

Abstract
Matrix metalloproteinase (MMP) -2 and -9 play important roles in the progression of atherosclerosis. This study aims to determine whether MMP-2 and -9 content in the fibrotic caps of atherosclerotic plaque is correlated with plaque autofluorescence. A time-resolved laser-induced fluorescence spectroscopy (TR-LIFS) system was used to measure the autofluorescence and assess the biochemical composition of human plaques obtained from carotid endarterectomy. Results presented here demonstrate for the first time the ability to characterize the biochemical composition as it relates to MMP-2 and -9 content in the atherosclerotic plaque cap using a label-free imaging technique implemented with a fiberoptic TR-LIFS system.
Keywords: Matrix metalloproteinases, time-resolved fluorescence spectroscopy, atherosclerosis, carotid plaque
Introduction
Sudden cardiac death and acute myocardial infarction are often the first symptoms of coronary artery disease[1] and are typically caused by the rupture of vulnerable atherosclerotic plaque[2]. Currently used clinical imaging determines narrowing of the artery, but the majority of acute coronary syndromes are shown to be caused by plaques that do not obstruct blood flow[3]. Thus the current focus of diagnostic imaging in atherosclerosis is to develop methods that characterize plaque burden and functional composition suggestive of vulnerability, including inflammatory cell infiltration, apoptosis, or endothelial activation[2, 4, 5]. Autofluorescence (steady-state and time-resolved) has been found to discriminate between normal and diseased arteries [6-9], and in particular, time-resolved laser-induced fluorescence spectroscopy (TR-LIFS) is a technique recently shown to be capable of recognizing several compositional and pathological features associated with plaque vulnerability (e.g., intimal infiltration of inflammatory cells and the presence of a necrotic/lipid core under a thin fibrous cap) with high specificity and sensitivity (up to 94 and 89%, respectively)[10]. TR-LIFS fluorescence emission derived parameters also correlated with individual plaque biochemical components including elastin, collagen, inflammatory cells, and necrosis[10]. Equally important is the fact that TR-LIFS interrogation of arteries can be conducted via fiberoptic catheters and it is currently in an advanced stage of development for in vivo intravascular use in conjunction with IVUS catheters[11].
One of the essential parameters in plaque stability is the thickness of the fibrous cap[2]. Its thinning and subsequent weakening is thought to occur due to the combined proteolytic degradation of the extracellular matrix scaffold and the apoptosis of smooth muscle cells (SMC). Specifically, active matrix metalloproteinase (MMP) -2 and -9, produced by activated SMCs and inflammatory cells, were shown to degrade the fibrous cap[12]. MMP-2 and -9 have been often studied in conjunction with atherosclerosis because of their capabilities of degrading extracellular matrix components such as collagen and elastin, promoting the migration and proliferation of SMCs and macrophages and signaling apoptosis in SMCs and macrophages, the latter of which plays a large role in the development of necrosis[13, 14]. These activities can cause weakening in the fibrotic cap and thus increase the risk of plaque rupture, responsible for over 75% of acute myocardial events[15]. MMP-2 and -9 are expressed at higher levels in atherosclerotic lesions than normal arterial wall, at higher levels in vulnerable compared to stable plaques[12, 13, 16-20], and are upregulated at arterial sites of high shear stress and cyclic strain[12, 18, 21]. Taken together all these findings suggest that detection of MMPs in plaque could provide important information regarding its functional composition and likelihood of rupture.
Structural proteins (e.g., elastin, collagens), lipid and lipoproteins are the main autofluorescent constituents of atherosclerotic plaques[9]. MMP activity directly affects the biomolecular structure of these proteins and the presence of lipid components in plaques. Thus because MMPs do not fluoresce themselves, we hypothesized that TR-LIFS may be used to detect their footprint, i.e., the effect of the enzymes on the plaque composition. Consequently, the goals of this study were (a) to determine if correlations exist between TR-LIFS derived variables and MMP-2 and -9 content in the fibrotic caps of carotid plaques since such correlations could enhance the sensitivity of the diagnosis of atherosclerosis using a TR-LIFS technique; and (b) to establish the initial framework for application of a label-free fluorescence spectroscopy method to nondestructive investigation of functional changes in atherosclerotic plaques.
Methods
The data and specimens presented here are a subset from a larger study that determined TR-LIFS parameters correlated with biochemical constituents of human carotid plaque (collagen, elastin, inflammatory cells, necrosis) and were capable of distinguishing markers of plaque vulnerability (macrophage infiltration, collagen degradation, necrotic core presence)[10]. This study aims to expand on those results to test the hypothesis that TR-LIFS is capable of identifying the biochemical signature of MMPs, i.e., changes in plaque composition that can be correlated to MMP content.
