


Abstract
Existing methods for orthologous gene mapping suffer from two general problems: (i) they are computationally too slow and their results are difficult to interpret for automated large-scale applications when based on phylogenetic analyses; or (ii) they are too prone to making mistakes in dealing with complex situations involving horizontal gene transfers and gene fusion due to the lack of a sound basis when based on sequence similarity information. We present a novel algorithm, Global Optimization Strategy (GOST), for orthologous gene mapping through combining sequence similarity and contextual (working partners) information, using a combinatorial optimization framework. Genome-scale applications of GOST show substantial improvements over the predictions by three popular sequence similarity-based orthology mapping programs. Our analysis indicates that our algorithm overcomes the intrinsic issues faced by sequence similarity-based methods, when orthology mapping involves gene fusions and horizontal gene transfers. Our program runs as efficiently as the most efficient sequence similarity-based algorithm in the public domain. GOST is freely downloadable at http://csbl.bmb.uga.edu/~maqin/GOST.
INTRODUCTION
Orthologous genes refer to genes that have evolved from a common ancestor through speciation only (1). A widely accepted corollary, especially for bacterial genomes, is that they are functional equivalents, i.e. they play the same functional role in the equivalent biological processes across different organisms. Identification of orthologous genes across genomes, or ‘orthologous gene mapping’, represents the most essential technique in comparative genomics, but the problem remains largely unsolved. One key issue is that the definition of orthologous genes is not operational unless phylogenetic trees could be accurately derivable and analysis methods are available for distinguishing orthologous from paralogous genes, which by themselves are very challenging and unsolved problems. Because of the nature of the problem, the majority of the computer programs developed for solving the problem have been generally empirical in nature and often lack a sound theoretical basis.
The current orthology-mapping programs generally fall into two categories, phylogeny-based and sequence similarity-based (2). In the first category, gene trees need to be constructed, followed by rather involved analyses of the constructed trees to derive orthologous gene relationships. Programs such as RIO (3), Orthostrapper (4), RSD (5), Mestortho (6), OMA (7) and QuartetS (8) fall into this category. The best example of sequence similarity-based methods is the Reciprocal Best Hit (RBH) program (9), which predicts the orthologous gene in a target genome for a given gene A in a query genome by finding a gene B in the target genome so that B is the best Blast hit in the target genome for A, and vice versa. Cluster of Orthologous Groups (COG)/eukaryotic Orthologous Groups (KOG) (10) ‘generalize’ RBH by considering three genomes instead of two, aimed to increase the prediction accuracy of RBH but suffered from low prediction coverage. In addition, there are several more recent developments aimed to further improve the prediction reliability, including INPARANOID (11) and OrthoMCL (12). However, systematic analyses indicate that RBH is still the one to beat in terms of prediction accuracy among all sequence similarity-based methods as long as two specific parameters, soft filtering and final alignment Smith–Waterman, are adjusted properly (13). In addition, RBH is the most time efficient compared to other orthology mapping methods (7).
Phylogeny-based methods are generally more reliable than sequence similarity-based methods, while the latter are generally orders of magnitude more efficient, and hence are capable of dealing with genome-scale applications. A recent survey (7) suggested that the substantially more time needed for phylogeny-based orthologous gene prediction may not be worthwhile for the limited increase in prediction accuracy over sequence similarity-based methods. It is fair to say that RBH represents the state of the art in orthologous gene prediction for large-scale applications. In their recent review, Chen et al. (2) reported that RBH tends to have high prediction accuracy (i.e. low false positive) but suffers from low prediction coverage (i.e. high false negatives) while programs like KOG and OrthoMCL tend to have the opposite behavior. One general issue with all these sequence similarity-based methods is that they all implicitly assume that orthologous relationship can be captured by sequence similarity information alone among homologous genes, which is clearly not true (14). There is no real biological reason of why two orthologous genes should have the best sequence alignment score among possible homologous alternatives, particularly when the similarity scores are close.
