

Abstract
Chapter summary
Interactions with endothelium are necessary for leukocytes to pass from the blood into extravascular tissues, and such interactions are facilitated in inflammation by the coordinated expression of endothelial adhesion molecules and chemoattractants. Although the general mechanisms and intracellular pathways of endothelial activation are now fairly well characterised in vitro, relatively little detailed information exists on how endothelial activation changes during the course of inflammatory responses and how such change influences the amount of leukocyte recruitment and the types of leukocytes recruited. Having developed a radiolabelled-antibody-uptake technique for quantifying the expression of endothelial adhesion molecules in relation to leukocyte trafficking, we have analysed the acute, self-limiting inflammatory response to injection of monosodium urate (MSU) crystals. Our studies have supported the view that endothelial activation is closely paralleled by leukocyte recruitment at the onset of the response and have highlighted separate vascular and extravascular stages of downregulation. More recent studies addressing the extravascular contribution to downregulation point to an important role for monocyte–macrophage differentiation in limiting further endothelial activation as a consequence of phagocytosis of MSU crystals.
Keywords: endothelium, gout, leukocyte trafficking, macrophage, monocyte
Introduction
This article discusses how the experimental study of gout provides a relatively simple, inflammatory-disease model with which to explore the relation between endothelial-cell activation, leukocyte trafficking, and perivascular activation of leukocytes. We have highlighted important recent advances in the in vivo study of endothelial-cell activation that have enabled the link between endothelial-cell activation and leukocyte trafficking to be investigated in detail in an animal model of acute gout. We also discuss recent evidence suggesting a protective role for macrophages in the resolution phase of inflammation.
Historical background
Gout appears to be a simple disease from an aetiological viewpoint, caused by the intra-articular deposition of monosodium urate monohydrate (MSU) crystals in individuals with elevated serum concentrations of uric acid. After the description of the clinical features of gout by Hippocrates in the fourth century BC, landmarks in understanding of the aetiology of gout were the detection of crystals in synovial fluid by Anton van Leeuwenhoek in the seventeenth century, the identification of the main ingredient of gout-associated stones and tophi as uric acid by Scheele and Wollaston, respectively, in the eighteenth century, and the association of gout with hyperuricaemia by Garrod in the nineteenth century. However, in the twentieth century the Framlingham study showed that hyperuricaemia does not necessarily lead to clinical gout [1]. Thus, the cumulative incidence of acute gouty arthritis over a 12-year period in hyperuricaemic men (serum uric acid concentrations greater than 8 mg/dl [0.48 mmol/l]) was only 36%. This relatively weak link between hyperuricaemia and gout may be explained at least in part by differences between individuals in the capacity to nucleate and grow MSU crystals. However, we know that other factors besides the presence of crystals are involved in triggering an acute attack of gout, since the presence of crystals can be readily detected in the synovial-fluid samples collected from asymptomatic joints [2]. A possible clue to the role of leukocytes in determining the inflammatory balance in hyperuricemia has come with the observation that the cellular infiltrate in acute gout is predominantly neutrophilic, whereas in asymptomatic gout the leukocytic infiltrate is almost exclusively mononuclear [3,4].
Regulation of expression of endothelial-cell adhesion molecules
Adhesion of leukocytes to vascular endothelium is a prerequisite for their emigration into the tissues in inflammation. The general mechanisms that allow leukocytes to adhere to vascular endothelial cells and migrate into tissues are now quite well understood: they involve sequential capture from free flow via selectins, activation of G-protein-coupled receptors, and subsequent integrin-mediated arrest on the endothelial-cell surface [5-7]. Critical to these interactions is the activation of endothelial cells, which leads to upregulated expression of selectins, chemokines, and integrin ligands on the endothelial-cell surface [8,9].
Activation of endothelial cells occurs in response to changes in the tissue microenvironment and may be classified according to requirement for de novo protein synthesis. Stimulation of endothelial cells with agonists such as histamine, C5a, or thrombin leads to the rapid translocation of Weibel–Palade bodies to the luminal surface, with incorporation of presynthesised P-selectin into the plasma membrane and release of IL-8 and von Willebrand factor [10-12]. This process, which is analogous to mast-cell degranulation, provides a rapid but transient mechanism for initiating leukocyte–endothelial-cell interactions within seconds of tissue perturbation. In contrast, a more delayed and sustained response occurs through the stimulation of a programme of gene transcription and de novo protein synthesis of adhesion molecules and chemokines that include E-selectin, intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), IL-8, and MCP-1. The agonists that have been best characterised as inducing this transcriptional response are the cytokines IL-1α/β and tumour necrosis factor (TNF)-α [8] Recently, evidence has been provided that IL-1 may increase the IL-8 content of Weibel–Palade bodies, providing a means by which endothelial cells can mount an augmented immediate response upon subsequent rechallenge [11,12].