Specimens
Carotid plaques from 29 patients undergoing carotid endarterectomy were included in this study. The tissue was spectroscopically evaluated (total: 77 locations) ex-vivo within 2 hours of surgery. Samples were not discriminated based on age or sex of the patient, all patients gave informed consent, and the study was approved by the institutional review board.
TR-LIFS measurements
The experiments were conducted using a point-spectroscopy TR-LIFS system described previously[22]. The location of each spectroscopic measurement was marked using India ink and the plaque specimen sent for histopathological analysis. The process of acquiring the data, deconvolving the laser from the tissue autofluorescence, and estimating fluorescence lifetime was developed by our group and was described in detail elsewhere [23-25]. The excitation source was a pulsed nitrogen laser (337 nm) and fluorescence emission was collected from 360 – 550 nm in 10 nm increments. A Laguerre expansion of kernels technique was used for the deconvolution of the fluorescence impulse response function[25]. In the present implementation, this analysis provides a set of 6 spectroscopic parameters for each of the emission wavelengths (λ) measured: average fluorescence lifetime (τλ) values, fluorescence intensity (Iλ) values, and 4 corresponding normalized Laguerre coefficient (LECλ) values that are retrieved during the deconvolution process and have been shown to be correlated with properties of the fluorescence decay curve[26].
Histopathology
TR-LIFS investigated segments were excised, fixed in 10% buffered formalin, processed routinely, and embedded in paraffin. 4 μm segments were stained with hematoxylin and eosin (H&E), elastic/trichrome (trichrome), CD68, CD45, smooth muscle cell (SMC) actin, picrosirius red (PR), and MMP -2 and -9 antibodies.
The composition of the artery wall was examined in a region of interest (ROI) directly beneath the ink mark. The size of the ROI was determined based on the fiber-optic excitation-collection geometry (~1.1 mm illuminated diameter area at the tissue surface) and the light penetration depth (~200-250 μm for 337 nm in arterial tissue)[23]. Relative percentages of collagen, elastin, SMCs, calcification, macrophages, and necrosis were determined using the specific methods described here. MMP-2 and -9 expression levels were determined based on the number of positively stained macrophages and SMCs within each ROI: level 0 (zero positive cells), level 1 (<20 positive cells), and level 2 (>20 positive cells).
Each investigated location was categorized into 5 groups based roughly on the current American Heart Association definitions[2, 27] to define the severity of plaque development: non-atherosclerotic intimal thickening (IT) with intimal thickness < 200 μm; fibrous plaque (FP) with intimal thickness > 200 μm, but no macrophages or necrosis; low-inflamed fibrous cap atheroma (FA) with intimal thickness > 200 μm and some necrosis/macrophages (<20%); inflamed fibrous cap atheroma (FA-inf) with >20% macrophages and <30% necrosis; thin cap fibrous atheroma (TCFA) with intimal thickness >200 μm, cap thickness < 200 μm, and >30% necrosis. These phenotypes can be listed in order of increasing vulnerability to rupture as: IT, FP, FA, FA-inf, TCFA. These categories allowed for broad generalizations to be examined regarding how MMP levels correlate with clinically relevant plaque phenotypes: IT (non-atherosclerotic, low risk of rupture), FP (collagen-rich, stable plaque, low risk of rupture), FA (increasing levels of inflammation, increased risk of rupture), FA-inf (high levels of inflammation, increased risk of rupture), and TCFA (high levels of necrosis, increased risk of rupture).
Statistics and Data Analysis
Three analyses were completed. 1) Average MMP-2 and -9 values were evaluated for each plaque phenotype defined above (IT, FP, FA, FA-inf, and TFCA) to determine if assessing MMP content would have clinical relevance and to compare with published information about MMP-2 and -9 in atherosclerotic plaques. Values are reported as mean ± standard error (SE). An unpaired t-test was computed to determine that the changes in MMP level between plaque phenotype were statistically significant. 2) Linear correlations were made between MMP content and the biological components that define the 5 plaque phenotypes (% collagen, elastin, SMC, necrosis, and macrophages) to determine whether changes in MMP levels between plaque phenotypes were correlated to the biology of the specimens. MMP-2 and -9 changes from level 0-to-1, level 1-to-2, and level 0-to-2 were analyzed separately because the biochemical changes associated with each level were not linearly related to the changes in MMP content. 3) MMP-2 and -9 levels were correlated with each corresponding set of spectroscopic parameters at four wavelength bands chosen based on the results from the previous study using this data that found discrimination between TR-LIFS measurements of plaques: 380-390 nm (385λ), 450-470 nm (460λ), 500-520 nm (510λ) and the intensity ratio 380-390 nm / 430-450 nm (IR). For this correlation analysis, MMP levels were averaged at 10% intervals of the spectroscopic parameters to account for the 0.2 ns time resolution of the TR-LIFS system. All analysis was performed with MATLAB (The MathWorks, Inc.). Pearson correlation coefficients (R) and 2-tailed p-values are reported for each set of correlations.