We here present a novel program Global Optimization Strategy (GOST) for orthologous gene mapping across bacterial genomes, which to a large extent overcomes the intrinsic issues faced by sequence similarity-based methods discussed above. The fundamental difference between GOST and the existing sequence similarity-based methods is that GOST is designed to find orthologous gene pairs across two genomes with a good ‘enough’ sequence similarity score under the condition that the two genes have homologous working partners in their respective genomes (throughout the article two genes are said to be homologous if their sequence similarity is below a specific E-value threshold by BLAST). Here two genes in a genome are considered as ‘working partners’ if they share a common ‘uber-operon’ (15), which generalizes our previous work where we defined such a relationship based on operons (14). We demonstrated the effectiveness of this strategy on a large set of bacterial genomes by showing that GOST outperforms three popular sequence similarity-based orthology mapping programs, RBH, INPARANOID and OrthoMCL by substantial margins in terms of prediction ‘coverage, mislabeling error rate’ and ‘missing rate’, which are commonly used to assess orthologous genes prediction programs (13). We further compared GOST with RBH, INPARANOID and OrthoMCL in their predictions when mapping Escherichia coli enzyme-encoding genes to all sequenced bacterial genomes against known orthologous relationships as documented in the SwissProt Enzyme database (16). Specifically GOST identified 665 more enzyme gene pairs than RBH, 1901 more than INPARANOID and 2354 more than OrthoMCL. We believe that the performance of GOST is actually better than what these numbers suggest as the Enzyme database contains only a small portion of all the orthology relationships among enzyme-encoding genes across these bacterial genomes. Overall, GOST is much more efficient than OrthoMCL and INPARANOID, and is as fast as RBH (see Supplementary Table S1 in the Supplementary Data).
MATERIALS AND METHODS
Data
We used E. coli K12 as the query genome and other 959 complete bacterial genomes from NCBI (release of April 2009) as the targets for orthologous gene mapping. The operon information was downloaded from the DOOR database (17) on 1 November 2009. The SwissProt database was downloaded from http://www.expasy.org/enzyme/ on 5 October 2010.
Orthologous gene mapping: problem formulation
We first introduce a few graph-theoretic definitions needed for our problem formulation and solution. A graph is called a ‘multi-graph’ if more than one edge is allowed between two vertices in the graph. A ‘bipartite’ graph B = (X, Y, E) is a graph whose vertex set can be partitioned into two subsets X and Y so that no edge exists between vertices of the same subset. A ‘matching’ M is a subset of E such that no two edges share a common vertex. A ‘maximum matching’ is a matching of the maximum cardinality. A graph is said to be ‘connected’ if for any pair of vertices, there is a path between the two vertices within the graph. A ‘component’ in a graph is a maximum connected sub-graph.
Let G1 and G2 be the gene sets of two given bacterial genomes. Define a bipartite graph B = (G1, G2, E), termed a ‘homology’ graph, where two vertices (one from each genome) are connected by an edge in E if and only if the Blast E-value between the corresponding genes is below a pre-defined threshold (see Method 1 in the Supplementary Data). One possible way to formulate the orthologous gene mapping problem is through finding a maximum matching in the bipartite graph B [we noticed that a article was just published using this formulation for the orthologous gene mapping problem (18) as we were writing this article] One way to include biological process information into the problem formulation is through application of operons as genes in the same operon generally work in the same biological process. Specifically we can constrain the bipartite matching problem by requiring that each gene pair in a matching has at least one additional pair of homologous genes sharing their operons. However our previous study showed that this constraint led to rather low mapping coverage. Hence we looked into a generalized form of operons, i.e. uber-operons (15). A ‘uber-operon’ is a group of operons in a genome whose operons are functionally related, and their union is conserved across multiple (reference) genomes more frequently than expected by chance [we refer the reader to (15) for details of the definition]. We believe that uber-operons are the evolutionary foot-prints of ancient operons, which were split in different ways into smaller operons along different evolutionary lineages (15). In this article we formulate the orthologous gene mapping problem through (implicitly) including the uber-operon information into sequence similarity-based-procedure.
