83:3104–3114. doi: 10.1128/JVI.01679-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walewski JL, Keller TR, Stump DD, Branch AD. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA. 2001;7:710–721. doi: 10.1017/s1355838201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMullan LK, Grakoui A, Evans MJ, Mihalik K, Puig M, Branch AD, Feinstone SM, Rice CM. Evidence for a functional RNA element in the hepatitis C virus core gene. Proc. Natl Acad. Sci. USA. 2007;104:2879–2884. doi: 10.1073/pnas.0611267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuksek K, Chen WL, Chien D, Ou JH. Ubiquitin-independent degradation of hepatitis C virus F protein. J. Virol. 2009;83:612–621. doi: 10.1128/JVI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simmonds P, Tuplin A, Evans DJ. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: implications for virus evolution and host persistence. RNA. 2004;10:1337–1351. doi: 10.1261/rna.7640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weill L, James L, Ulryck N, Chamond N, Herbreteau CH, Ohlmann T, Sargueil B. A new type of IRES within gag coding region recruits three initiation complexes on HIV-2 genomic RNA. Nucleic Acids Res. 2010;38:1367–1381. doi: 10.1093/nar/gkp1109. [DOI] [PMC free article] [PubMed] [Google Scholar]