Studying endothelial activation in vivo
Injection of IL-1 or TNF-α into the skin in vivo stimulates a subacute inflammatory response associated with marked leukocyte emigration, which can be quantified by measuring the uptake of intravenously injected radiolabelled leukocytes [13,14]. In order to relate the uptake of leukocytes to endothelial activation, we developed a technique in the pig for quantifying expression of adhesion molecules by measuring the uptake of radiolabelled antibodies, using a differentially radiolabelled nonspecific antibody as an internal control [15]. This allowed us to demonstrate a close relation between the onset of neutrophil recruitment and the expression of E-selectin, both in response to injection of cytokines such as IL-1 or TNF-α and during delayed-hypersensitivity responses [16-18]. A similar approach has since been adopted for measuring endothelial activation and adhesion-molecule expression in models of inflammation in rats [19] and mice [20-24].
Gout as a model for studying endothelial activation and leukocyte trafficking
Developments in understanding of the general mechanisms of leukocyte trafficking now allow us to start dissecting in detail the relation between endothelial activation and leukocyte trafficking in inflammatory rheumatic diseases. Because the aetiology is known, acute gout presents a relatively accessible, self-limiting inflammatory condition upon which to model mechanisms that may underlie other relapsing–remitting diseases. We have particularly addressed the questions of how leukocytes are recruited into the tissues during the amplification phase of the acute attack, and then how the attack spontaneously resolves.
Leukocyte trafficking and endothelial activation during experimental inflammation induced by monosodium urate crystals
Injection of MSU crystals into human skin leads to an erythematous reaction that is maximal at 24 hours and then spontaneously subsides [25]. The response is very similar in pig skin, providing a good model for studying how endothelial activation and leukocyte recruitment relate to the time course of MSU-crystal-induced inflammation. We analysed endothelial activation and leukocyte trafficking by measuring the uptake of differentially radiolabelled anti-E-selectin, neutrophils, and/or mononuclear cells at various times after intracutaneous injection of MSU crystals [26].
Leukocyte recruitment commenced between 1 and 2 hours after injection of MSU crystals, in close parallel with the onset of E-selectin expression (Fig. 1). Unexpectedly, the phase of E-selectin expression and leukocyte recruitment was quite brief, and both had returned to baseline by the peak of erythema at 24 hours. After 24 hours, erythema resolved in spite of the continued presence of MSU crystals in the skin. When the relations between endothelial activation and leukocyte trafficking and the kinetics of the inflammatory response are considered, three conclusions can be drawn. Firstly, initial endothelial activation and leukocyte recruitment were closely connected. Secondly, the transient nature of endothelial E-selectin expression and leukocyte recruitment appeared to limit the entry of leukocytes into the tissues to a relatively early stage of the response. Finally, assuming erythema is a reflection of postmigratory leukocyte activation, mechanisms must exist that reduce the responsiveness of leukocytes to MSU crystals and thereby terminate further endothelial stimulation.
Figure 1.

Kinetics of endothelial E-selectin expression, entry of neutrophils and mononuclear cells into the tissues, and erythema in pig skin after injection of monosodium urate (MSU) crystals. Note that E-selectin expression and leukocyte trafficking have returned to near baseline before maximal erythema is seen at 24 hours, indicating a vascular downregulation of the inflammatory response. Erythema subsides spontaneously after 24 hours, in spite of the continued presence of MSU crystals in the tissues, suggesting extravascular downregulating mechanisms (modified from [26], with kind permission from the British Journal of Rheumatology).