Consider two genomes for orthologous gene mapping. Define a new graph G = (O, M), with vertex set O consisting of operons of the two genomes, and with the edge set being the current matching M of B (not necessarily maximum). Clearly G is a multi-graph since there might be multiple edges of M between two vertices (operons) in O. Let c(O, M) denote the number of connected components in G. Based on the above discussion, we found that a maximum matching M in B gives rise to an orthologous gene mapping if and only if M maximizes c(O, M). Let 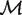 * be the set of all the maximum matchings in B. Hence the orthologous gene mapping between the two genomes is to find a maximum matching M in
* be the set of all the maximum matchings in B. Hence the orthologous gene mapping between the two genomes is to find a maximum matching M in 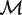 * such that c(O, M) is maximized. Let
* such that c(O, M) is maximized. Let 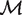 be the set of all the matchings in B. It is easy to check that the following two optimization problems
be the set of all the matchings in B. It is easy to check that the following two optimization problems
have the same optimum solution. Therefore, the problem of finding orthologous genes mapping can be modeled as to find an optimum solution to the following problem:
Although this optimization problem is theoretically intractable (it is relatively simple to prove that it is NP-hard), it is easy to get an optimum solution virtually with probability = 1.0 in this particular context because of the following observation: the orthologous gene pairs (or matched edges in B) in two conserved uber-operons (one from each genome) are always denser than those in two unrelated uber-operons. We predict each edge in the calculated optimum matching as a pair of orthologous genes across the two genomes under consideration. The following gives a high-level description of our algorithm.
For the simplicity of presentation, we introduce another weighted complete graph, G* = (V, E), with vertex set V consisting of all the connected components of G and edge set E consisting of all the pairs of vertices in V. For each edge e = (C1, C2), its weight is defined as w(e) = |M12| − |M1| − |M2|, where M1 and M2 are the restrictions of M on C1 and C2, respectively, and M12 the maximum matching of the subgraph C12 which is induced in B by the union of C1 and C2. It is obvious from the definition of G* that G* depends on the current matching M. The algorithm starts with M being empty. At this moment the adjoining graph G is a graph with vertex set O consisting of all the operons from G1 and G2 and with edge set being empty, and the graph G* a weighted complete graph with vertex set V the same as O and the weight of an edge being the cardinality of a maximum matching between the two operons connected by the edge.
Input: query genome G1 and target genome G2.
Output: a maximal set of orthologous gene pairs between the two genomes.
Step 1. Construct graphs B = (G1, G2), G = (O, M) and G* = (V, E) with M =
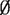 .
.Step 2. Finding an edge e = (C1, C2) of G* with weight w(e) biggest.
If w(e) = 0, go to Step 4.
Step 3. Merge the two components C1 and C2 into one, reset M = M − M1 − M2 + M12 and modify G and G* accordingly; return to Step 2.
Step 4. Output the current matching M, i.e. a maximal orthologous mapping.
RESULTS AND DISCUSSION
A challenge in assessing orthologous gene mapping programs, particularly on large scale applications, is that there is no widely accepted benchmark dataset of orthologous genes across different genomes. We used the following methods to evaluate GOST and other three programs (RBH, INPARANOID and OrthoMCL) across 959 bacterial genomes in terms of (i) whether the predicted gene pairs have their working partners being homologous; and (ii) whether the predicted orthologous enzyme-encoding genes have the same enzymatic functions according to the enzyme database, commonly used to assess orthology mapping programs (7,13). In addition, we have examined the performance of the programs on a selected set of challenging cases that involve horizontal gene transfers and gene fusions.
Prediction assessment on genome-scale predictions
Prediction ‘coverage’ is defined as the number of the predicted orthologous gene pairs between a query and a target genome. Two genes in a genome are called ‘operon-based working partners’ if and only if they are in the same operon. Two homologous genes, one in the query and one in the target genome, are considered to be correctly predicted if they each have an operon-based working partner, which are homologous. A gene in a target genome is called a ‘supported’ gene if one of its operon-based working partners is a homologous gene of some query gene. We use the following measures to assess the prediction programs. A ‘missing error’ is made for a gene in the query genome if this gene has homologous supported genes in the target genome as detected by BLAST and this gene itself is not predicted to be an ortholog of any gene in the target genome. A ‘mislabeling error’ is made for a gene in the query genome, if this gene has at least two homologues x and x′ in a target genome with x predicted to be its ortholog, and a gene sharing an operon with it has a homolog y with y and x′ being in the same operon in the target genome [our definitions of missing error and mislabeling error are different from the similar definitions given in (13) as we used operons in our definition. However our performance assessment is done using their definitions which is well explained in Method 2 and Supplementary Figure S1 in the Supplementary Data, and see Supplementary Figure S2 for detailed performance]. Together they are referred to as ‘errors’. The ‘missing error rate’ is defined as the ratio between the number of ‘missing errors’ and the number of ‘errors’ plus the number of correctly predicted orthologous gene pairs; and the ‘mislabeling error rate’ is defined similarly. Supplementary Table S2 lists the prediction coverage, missing error rate and mislabeling error rate for the four programs across 959 bacterial genomes. The details about comparisons of the distributions of coverage, missing error rate and mislabeling error rate against genomic similarity score (GSS) (13) are in Figure 1.