Results
All correlations reported are statistically significant (P<0.05) unless noted otherwise.
MMP Expression Levels Increase with Likelihood of Plaque Rupture
Each investigated region of plaque was histopathologically classified by MMP-9 level (0, n=33; 1, n=22; 2, n=22) and MMP-2 level (0, n=27; 1, n=27; 2, n=23). Representative micrographs of several MMP stained specimens are shown in figure 1; the left panel is a typical non-atherosclerotic artery with MMP-2 staining level 2 and MMP-9 staining level 0, demonstrating that normal artery stains for MMP-2, but not MMP-9; the right panel is a typical necrotic core atheroma with MMP-2 and -9 staining level 2, demonstrating that MMP-2 and -9 are both elevated in plaques with high risk of rupture. IT expressed MMP-9 level 0 (0.0 ± 0) and MMP-2 level 1 (0.6 ± 0.2), FP and FA expressed MMP-9 and -2 level 1 (FP: 0.5 ± 0.1 and 0.6 ± 0.1, FA: 0.8 ± 0.2 and 0.8 ± 0.2, respectively), FA-inf and TCFA expressed MMP-2 and -9 level 2 (FA-inf: 1.6 ± 0.2 and 1.6 ± 0.2, TCFA: 1.6 ± 0.3 and 1.6 ± 0.3, respectively), (Figure 2).
Figure 1.
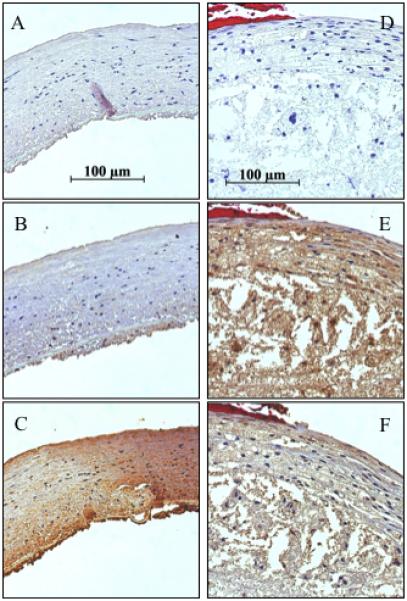
Representative photomicrographs of carotid plaque specimens; 40x objective; MMP antibody staining. Left panel: Normal artery. (A) Control, (B) MMP-9, and (C) MMP-2. Right panel: Necrotic plaque. (D) Control, (E) MMP-9, (F) MMP-2.
Figure 2.
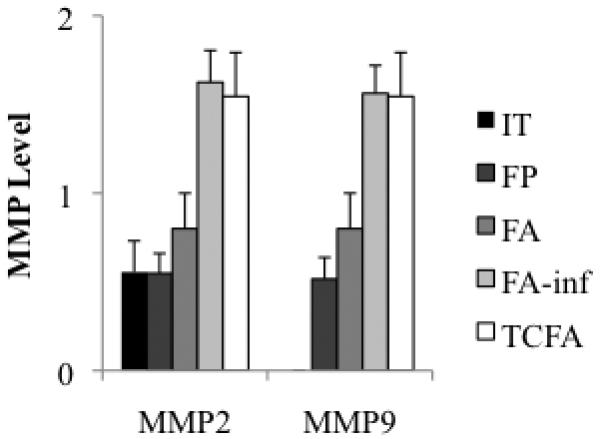
Average MMP-2 and -9 levels for each plaque phenotype.
Changes in MMP Expression Levels Correlate with Changes in Levels of Specific Plaque Components
Between MMP-9 level 0-to-1, there was an increase in collagen and a decrease in elastin, however the decrease in elastin was the only component to have a significant Pearson correlation with the changing MMP-9 values between these levels (supplemental table 1); a further increase from level 1-to-2 correlated with a decrease in collagen and SMCs and an increase in macrophages and necrosis. The increase in MMP-9 level from 0-to-2 was correlated with a decrease in elastin and SMCs and a dramatic increase in macrophages and necrosis (Figure 3A, supplement: table 1). The biological composition of the specimens did not change as MMP-2 increased from level 0-to-1. The increase in MMP-2 from level 1-to-2 correlated with a decrease in collagen, elastin, and SMCs and an increase in macrophages and necrosis. The increase in MMP-2 from level 0-to-2 correlated with a decrease in elastin and SMCs and an increase in macrophages and necrosis (Figure 3B, supplement: table 1).