The role of the monocyte in endothelial activation during the onset of acute gout
MSU crystals are able to activate a number of acute inflammatory pathways, which may induce and/or amplify an acute attack of gout. These include the alternative pathway of complement and the kallikrein system, and stimulation of mast-cell degranulation with rapid release of vasoactive mediators and TNF-α [27]. We have focused particularly on the monocyte, because of its potential for the sustained release of endothelial activating factors. Monocytes are known to respond to the phagocytosis of MSU crystals by activating expression of a number of proinflammatory genes, including those encoding interleukin (IL)-1 [28], IL-6 [29], IL-8 [30], TNF-α [31], and Cox-2 [32]. In the case of IL-8, gene transcription follows signalling via tyrosine phosphorylation of extracellularly regulated kinase (ERK)1/ERK2, p38 MAPK (mitogen-activated protein kinase), and JNK (c-Jun N-terminal kinase) [30]. Promoter analysis has demonstrated that transcriptional activation of the IL-8 gene by MSU crystals involves binding of activator protein-1 and the NF-κB complex c-Rel/RelA to the IL-8 promoter [33].
In order to determine which monocyte-derived factors are responsible for activation of expression of endothelial-cell adhesion molecule, we established an in vitro model in which MSU-crystal-stimulated monocyte supernatants were transferred to endothelial-cell cultures in the presence of neutralising antibodies to candidate cytokines [34]. These experiments showed that the capacity of MSU-stimulated monocytes to induce expression of the adhesion molecules E-selectin, ICAM-1, and VCAM-1 was entirely attributable to release of TNF-α and IL-1β. Furthermore, when the time course of production of these two cytokines was studied in more detail, IL-1β secretion was found to precede that of TNF-α.
Cytokine-mediated activation of endothelium in vivo can be demonstrated by imaging the uptake of intravenously injected anti-E-selectin monoclonal antibody, as shown in rheumatoid arthritis (RA) [35] and inflammatory bowel disease [36]. Using a pig model of MSU-crystal-induced arthritis [37], we found that E-selectin expression and neutrophil recruitment were inhibited approximately 50% in the presence of neutralizing antibodies to TNF-α, a finding consistent with the data found in vitro[34] (Fig. 2).
Figure 2.
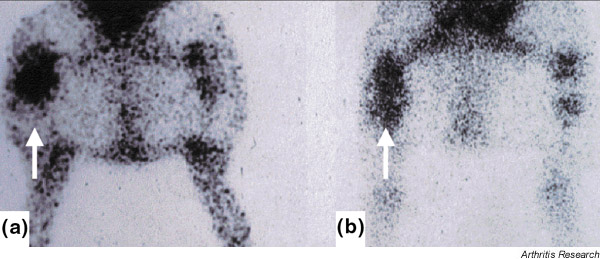
Imaging E-selectin expression in MSU crystal-induced arthritis in the pig. Scintigraphic images of anti-E-selectin monoclonal antibody (mAb) uptake in untreated and anti-TNF-α-treated pigs. Scintigraphic images of the hind limbs and abdomen of an untreated (a) and an anti-TNF-α-treated animal (b) were taken 24 hours after the intra-articular injection of MSU crystals into the right knee and saline solution into the left knee. There is marked uptake of anti-E-selectin mAb into the inflamed joint of the untreated animal, particularly in the region of the joint space (a, arrow). In contrast, anti-E-selectin mAb uptake in the injected knee of an anti-TNF-α treated animal demonstrates a pattern of uptake that is both less intense and less focal (reproduced from [34], with kind permission from Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.).
Possible role of the macrophage in downregulating the tissue response
A number of mechanisms have been proposed to account for the spontaneous resolution of acute gout. These include coating of crystals with protective proteins [38,39], and anti-inflammatory effects of the hypothalmic–pituitary axis [40,41]. Also, inflammation will decline upon neutrophil apoptosis in the tissues, although neutrophil apoptosis may be delayed by uptake of MSU crystals [42].
Apoptotic neutrophils are rapidly and efficiently phagocytosed by tissue macrophages and possibly also by other resident cells [43]. This is thought to protect tissues from damage due to autolysis and spillage of the apoptotic neutrophil contents, and no doubt reduces the duration and extent of neutrophil-mediated inflammation. Importantly, clearance of apoptotic neutrophils by macrophages occurs without the elaboration of proinflammatory cytokines. Instead, macrophages that have taken up apoptotic neutrophils may generate factors with anti-inflammatory properties, including transforming growth factor-β, platelet-activating factor, and prostaglandin E2[44]. Evidence for the uptake of apoptotic neutrophils by macrophages in gout exists in the form of the Reiter cell, which can be found in synovial fluid during an acute gout attack [45,46].