Figure 1.

A comparison of distributions of (a) prediction coverage, (b) missing error rates and (c) mislabeling error rates by RBH, INPARANOID, OrthoMCL and GOST against GSS. Since RBH and GOST do not consider co-orthologs, we count each group of predicted co-orthologous genes as one for INPARANOID and OrthoMCL.
By comparing the detailed prediction results given in Figure 1, we noted that GOST consistently outperforms the three other programs across the above three measures, especially when GSS is low, i.e. in (0.2–0.5), which accounts for 85% of the 959 target genomes. On both types of errors, GOST outperforms the other three algorithms by a significant margin although all four programs have low mislabeling error rates.
Prediction performance on enzyme-encoding genes
While assessing the performance of orthologous gene mapping programs remains an unsettled problem due to the reality that there is no widely accepted benchmark dataset, performance assessment on a special class of genes, i.e. enzyme-encoding genes, can be readily made. Specifically, we consider orthologous enzymes in the SwissProt database (16) as true orthologs, which can be used to assess the performance of the four prediction programs on enzyme-encoding genes. We noted that some E. coli enzymes have gene assignments only in a few genomes, which could lead to incorrect conclusions about the performance of the four programs under testing if including such enzymes when assessing performance statistics. Hence we consider only enzymes with gene assignments in at least 5% of the 959 target genomes, i.e. 48 genomes, which leaves 419 E. coli enzyme-encoding genes for orthology mapping. In the remaining of this section, an E. coli gene refers to one of these 419 genes unless stated otherwise. Note that our evaluation method could have limitations knowing that (i) some of the expert-curated enzymes could possibly have errors; and (ii) SwissProt Enzyme database contains only a small portion of all the orthology relationships among enzyme-encoding genes under consideration. So the reader may need to take caution in interpreting the comparison results.
For each E. coli K12 gene e, let Ne(X) be the number of genes predicted to be an ortholog of e across all bacterial genomes under consideration by program X. For each target genome g, let Ng(X) be the number of orthologous genes of E. coli genes predicted by X. Programs A and B are considered to have the same level of performance over an enzyme e if |Ne(A) – Ne(B)| ≤ K, and the same level of performance over a genome g if |Ng(A) – Ng(B)| ≤ K for some positive number K (in our current study, K = 5). A is said to perform better than program B over enzyme e (respectively genome g) if Ne(A) – Ne(B) > K (respectively Ng(A) – Ng(B) > K). The frequency distributions of Ne(GOST)-Ne(RBH), Ne(GOST)-Ne(INPARANOID) and Ne(GOST)-Ne(OrthoMCL) binned into a specific range, say between 5 and 10, are given in Supplementary Figure S3. We can see from the figure that GOST performs at least as well as the three other programs for the majority of the 419 enzymes (385 enzymes for RBH, 358 enzymes for INPARANOID and 347 enzymes for OrthoMCL). Interestingly, GOST performs substantially better than the other programs over a few enzymes, e.g. it predicts 172 more orthologous genes for EC 2.7.2.8 (N-acetylglutamate kinase) and 99 more genes for EC 2.7.13.3 (histidine kinase) than RBH. Further inspection indicates that gene fusions or horizontal gene transfers are the main reasons that have affected the performance of the other three programs substantially more than GOST as shown in the analysis below.