Figure 3.
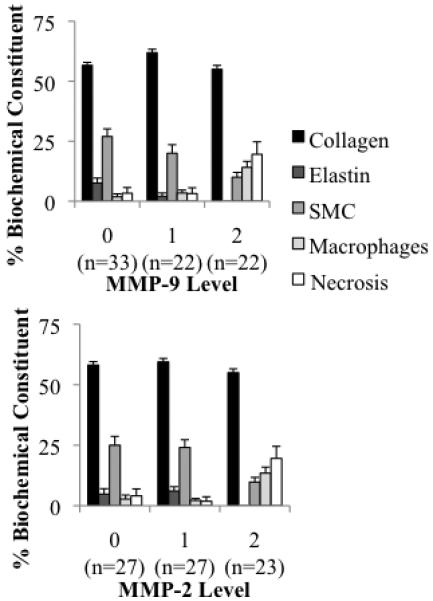
Average plaque biochemical constituent (determined by histopathological analysis) for each MMP level.
MMP Level Changes Correlate with TR-LIFS Parameters
MMP-9
The MMP-9 level change from 0-to-1 correlated with an increase in τ385λ, IR, and LEC-0385λ and a decrease in LEC-1385λ and LEC-1510λ. These parameters also correlated with a decrease in elastin and an increase in collagen. The MMP-9 level change from 1-to-2 was correlated with an increase in LEC-1385λ, LEC-1460λ, LEC-2460λ, LEC-2510λ, and LEC-3460λ, and a decrease in τ460λ, τ510λ, and LEC-0460λ. These parameters also correlated with a decrease in collagen and an increase in macrophages and necrosis. The MMP-9 level change from 0-to-2 was correlated with an increase in IR, LEC-1460λ, LEC-2460λ, LEC-2510λ, and LEC-3460λ, and a decrease in τ460λ, τ510λ, and LEC-0460λ. These parameters also correlated with a decrease in collagen and elastin and an increase in macrophages and necrosis (Figure 4: parameters with strongest correlations, supplement table 2, figures 1 and 2: entire dataset).
Figure 4.
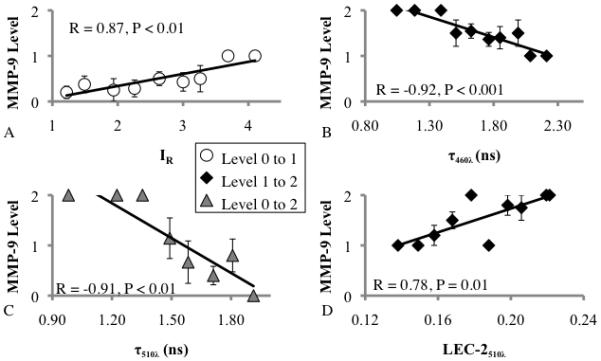
Linear correlation between MMP-9 level vs. (A) IR, (B) τ460λ, (C) τ510λ, and (D) LEC-2510λ. TR-LIFS variables (x-axis) were grouped in 10% steps with MMP-9 levels averaged at each step. Steps without data are not represented.
MMP-2
The changes in TR-LIFS variables between MMP-2 levels 0-to-1 were not significant. The MMP-2 level change from 1-to-2 was correlated with an increase in LEC-0460λ, LEC-1460λ, LEC-2460λ, LEC-2510λ, and LEC-3460λ and a decrease in τ460λ and LEC-0510λ. These parameters were also correlated with a decrease in collagen and elastin and an increase in macrophages and necrosis. The MMP-2 level change from 0-to-2 was correlated with an increase in LEC-1460λ, LEC-2460λ, LEC-2510λ, and LEC-3460λ and a decrease in τ460λ, LEC-0510λ, and LEC-0510λ. These parameters were also correlated with a decrease in collagen and elastin and an increase in macrophages and necrosis (Figure 5: parameters with strongest correlations, supplement table 2, supplement figures 1 and 2: entire dataset).
Figure 5.
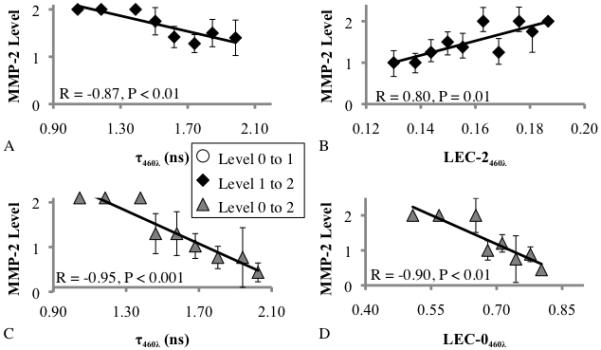
Linear correlation between MMP-2 level vs. (A) τ460λ, level 1-to-2, (B) LEC-2460λ, C) τ460λ, level 0-to-2, and (D) LEC-0460λ. TR-LIFS variables (x-axis) were grouped by 10% steps with MMP-2 levels averaged at each step. Steps without data are not represented.