It has been known since the 1970s that MSU crystals can be found in asymptomatic joints of hyperuricaemic individuals. When synovial fluid leukocytes associated with crystals were examined, it was observed that >99.5% of internalised crystals were contained within mononuclear cells but almost never within neutrophils [3,4]. This observation raised the possibility that whereas monocytes elicit a proinflammatory response upon uptake of MSU crystals, macrophages might clear crystals without the induction of proinflammatory activity, in a manner analogous to the clearance of apoptotic neutrophils. We therefore addressed directly the possibility that the inflammatory response to MSU may be influenced by the state of monocyte-to-macrophage differentiation.
We studied a panel of mouse monocyte–macrophage cell lines, representing different stages of macrophage maturation [47]. The order of the cell lines in the differentiation line-up was established by studying expression of the macrophage markers F4/80 and BM 8. The cell lines revealed a close correlation between level of expression of these surface markers and the capacity to ingest MSU crystals. TNF-α production in response to MSU crystals, however, was not linked to phagocytic capacity, in that the most TNF-α was synthesised by cells at an intermediate stage of differentiation (Fig. 3). In contrast, the two most mature macrophage cell types, MH-S and IC21, failed to secrete TNF-α in spite of their being the most efficient at phagocytosis of MSU crystals. Furthermore, after uptake of MSU crystals, supernatants from these mature macrophage cell lines failed to activate endothelial cells. In contrast, supernatants from the partially differentiated cell line RAW264.7 induced endothelial-cell ICAM-1 expression through a TNF-α- and IL-1β-dependent mechanism, in accord with our previous study [34].
Figure 3.

Secretion of TNF-α by macrophage cell lines in response to MSU crystals or zymosan. TNF-α as measured by ELISA in culture supernatants collected from five phagocytically competent macrophage cell lines cultured for 16 hours in the presence of media alone, MSU crystals (0.5 mg/ml), or unopsonized zymosan particles (400 g/ml). Note that the mature macrophage cell lines IC-21 and MH-S secreted TNF-α in response to zymosan but not to MSU crystals. All these cell lines efficiently phagocytosed MSU crystals. Values are means ± SD of triplicates. (Reproduced from [47], with kind permission from Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.).
Stimulation of the macrophage cell line IC21 with lipopolysaccharide or zymosan led to readily detectable TNF-α production, signifying that the lack of response to MSU crystals was not due to an inability to make proinflammatory cytokines. Moreover, incubation of IC21 cells with both zymosan and MSU together resulted in suppression of the zymosan response, suggesting that MSU crystals may stimulate an active suppressive response. Since suppressor activity passed across a semipermeable filter, this appears to involve the release of as-yet-uncharacterised soluble factors.
We have recently extended these experiments to human monocytes and to macrophages differentiated in vitro and obtained very similar results (unpublished observations). Whereas freshly isolated monocytes responded to MSU crystals by releasing IL-1β, TNF-α, and IL-6, differentiated macrophages from the same individual internalised crystals but failed to generate these cytokines or any other factor capable of activating endothelial-cell adhesion molecule expression [48]. Again, human macrophages were as responsive as monocytes in terms of TNF-α release following zymosan stimulation. Ongoing work is establishing in more detail the profile of proinflammatory and anti-inflammatory genes activated in monocytes and macrophages after uptake of MSU crystals and characterising the receptors and signalling mechanisms involved.
Monocytes and macrophages as partners in the orchestration of acute gout
On the basis of this recent work, we propose a model of gout in which the critical determinant of an acute attack is not just the presence of free MSU crystals, but also the availability in the extravascular tissues of recently recruited blood monocytes (Fig. 4). In individuals with hyperuricaemia, the asymptomatic state may be maintained by the silent removal by tissue macrophages of small quantities of crystals as and when they precipitate. However, fresh monocyte recruitment may occur in response to any of the well-established precipitants of acute gout (e.g. trauma, infection), perhaps ensuing from initial endothelial activation by mast-cell degranulation and release of TNF-α. Uptake of crystals by monocytes leads to the elaboration of IL-1β and TNF-α, which in turn activates endothelium and amplifies the inflammatory response through the recruitment of neutrophils and further monocytes. Our observations in pig skin suggest that the positive feedback loop is terminated initially at the level of vascular endothelium, by mechanisms shutting off further leukocyte entry into the tissues. Subsequently, the downregulation of postmigratory tissue leukocyte activation and the further elaboration of endothelial activating factors may be achieved by the noninflammatory removal of free crystals by macrophages that have differentiated from recruited monocytes, possibly involving the release of anti-inflammatory mediators. However, the macrophage may not be a completely innocent partner, as it remains possible that the resolution mechanisms induced by MSU crystals could include factors involved in tissue repair (such as proteases and growth factors) that may contribute to the destructive changes associated with tophi.