Figure 2 shows a genome-centric perspective of the predictions by the four programs. Specifically, GOST performs at as the same level of the three other programs for most organisms (945 genomes for RBH, 879 genomes for INPARANOID and 877 for OrthoMCL) and it outperforms RBH on 13 genomes, INPARANOID on 77 genomes, and OrthoMCL on 79 genomes while GOST was outperformed by RBH on zero genome, by INPARANOID on two genomes and by OrthoMCL on two genomes.
Figure 2.

Performance comparison between GOST and the other three programs. The x-axis represents the difference between the number of mapped enzyme genes by GOST and one of the other three programs, grouped into bins, where [X, Y] represents enzymes over which GOST predicted X–Y more orthologous genes across all the target genomes. The height along the y-axis of a bar represents the number of enzymes within each range. Ng(X) represents the number of orthologous genes of E. coli genes predicted by program X for each target genome g.
In the following analyses, we compare only between GOST and RBH since Figure 2 shows that RBH is the best among the three programs that we compare against.
GOST has a higher sensitivity than RBH
Overall GOST and RBH predicted 1 263 642 (circle A in Figure 3a) and 1 165 246 orthologous gene pairs (circle B), respectively, from the 4124 E. coli genes to the 959 target genomes. 1 113 013 of these gene pairs are predicted by both programs, shown as the interaction C of A and B in Figure 3a. GOST has X = 150 629 unique predictions and RBH has Y = 52 233 unique ones; and GOST predicts ∼100 000 more orthologous gene pairs than RBH.
Figure 3.

(a) Comparison of orthologous predictions between GOST (circle A) and RBH (circle B), X represents unique predictions by GOST and consists of two non-overlapping subsets X1 and X2 which involve E. coli genes not covered and covered, respectively, by RBH for the current target genome. Y, Y1 and Y2 are similarly defined. (b) An example in X1 and (c) an example in X2. Each block arrow represents a gene, and each rectangular box represents an operon. A directed edge represents a pair of homologous genes and the directed edge pointing two ways corresponds to a bidirectional best hit between two genes. The gene pair in red is identified by GOST, the one in yellow is supporting information to the red gene pair and the gene in green is the ortholog predicted by RBH.
X consists of two types of GOST predictions for each target genome: X1 and X2 involving E. coli genes not covered and covered by RBH for the current target genome, respectively. Y1 and Y2 are defined similarly for RBH (more details can be found in Supplementary Figure S4 in the Supplementary Data). Figure 3b shows an example of X1, with the (g, t) gene pair predicted by GOST while z is the best BLAST hit of t. Here GOSTs prediction is supported by the information that there is a second homologous gene pair (g′, t′), which share two operons with (g, t) in the two genomes. RBH fails to identify an orthologous gene of g as there exist no bi-directional best hits in the target genome. Overall, there are 125 831 such cases missed by RBH.
Figure 3c shows an example of X2, in which GOST predicted (g, t) as an orthologous pair and RBH predicted (g, m) as the orthologous pair for the same pair of genomes. We noted that GOSTs prediction is supported by a second homologous gene pair (g′, t′) sharing the two operons. Overall, among the 24 798 orthologous pairs in X2, 7800 (31.5%) pairs have been found to have supporting evidence similar to the above. In contrast, only 182 (0.7%) such cases in Y2 have been found to have this type of supporting evidence.
GOST can resolve complicated orthology-mapping problems involving gene fusions
Separated genes in one organism can be fused into a single gene in another organism, which is known as ‘gene fusion’ (19). Gene fusion can make a sequence similarity-based orthology mapping program fail. Figure 4a shows an example highlighting the challenge faced by programs like RBH. GI:16128036 (shown in red) is an E. coli gene and its orthologous gene is fused with another gene (its E. coli ortholog is GI:16129654) in Slackia. heliotrinireducens DSM 20476, forming gene GI:257063013. The best BLAST hit of E. coli GI:16128036 is GI:257063013 in S. heliotrinireducens while the reciprocal best hit of GI:257063013 in E. coli is GI:16129654 rather than GI:16128036, hence RBH failed to call this orthologous gene pair while GOST is able to make the correct call. Overall out of the 125 831 mapped orthologous genes in X1 (Figure 3), 13 841 (11%) encode multi-domain proteins based on searches against the Conserved Domains Database (20).
Figure 4.