Discussion
Several techniques currently researched to image MMPs use molecular imaging methods, tagging the MMPs with contrast-enhancing probes identifiable, for example, by NIR fluorescence imaging, PET, or MRI[28, 29]. Targeted-contrast agents allow for specificity in the detection of particular MMPs or other plaque components, on the other hand, we found that TR-LIFS is able to detect the specific footprint associated with MMP-2 and -9, via correlations with plaque composition, specifically of its fibrous cap. Also, this method has the advantage that it does not require the administration of any exogenous probes, however it does require detection from close proximity to the plaque, as obtainable by intravascular catheter. This study is the first to develop methodologies that allow for the study of MMP content in atherosclerotic plaques based on endogenous plaque fluorescence contrast. Our findings demonstrate the diagnostic potential of TR-LIFS for identifying functional features of plaque vulnerability.
MMP Content vs. Plaque Phenotype
The finding that all IT specimens were categorized as MMP-9 level 0 and MMP-2 level 0 or 1 is in agreement with earlier reports that show MMP-2 is present in non-atherosclerotic and atherosclerotic arteries compared to MMP-9 that is only present in atherosclerotic arteries[12-14, 30]. Thus an IT specimen could be identified by an absence of MMP-9. The difference between IT (non-atherosclerotic) and FP (stable plaques) was not apparent based on MMP-2 but was characterized by an increase in MMP-9. The increase in both MMP-2 and -9 in FA specimens, particularly in FA-inf and TCFA specimens, was expected based on previous studies that show MMP-9 and -2 to be correlated with the presence of lipids and macrophages[12, 16-18]. Conflicting results are published regarding MMP-2 content in plaques. Some studies found MMP-2 to be correlated with a stable plaque phenotype[19, 31], another found MMP-2 to be highest in fatty streaks[30] and yet another found MMP-2 to be higher in atherosclerotic plaque than in fatty streaks and lowest, but still present, in normal artery[32]. Our finding that MMP-2 levels were highest in FA-inf and TCFA plaques (figure 2) is in agreement with the general idea that MMP levels increase with vulnerability[12, 18]. A detailed discussion regarding the specific relationship between the MMPs and the plaque biochemical constituents can be found in the supplementary document.
MMP Levels vs. TR-LIFS Parameters
Our approach to the analysis of TR-LIFS data allows for the derivation of a set of multiple parameters that describe tissue fluorescence response in great detail. Parameters derived are direct descriptors of the underlying physics of the fluorescence measurements (e.g. average fluorescence lifetime and fluorescence intensity) as well as parameters that provide a more complete mathematical representation of the fluorescence excited state decay dynamics (e.g. Laguerre coefficients) that do not entail a direct physical representation of the fluorescence system. The Laguerre coefficients in particular were found to provide an added benefit in the delineation of distinct plaque composition and correlations with MMP levels in plaques. Time-resolved fluorescence measurements are independent of fluorescence intensity and thus the attenuation of fluorescence intensity caused by the presence of blood in an intravascular setting would not affect the ability to retrieve fluorescence decay characteristics. In a previous study we demonstrated that TR-LIFS implemented in a catheter-based system can retrieve robust fluorescence lifetime data in blood-flow mimicking conditions. The presence of blood affected the fluorescence intensity but not the fluorescence lifetime[11].
MMP-9
The increase in average fluorescence lifetime between MMP-9 levels 0-to-1 was attributed to an increase in collagen and decrease in elastin as confirmed by histopathology and the fluorescence properties of these two molecules that are a narrow (40 nm) fluorescence emission with a peak at 385 nm and lifetime of ~3-4 ns for collagen and a wide emission band (70 nm) with a peak at 400 nm and average lifetime of ~2 ns for elastin[10]. These properties also explain the increase in IR: a decrease in elastin would cause a decrease in intensity throughout the band of elastin emission (365-435 nm) and an increase in collagen would cause an increase in intensity throughout the band of emission of collagen (365-405 nm) and thus an increase in the intensity ratio between 380-390 nm and 430-450 nm. The reason for assessing a ratio is because fluorescence often varies based on properties of the sample such as surface texture or the presence of blood (which exhibits significant absorption at 410 nm) and taking the ratio eliminates the possibility that any structural properties of the specimen effected the correlations. From MMP-9 level 1-to-2, the decrease in lifetime was attributed to the decrease of collagen and increase of macrophages and necrosis. Macrophages and necrosis are rich in lipids and lipoproteins, which have shorter average fluorescence lifetimes than collagen or elastin, particularly at longer wavelengths[10]. The fact that lipids tend to fluoresce at longer emission wavelengths also explains the fact that the significant decreases in lifetime are at the 460λ and 510λ bands as opposed to the 385λ band that was more affected by changes in collagen and elastin[10]. From MMP-9 level 0-to-2, similar changes in spectroscopy exist that indicate loss of collagen and increase of lipids (decrease in lifetime) but in this case the significant change in IR is also indicative of the decrease in elastin between these levels.