Figure 4.

Model of the differential roles of monocytes and macrophages in the inflammatory response to MSU crystals. The model proposes that monocytes play a central role in stimulating an acute attack of gout, whereas differentiated macrophages may play an anti-inflammatory role in terminating an acute attack and in preserving the asymptomatic state (modified from [48], with kind permission from Current Science Ltd). Mo, monocyte; Mφ, macrophage; PMN, polymorphonuclear leukocyte.
Acute versus chronic rheumatic diseases
The relation between endothelial activation and monocyte–macrophage differentiation identified in gout provides a platform on which to base an understanding of the kinetics of inflammation in rheumatic diseases that are not self-limiting. In immune-mediated conditions such as RA, a number of influences may limit the downregulating activities of endothelium and macrophages. First, in RA synovium, postcapillary venules come to resemble high endothelial venules found in peripheral lymphoid organs [49]. These high endothelial venules have plump, cuboidal/columnar endothelial cells and are adapted to support sustained rather than self-limited leukocyte recruitment [50,51]. This and other morphological changes characteristic of lymphoid neogenesis are most probably under the control of the B lymphocyte chemoattractant (BLC/CXCL13) [52,53].
A second important difference in RA is the presence of immune complexes and/or complement, which may subvert the noninflammatory properties of differentiated macrophages by promoting phagocytosis through different receptors. Thus, opsonic serum has been shown to reverse the noninflammatory program of apoptotic neutrophil removal by macrophages, instead rendering the process proinflammatory [54]. Immune complexes in RA may directly trigger the release of proinflammatory cytokines, such as TNF-α, by binding to the immunoglobulin receptor, Fc gamma receptor IIIA [55].
A third difference between the self-limiting inflammatory response in gout and RA is that the prevailing chemokine/cytokine milieu of rheumatoid synovium may favour precursor differentiation to a dendritic cell rather than a tissue macrophage phenotype. Rheumatoid synovium is rich in cytokines, such as IL-4 and IL-15 [56], that skew monocyte differentiation towards dendritic cells [57], but is poor in cytokines, such as M-CSF, that promote the macrophagic end-point [58].
Concluding remarks
The model outlined above can now act as a template for addressing the triangular interactions between monocytes, macrophages, and endothelial cells, and for determining the influence that monocyte–macrophage differentiation has on the control of other inflammatory responses caused by potentially harmful particles. It is clear from the variety of rheumatological syndromes associated with different crystals that the biological effects of crystal deposition vary with the species of crystal involved. This in turn is due to differential cellular responses [31-33], perhaps related to distinct utilisation of cell-surface receptors. The detailed analysis of the various receptor and signalling pathways involved in cellular responses to different crystals may provide important insights that will help us understand the mechanisms underlying the heterogeneity of crystal-related rheumatic diseases.