An illustration of impact of gene fusion on ortholog identification. (a) and (b) are two real examples corresponding to Figure 3(b) and (c), respectively. (a) The block arrow consisting of a red and a blue arrow represents a candidate ortholog of the red gene in E. coli recognized by GOST; and the two yellow arrows represent a pair of working partners which RBH failed to call a candidate ortholog. (b) The red-blue mixed (respectively green) block arrow represents a candidate ortholog of the red gene in E. coli recognized by GOST (resp. RBH) and the two yellow arrows represent a pair of working partners. The meanings of directed edges are same to those in Figure 3.
In addition, 5896 (20.4%) mapped genes out of the 24 798 X2 genes encode multi-domain proteins, again highlighting the general issue faced by RBH. We provided one such example in Figure 4b (see corresponding gene tree in Supplementary Figure S5 in the Supplementary Data and more examples can be found in Supplementary Table S3). In this case, E. coli gene GI:145698337s ortholog is GI:220915733, formed by a fusion of E. coli gene GI:145698337 and GI:16130722, in the Anaeromyxobacter dehalogenans 2CP-1 genome. RBH incorrectly predicted GI:220915313 to be the ortholog of GI:145698337 as they are the reciprocal best hits of each other. However, GI:145698337 hits GI:220915733 and GI:220915313 with the same BLAST P-values 2e−19 (see details in Supplementary Table S4), and the orthologous pair (GI:16131796, GI:220915734) provides an additional support for the GOST call (GI:145698337, GI:220915733) to be the correct ortholog pair. Hence the real ortholog may not always have the highest sequence similarity to the query especially when several homologs have similar BLAST P-values. Overall, GOST can overcome the general issue caused by gene fusions, while sequence similarity-based orthology mapping programs such as RBH have intrinsic difficulties.
GOST is capable to identify orthologs in the presence of horizontal gene transfers
Horizontally transferred genes (HTGs) (21) could affect orthologous gene mapping results because genes acquired by horizontal gene transfers may show higher sequence similarities to homologous genes in the donor or closely related organisms than the actual orthologs in pair-wise genome comparisons. While it is difficult to derive the detailed statistics of the impact of HGTs on sequence similarity-based orthology mapping programs due to the lack of large set of HTGs, we provide the following case studies to illustrate why GOST fares better than RBH in the presence of HTGs.
Keeling et al. (22) recently classified HTGs into six categories, namely duplicative transfer, recent homologous replacement, ancient homologous replacement, duplicative transfer with differential loss, sequential transfer and transfer of new gene. These six categories largely represent two types of gene transfers: ‘duplicative transfers’ and ‘orthologous replacement transfers’. The former may cause incorrect calls of orthologs, while the latter generally will not as the original copy is lost. Supplementary Table S5 summarizes the orthologous gene mapping performance by GOST and RBH on five examples that involve HTGs, covering different scenarios. We highlight one case and refer the reader to Supplementary Figure S6 for the others.
Consider orthology mapping from E. coli K12 to Halothermothrix orenii H 168, an evolutionarily distant organism from E. coli. RBH mapped GI:16129840 of E. coli to GI:220933093 of H. orenii since they have the best reciprocal BLAST hits while GOST mapped the gene to GI:220931604. Comparing the species tree and the gene tree in Figure 5a, we note that the locations of GI:220931604 are consistent between the two trees while the locations of GI:220933093 are clearly not, implying that GI:220933093 is a recent HTG from an organism close to E. coli.
Figure 5.

(a) A species tree containing E. coli (red) and H. orenii H 168 (green); and the corresponding gene tree contains the gene GI:16129840 (in red) from E.coli and its two predicted orthologous genes GI:220931604 and GI:220933093 (in green) from H. orenii by GOST and RBH, respectively. (b) An illustration of orthologous gene mappings for both RBH and GOST, where the red (respectively green) block arrow in H. orenii represents the candidate ortholog 220933093 (respectively 220931604) of 16129840 recognized by GOST (respectively RBH) and the two yellow arrows represent a pair of working partners.
Again, the key reason that GOST is generally not affected by HTGs is that it relies on both sequence similarity and contextual information to derive orthology relationships. This again highlights that a real ortholog does not always have the highest sequence similarity to the query.