Thus MMP-9 level 0 and a likely IT specimen can be identified by low IR and τ385λ values; MMP-9 level 1 and a likely FP or FA specimen has high IR, τ385λ, and τ460λ values; MMP-9 level 2 and an increased risk of plaque rupture can be identified by high IR, and low τ460λ, and τ510λ values.
MMP-2
There are no significant biological changes between MMP-2 levels 0-to-1, but there is a significant decrease in τ460λ values that possibly indicates a decrease in collagen or an increase in lipids or elastin. But, because there is no significant change in the pathology of the samples at this level, no definitive reason can be given for this decrease in lifetime. Between levels 1-to-2, lifetime is again found to decrease, correlating with a decrease in collagen and elastin and an increase in macrophages and necrosis.
Thus MMP-2 level 2 and the increased risk of vulnerability associated with this level can be identified by short lifetime values at τ460λ, which were also shown to identify increased MMP-9 levels.
It is important to note several limitations to this work: 1) In this study we assessed MMP expression, but we did not investigate the level of activated MMPs, which requires different tissue processing (not compatible with the other histological analyses), thus would have to be done in a separate study. However, the biochemical composition observed in our study to correlate with increased MMP expression levels was in agreement with studies that did examine activity level, validating our finding that TR-LIFS can investigate MMP expression based on the biochemical composition of the tissue. 2) This study focused on establishing the methodology and demonstrating correlations with MMP-2 and -9 because of their acknowledged roles in the inflammatory responses associated with increased risk of plaque rupture, correlations between TR-LIFS derived parameters and other MMPs, e.g., MMP-1, -8, and -13,will be investigated in future work. 3) This study was completed ex-vivo, however, based on our experience, the composition (e.g. structural proteins including collagen, and lipids) and morphology of atherosclerotic plaque does not change significantly, except for small geometrical changes due to tissue shrinkage post-excision. Since information related to MMP content in plaque is measured indirectly via measurements of changes in collagen fluorescence properties the fluorescence features retrieved from ex-vivo measurements are expected to follow similar trends with those obtained ex-vivo [10].
In conclusion, this study found distinct levels of MMP-2 and -9 discernable in the fibrotic caps of human carotid plaques using TR-LIFS derived spectroscopic variables in wavelength bands correlated to changes in collagen, elastin, and lipid content in plaques (385λ, 460λ, and 510λ). To our knowledge, this work represents the first study to successfully examine MMP content in human plaques without labeling or destroying the measured tissue, both important when designing a clinically relevant method of in vivo MMP assessment. Moreover, efforts are underway in our laboratory to develop an intravascular catheter to translate this technology to the clinic for use during heart catheterizations. While the current study did not interrogate the relationships between active MMPs and TR-LIFS parameters, it established the framework that will allow for the study of such relationships in subsequent studies.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge discussions with Dr. Seemantini Nadkarni regarding the quantification of collagen from picrosirius red stained slides. We would also like to thank Ms. Caitlin McDonnell for her help with developing the PSR collagen quantification code; the vascular surgeons, residents (Drs. Todd Reil and Amir Dorafshar), and nurses at UCLA for their support in carotid sample collection; Drs. Javier Jo, Thanassis Papaioannou, and Qiyin Fang for their work on the collection and pre-processing of the initial experimental data; and Dr. Jian-Hua Qiao for his early contributions to the histopathology analysis. This study was supported by the National Institute of Health grant R01 HL 67377 (to L.M.) and a Howard Hughes Medical Institution Med-into-Grad Fellowship, University of California, Davis (to J.E.P.).
Biography

Jennifer Phipps obtained a PhD in Biomedical Engineering from the University of California, Davis. Her main research focus is on signal and image processing of novel optical techniques for imaging atherosclerotic cardiovascular disease.

Nisa Hatami obtained her B.S. in Biomedical Engineering at UC Davis, and her M.S. in Biomedical Sciences at Tufts University. She expects to obtain an M.D. and continue researching in the field of cardiovascular disease as a physician.