Glossary of terms
MSU = monosodium urate
London, UK. 24-26 June 2002
References
- Hall AP, Barry PE, Dawber TR, McNamara PM. Epidemiology of gout and hyperuricemia. A long-term population study. Am J Med. 1967;42:27–37. doi: 10.1016/0002-9343(67)90004-6. [DOI] [PubMed] [Google Scholar]
- Weinberger A, Schumacher HR, Agudelo CA. Urate crystals in asymptomatic metatarsophalangeal joints. Ann Intern Med. 1979;91:56–57. doi: 10.7326/0003-4819-91-1-56. [DOI] [PubMed] [Google Scholar]
- Louthrenoo W, Sieck M, Clayburne G, Rothfuss S, Schumacher HRJ. Supravital staining of cells in noninflammatory synovial fluids: analysis of the effect of crystals on cell populations. J Rheumatol. 1991;18:409–413. [PubMed] [Google Scholar]
- Pascual E, Jovani V. A quantitative study of the phagocytosis of urate crystals in the synovial fluid of asymptomatic joints of patients with gout. Br J Rheumatol. 1995;34:724–726. doi: 10.1093/rheumatology/34.8.724. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration . Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- Frenette PS, Wagner DD. Adhesion Molecules – Part I. N Engl J Med. 1996;334:1526–1529. doi: 10.1056/NEJM199606063342308. [DOI] [PubMed] [Google Scholar]
- Frenette PS, Wagner DD. Adhesion molecules – Part II: Blood vessels and blood cells. N Engl J Med. 1996;335:43–45. doi: 10.1056/NEJM199607043350108. [DOI] [PubMed] [Google Scholar]
- Pober JS, Cotran RS. Cytokines and endothelial cell biology. Physiol Rev. 1990;70:427–451. doi: 10.1152/physrev.1990.70.2.427. [DOI] [PubMed] [Google Scholar]
- Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- Wagner DD. The Weibel-Palade body: the storage granule for von Willebrand factor and P-selectin. Thromb Haemost. 1993;70:105–110. [PubMed] [Google Scholar]
- Utgaard JO, Jahnsen FL, Bakka A, Brandtzaeg P, Haraldsen G. Rapid secretion of prestored interleukin 8 from Weibel-Palade bodies of microvascular endothelial cells. J Exp Med. 1998;188:1751–1756. doi: 10.1084/jem.188.9.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff B, Burns AR, Middleton J, Rot A. Endothelial cell "memory" of inflammatory stimulation: human venular endothelial cells store interleukin 8 in Weibel-Palade bodies. J Exp Med. 1998;188:1757–1762. doi: 10.1084/jem.188.9.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulsky MI, Chan MKW, Movat HZ. Acute inflammation and microthrombosis induced by endotoxin, interleukin-1, and tumour necrosis factor and their implication in Gram-negative infection. Lab Invest. 1988;58:365–378. [PubMed] [Google Scholar]
- Binns RM, Licence ST, Wooding FBP, Duffus WPH. Active lymphocyte traffic induced in the periphery by cytokines and phytohemagglutinin: three different mechanisms? Eur J Immunol. 1992;22:2195–2203. doi: 10.1002/eji.1830220903. [DOI] [PubMed] [Google Scholar]
- Keelan ETM, Licence ST, Peters AM, Binns RM, Haskard DO. Characterization of E-selectin expression in vivo using a radi-olabelled monoclonal antibody. Am J Physiol. 1994;266:H279–H290. doi: 10.1152/ajpheart.1994.266.1.H279. [DOI] [PubMed] [Google Scholar]
- Binns RM, Licence ST, Harrison AA, Keelan ETD, Robinson MK, Haskard DO. In vivo E-selectin upregulation correlates with early infiltration of PMN, later with PBL-entry: mAbs block both. Am J Physiol. 1996;266:H183–H193. doi: 10.1152/ajpheart.1996.270.1.H183. [DOI] [PubMed] [Google Scholar]
- Binns RM, Whyte A, Licence ST, Harrison AA, Tsang Y, Haskard DO, Robinson MK. The role of E-selectin in lymphocyte and polymorphonuclear cell recruitment into cutaneous delayed hypersensitivity reactions in sensitized pigs. J Immunol. 1996;157:4094–4099. [PubMed] [Google Scholar]
- Harrison AA, Stocker CJ, Chapman PT, Tsang YT, Huehns TY, Gundel RH, Peters AM, Davies KA, George AJ, Robinson MK, Haskard DO. Expression of VCAM-1 by vascular endothelial cells in immune- and non-immune inflammatory reactions in the skin. J Immunol. 1997;159:4546–4554. [PubMed] [Google Scholar]
- Panes J, Perry MA, Anderson DC, Manning A, Leone B, Cepinskas G, Rosenbloom CL, Miyasaka M, Kvietys PR, Granger DN. Regional differences in constitutive and induced ICAM-1 expression in vivo. Am J Physiol. 1995;269:H1955–1964. doi: 10.1152/ajpheart.1995.269.6.H1955. [DOI] [PubMed] [Google Scholar]