CONCLUDING REMARK
Sequence similarity-based methods remain the dominating technique for large-scale orthologous gene mapping because its computational efficiency and generally acceptable prediction accuracy but sequence similarity alone could not guarantee orthologous relationship both theoretically and practically. In this article, we presented a novel gene mapping procedure through integration of contextual and sequence similarity information. The combination of these two types of information clearly makes our program more reliable with higher coverage in complex situations as shown above. This new tool provides an orthology mapping capability at an accuracy level comparable to those of phylogeny-based approaches and yet efficient enough for large-scale applications.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online: Supplementary Methods 1–2, Supplementary Figures 1–6 and Supplementary Tables 1–5.
FUNDING
National Science Foundation (NSF/MCB-0958172, NSF/DEB–0830024 and USG Inter-Institutional Collaborative Grant, in part); US Department of Energy's BioEnergy Science Center (BESC) grant through the Office of Biological and Environmental Research (in part); NSFC (grants 61070095 and 60873207, in part, to G.L.). Funding for open access charge: US Department of Energy's BioEnergy Science Center (BESC).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
G.L. conceived the basic idea and designed the algorithm and wrote the ‘Introduction’ and ‘Materials and Methods’ sections. Q.M. developed the software, carried out the computational experiments and wrote ‘Results and Discussion’ section and cooperated with Y.Y. and X.Z. X.M. evaluated the prediction performance with enzyme-encoding genes. Y.X. proofread and revised the manuscript. All authors read and approved the final manuscript.
REFERENCES
- 1.Fitch WM. Distinguishing homologous from analogous proteins. Syst. Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 2.Chen F, Mackey AJ, Vermunt JK, Roos DS. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS One. 2007;2:e383. doi: 10.1371/journal.pone.0000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zmasek CM, Eddy SR. RIO: analyzing proteomes by automated phylogenomics using resampled inference of orthologs. BMC Bioinformatics. 2002;3:14. doi: 10.1186/1471-2105-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Storm CE, Sonnhammer EL. Automated ortholog inference from phylogenetic trees and calculation of orthology reliability. Bioinformatics. 2002;18:92–99. doi: 10.1093/bioinformatics/18.1.92. [DOI] [PubMed] [Google Scholar]
- 5.Wall DP, Deluca T. Ortholog detection using the reciprocal smallest distance algorithm. Methods Mol. Biol. 2007;396:95–110. doi: 10.1007/978-1-59745-515-2_7. [DOI] [PubMed] [Google Scholar]
- 6.Kim KM, Sung S, Caetano-Anolles G, Han JY, Kim H. An approach of orthology detection from homologous sequences under minimum evolution. Nucleic Acids Res. 2008;36:e110. doi: 10.1093/nar/gkn485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altenhoff AM, Dessimoz C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput. Biol. 2009;5:e1000262. doi: 10.1371/journal.pcbi.1000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu C, Zavaljevski N, Desai V, Reifman J. QuartetS: a fast and accurate algorithm for large-scale orthology detection. Nucleic Acids Res. 2011;39:e88. doi: 10.1093/nar/gkr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc. Natl Acad. Sci. USA. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 11.Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics. 2008;24:319–324. doi: 10.1093/bioinformatics/btm585. [DOI] [PubMed] [Google Scholar]
- 14.Mao F, Su Z, Olman V, Dam P, Liu Z, Xu Y. Mapping of orthologous genes in the context of biological pathways: an application of integer programming. Proc. Natl Acad. Sci. USA. 2006;103:129–134. doi: 10.1073/pnas.0509737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Che D, Li G, Mao F, Wu H, Xu Y. Detecting uber-operons in prokaryotic genomes. Nucleic Acids Res. 2006;34:2418–2427. doi: 10.1093/nar/gkl294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000;28:304–305. doi: 10.1093/nar/28.1.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 2009;37:D459–D463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahmood K, Konagurthu AS, Song J, Buckle AM, Webb GI, Whisstock JC. EGM: encapsulated gene-by-gene matching to identify gene orthologs and homologous segments in genomes. Bioinformatics. 2010;26:2076–2084. doi: 10.1093/bioinformatics/btq339. [DOI] [PubMed] [Google Scholar]
- 19.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 20.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2010;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 22.Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008;9:605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]