Dr. Galis is currently the Chief of the Vascular Biology and Hypertension branch within the National Heart Lung and Blood Institute, at the National Institutes of Health. Previously, Dr. Galis held faculty positions in Cardiology, Biomedical Engineering, and Surgery and served as the Chief Scientific Officer, Cardiovascular R&D at Eli Lilly and Co. She obtained graduate or post-graduate degrees in physics, biophysics, cell biology, pathology and vascular medicine from the University of Bucharest, Romania, the McGill University School of Medicine, Canada, and the Harvard School of Medicine, USA. Her scientific/technical expertise encompasses: vascular biology and pathology, cardiovascular medicine, atherosclerosis, biomarkers, imaging, genomics, bioengineering, drug discovery and development.

Dr. J. Dennis Baker is a Professor in the Division of Vascular Surgery at the David Geffen School of Medicine at the University of California, Los Angeles. He is also the Chief of the Vascular Surgery Section at the West Los Angeles VA Medical Center. In 2010 he received a Lifetime Achievement Award from the Society for Clinical Vascular Surgery.

Dr. Michael C. Fishbein is a Piansky Professor of Pathology and Laboratory Medicine at the University of California, Los Angeles. He is the Chief of Autopsy, Cardiovascular and Pulmonary Pathology and a Member of the Jonsson Comprehensive Cancer Center Thoracic Oncology Program Area, Cardiology. He specializes in autopsy, cardiovascular and pulmonary pathology, as well as surgical pathology.

Laura Marcu is a professor of Biomedical Engineering at the University of California, Davis. Center of her research program is the development of clinically compatible laser spectroscopy and imaging systems that facilitate in-vivo investigations of the relationship between tissue pathology and measured optical responses. Projects include development of fluorescence lifetime spectroscopy and imaging techniques for the recognition of major classes of fluorescent biomolecules in biological tissue, diagnosis of atherosclerotic cardiovascular disease, and intraoperative delineation of brain tumors and head and neck tumors.
Footnotes
JE Phipps, et al.: A fluorescence lifetime study of MMPs in carotid plaque
REFERENCES
- 1.Narula J, Garg P, Achenbach S, Motoyama S, Virmani R, Strauss HW. Nat Clin Pract Cardiovasc Med. 2008;5(Suppl 2):S2–10. doi: 10.1038/ncpcardio1247. [DOI] [PubMed] [Google Scholar]
- 2.Virmani R, Burke AP, Farb A, Kolodgie FD. J Am Coll Cardiol. 2006;47:C13–18. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 3.Sharif F, Murphy RT. Catheter Cardiovasc Interv. 2010;75:135–144. doi: 10.1002/ccd.22164. [DOI] [PubMed] [Google Scholar]
- 4.Libby P, DiCarli M, Weissleder R. J Nucl Med. 2010;51(Suppl 1):33S–37S. doi: 10.2967/jnumed.109.069633. [DOI] [PubMed] [Google Scholar]
- 5.Lindsay AC, Choudhury RP. Nat Rev Drug Discov. 2008;7:517–529. doi: 10.1038/nrd2588. [DOI] [PubMed] [Google Scholar]
- 6.Fitzmaurice M, Bordagaray JO, Engelmann GL, Richards-Kortum R, Kolubayev T, Feld MS, Ratliff NB, Kramer JR. Am Heart J. 1989;118:1028–1038. doi: 10.1016/0002-8703(89)90239-1. [DOI] [PubMed] [Google Scholar]
- 7.Hegyi L, Talbot C, Monaco C, Sandison A, Davies AH, Peston D, Requejo-Isidro J, Elson DS, Dunsby C, Munro I, Neil MA, French PM, Stamp GW, Lever M. J. Heart. 2007;93 [Google Scholar]
- 8.Thomas P, Pande P, Clubb F, Adame J, Jo JA. Photochem Photobiol. 2010;86:727–731. doi: 10.1111/j.1751-1097.2010.00707.x. [DOI] [PubMed] [Google Scholar]
- 9.Arakawa K, Isoda K, Ito T, Nakajima K, Shibuya T, Ohsuzu F. Arteriosclerosis Thrombosis and Vascular Biology. 2002;22:1002–1007. doi: 10.1161/01.atv.0000017461.79231.3d. [DOI] [PubMed] [Google Scholar]