- Henninger DD, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, Granger DN. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs in the mouse. J Immunol. 1997;158:1825–1832. [PubMed] [Google Scholar]
- Hickey MJ, Kanwar S, McCafferty DM, Granger DN, Eppihimer MJ, Kubes P. Varying roles of E-selectin and P-selectin in different microvascular beds in response to antigen. J Immunol. 1999;162:1137–1143. [PubMed] [Google Scholar]
- Harari O, McHale J, Marshall D, Ahmed S, Brown D, Askenase PW, Haskard DO. Endothelial cell E- and P-selectin up-regulation in murine contact sensitivity is prolonged by distinct mechanisms occurring in sequence. J Immunol. 1999;163:6860–6866. [PubMed] [Google Scholar]
- McHale JF, Harari OA, Marshall D, Haskard DO. TNF-α and IL-1 sequentially induce endothelial ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J Immunol. 1999;163:3993–4000. [PubMed] [Google Scholar]
- Harari O, Marshall D, McHale J, Ahmed S, Haskard DO. Limited endothelial E- and P-selectin expression in MRL/lpr lupus-prone mice. Rheumatology. 2001;40:889–895. doi: 10.1093/rheumatology/40.8.889. [DOI] [PubMed] [Google Scholar]
- Dieppe PA, Doherty M, Papadimitriou GM. Inflammatory responses to intradermal crystals in healthy volunteers and patients with rheumatic diseases. Rheumatol Int. 1982;2:55–58. doi: 10.1007/BF00541246. [DOI] [PubMed] [Google Scholar]
- Chapman PT, Jamar F, Harrison AA, Schofield JB, Peters AM, Binns RM, Haskard DO. Characterization of E-selectin expression, leukocyte traffic and clinical sequelae in urate crystal-induced inflammation: an insight into gout. Brit J Rheumatol. 1996;35:323–334. doi: 10.1093/rheumatology/35.4.323. [DOI] [PubMed] [Google Scholar]
- Terkeltaub R. In: Gout, Hyperuricaemia, and Other Crystal-associated Arthropathies. Smyth CJ, Holers VM, editor. New York: Marcel Dekker; 1999. Pathogenesis of inflammatory manifestations caused by crystals. pp. 1–14. [Google Scholar]
- Di Giovine FS, Malawista SE, Nuki G, Duff GW. Interleukin 1 (IL 1) as a mediator of crystal arthritis: stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL-1. J Immunol. 1987;38:3213–3218. [PubMed] [Google Scholar]
- Guerne PA, Terkeltaub R, Zuraw B, Lotz M. Inflammatory microcrystals stimulate interleukin-6 production and secretion by human monocytes and synoviocytes. Arthritis Rheum. 1989;32:1443–1452. doi: 10.1002/anr.1780321114. [DOI] [PubMed] [Google Scholar]
- Terkeltaub R, Zachariae C, Santoro D, Martin J, Peveri P, Matsushima K. Monocyte-derived neutrophil chemotactic factor/interleukin-8 is a potential mediator of crystal-induced inflammation. Arthritis Rheum. 1991;34:894–903. doi: 10.1002/art.1780340716. [DOI] [PubMed] [Google Scholar]
- Di Giovine FS, Malawista SE, Thornton E, Duff GW. Urate crystals stimulate production of tumor necrosis factor alpha from human blood monocytes and synovial cells. Cytokine mRNA and protein kinetics, and cellular distribution. J Clin Invest. 1991;87:1375–1381. doi: 10.1172/JCI115142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouliot M, James MJ, McColl SR, Naccache PH, Cleland LG. Monosodium urate microcrystals induce cyclooxygenase-2 in human monocytes. Blood. 1998;91:1769–1776. [PubMed] [Google Scholar]
- Liu R, O'Connell M, Johnson K, Pritzker K, Mackman N, Terkeltaub R. Extracellular signal-regulated kinase 1/extracellular signal-regulated kinase 2 mitogen-activated protein kinase signaling and activation of activator protein 1 and nuclear factor kappaB transcription factors play central roles in interleukin-8 expression stimulated by monosodium urate monohydrate and calcium pyrophosphate crystals in monocytic cells. Arthritis Rheum. 2000;43:1145–1155. doi: 10.1002/1529-0131(200005)43:5<1145::AID-ANR25>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Chapman PT, Yarwood H, Harrison AA, Stocker CJ, Jamar F, Gundel RH, Peters AM, Haskard DO. Endothelial activation in monosodium urate monohydrate crystal-induced inflammation: in vitro and in vivo studies on the roles of tumor necrosis factor-alpha and interleukin-1. Arthritis Rheum. 1997;40:955–965. doi: 10.1002/art.1780400525. [DOI] [PubMed] [Google Scholar]