- 10.Marcu L, Jo JA, Fang Q, Papaioannou T, Reil T, Qiao JH, Baker JD, Freischlag JA, Fishbein MC. Atherosclerosis. 2009;204:156–164. doi: 10.1016/j.atherosclerosis.2008.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephens DN, Park J, Sun Y, Papaioannou T, Marcu L. J Biomed Opt. 2009;14:030505. doi: 10.1117/1.3146813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galis ZS, Sukhova GK, Lark MW, Libby P. Journal of Clinical Investigation. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newby AC. Trends Cardiovasc Med. 2007;17:253–258. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galis ZS, Khatri JJ. Circulation Research. 2002;90:251–262. [PubMed] [Google Scholar]
- 15.Davies M. J. Heart. 2000;83:361–366. doi: 10.1136/heart.83.3.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newby AC. Arterioscler Thromb Vasc Biol. 2008;28:2108–2114. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- 17.Shah PK, Galis ZS. Circulation. 2001;104:1878–1880. [PubMed] [Google Scholar]
- 18.Krams R, Cheng C, Helderman F, Verheye S, van Damme LC, Mousavi Gourabi B, Tempel D, Segers D, de Feyter P, Pasterkamp G, De Klein D, de Crom R, van der Steen AF, Serruys PW. EuroIntervention. 2006;2:250–256. [PubMed] [Google Scholar]
- 19.Sluijter JP, Pulskens WP, Schoneveld AH, Velema E, Strijder CF, Moll F, de Vries JP, Verheijen J, Hanemaaijer R, de Kleijn DP, Pasterkamp G. Stroke. 2006;37:235–239. doi: 10.1161/01.STR.0000196986.50059.e0. [DOI] [PubMed] [Google Scholar]
- 20.Muzzio ML, Miksztowicz V, Brites F, Aguilar D, Repetto EM, Wikinski R, Tavella M, Schreier L, Berg GA. Arch Med Res. 2009;40:48–53. doi: 10.1016/j.arcmed.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Cummins PM, von Offenberg Sweeney N, Killeen MT, Birney YA, Redmond EM, Cahill PA. Am J Physiol Heart Circ Physiol. 2007;292:H28–42. doi: 10.1152/ajpheart.00304.2006. [DOI] [PubMed] [Google Scholar]
- 22.Fang QY, Papaioannou T, Jo JA, Vaitha R, Shastry K, Marcu L. Review of Scientific Instruments. 2004;75:151–162. doi: 10.1063/1.1634354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcu L, Fishbein MC, Maarek JMI, Grundfest WS. Arteriosclerosis Thrombosis and Vascular Biology. 2001;21:1244–1250. doi: 10.1161/hq0701.092091. [DOI] [PubMed] [Google Scholar]
- 24.Marmarelis VZ. Ann Biomed Eng. 1993;21:573–589. doi: 10.1007/BF02368639. [DOI] [PubMed] [Google Scholar]
- 25.Jo JA, Fang QY, Papaioannou T, Marcu L. Journal of Biomedical Optics. 2004;9:743–752. doi: 10.1117/1.1752919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo JA, Fang Q, Papaioannou T, Baker JD, Dorafshar AH, Reil T, Qiao JH, Fishbein MC, Freischlag JA, Marcu L. Journal of Biomedical Optics. 2006;11:021004. doi: 10.1117/1.2186045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Arteriosclerosis Thrombosis and Vascular Biology. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 28.Schafers M, Schober O, Hermann S. J Nucl Med. 2010;51:663–666. doi: 10.2967/jnumed.109.065698. [DOI] [PubMed] [Google Scholar]
- 29.Kim DE, Kim JY, Schellingerhout D, Kim EJ, Kim HK, Lee S, Kim K, Kwon IC, Shon SM, Jeong SW, Im SH, Lee DK, Lee MM, Kim GE. Arterioscler Thromb Vasc Biol. 2010;30:449–456. doi: 10.1161/ATVBAHA.109.194613. [DOI] [PubMed] [Google Scholar]
- 30.Choudhary S, Higgins CL, Chen IY, Reardon M, Lawrie G, Vick GW, 3rd, Karmonik C, Via DP, Morrisett JD. Arterioscler Thromb Vasc Biol. 2006;26:2351–2358. doi: 10.1161/01.ATV.0000239461.87113.0b. [DOI] [PubMed] [Google Scholar]
- 31.Kuzuya M, Nakamura K, Sasaki T, Cheng XW, Itohara S, Iguchi A. Arteriosclerosis Thrombosis and Vascular Biology. 2006;26:1120–1125. doi: 10.1161/01.ATV.0000218496.60097.e0. [DOI] [PubMed] [Google Scholar]
- 32.Li ZH, Li L, Zielke R, Cheng L, Xiao RP, Crow MT, Stetler-Stevenson G, Froehlich J, Lakatta EG. American Journal of Pathology. 1996;148:121–128. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.