- Chapman PT, Jamar F, Keelan ETM, Peters AM, Haskard DO. Use of a radiolabeled monoclonal antibody against E-selectin for imaging endothelial activation in rheumatoid arthritis. Arthritis Rheum. 1996;39:1371–1375. doi: 10.1002/art.1780390815. [DOI] [PubMed] [Google Scholar]
- Bhatti M, Chapman P, Peters AM, Haskard DO, Hodgson H. Visualizing E-selectin in the detection and evaluation of inflammatory bowel disease. Gut. 1998;43:40–47. doi: 10.1136/gut.43.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PT, Jamar F, Harrison AA, Binns RM, Peters AM, Haskard DO. Non-invasive imaging of E-selectin expression by activated endothelium in urate crystal-induced arthritis. Arthritis Rheum. 1994;37:1752–1756. doi: 10.1002/art.1780371207. [DOI] [PubMed] [Google Scholar]
- Terkeltaub R, Martin J, Curtiss LK, Ginsberg MH. Apolipoprotein B mediates the capacity of low density lipoprotein to suppress neutrophil stimulation by particulates. J Biol Chem. 1986;261:15662–15667. [PubMed] [Google Scholar]
- Terkeltaub RA, Dyer CA, Martin J, Curtiss LK. Apolipoprotein (apo) E inhibits the capacity of monosodium urate crystals to stimulate neutrophils. Characterization of intraarticular apo E and demonstration of apo E binding to urate crystals in vivo. J Clin Invest. 1991;87:20–26. doi: 10.1172/JCI114971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Bravo E, Sieck MS, Schumacher HRJ. Changes in the proteins coating monosodium urate crystals during active and subsiding inflammation. Immunogold studies of synovial fluid from patients with gout and of fluid obtained using the rat subcutaneous air pouch model. Arthritis Rheum. 1993;36:1274–1285. doi: 10.1002/art.1780360912. [DOI] [PubMed] [Google Scholar]
- Getting SJ, Gibbs L, Clark AJ, Flower RJ, Perretti M. POMC gene-derived peptides activate melanocortin type 3 receptor on murine macrophages, suppress cytokine release, and inhibit neutrophil migration in acute experimental inflammation. J Immunol. 1999;162:7446–7453. [PubMed] [Google Scholar]
- Akahoshi T, Nagaoka T, Namai R, Sekiyama N, Kondo H. Prevention of neutrophil apoptosis by monosodium urate crystals. Rheumatol Int. 1997;16:231–235. doi: 10.1007/BF01375654. [DOI] [PubMed] [Google Scholar]
- Haslett C, Savill JS, Whyte MK, Stern M, Dransfield I, Meagher LC. Granulocyte apoptosis and the control of inflammation. Philos Trans R Soc Lond B Biol Sci. 1994;345:327–333. doi: 10.1098/rstb.1994.0113. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation: programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvi E, Manganelli S, De Stefano R, Frati E, Marcolongo R. CD36 and CD14 immunoreactivity of Reiter cells in inflammatory synovial fluids. Ann Rheum Dis. 2000;59:399–400. doi: 10.1136/ard.59.5.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagnik DR, Hillyer P, Marshall D, Krausz T, Haskard DO, Landis RC. Non-inflammatory phagocytosis of monosodium urate monohydrate crystals by macrophages: implications for the control of joint inflammation in gout. Arthritis Rheum. 2000;43:1779–1789. doi: 10.1002/1529-0131(200008)43:8<1779::AID-ANR14>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Landis RC, Yagnik DR, Emons V, Mason J, Haskard DO. Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages. Arthritis Rheum. 2001;44:S162. doi: 10.1002/art.10614. [DOI] [PubMed] [Google Scholar]
- Iguchi T, Ziff M. Electron microscopic study of rheumatoid synovial vasculature. Intimate relationship between tall endothelium and lymphoid aggregation. J Clin Invest. 1986;77:355–361. doi: 10.1172/JCI112312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemont AJ. Molecules controlling lymphocyte-endothelial interactions in lymph nodes are produced in vessels of inflamed synovium. Ann Rheum Dis. 1987;46:924–928. doi: 10.1136/ard.46.12.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie SA, Streeter PR, Bolt PA, Butcher EC, Picker LJ. The human peripheral lymph node vascular addressin: an inducible endothelial antigen involved in lymphocyte homing. Am J Pathol. 1993;143:1688–1698. [PMC free article] [PubMed] [Google Scholar]
- Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–481. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- Shi K, Hayashida K, Kaneko M, Hashimoto J, Tomita T, Lipsky PE, Yoshikawa H, Ochi T. Lymphoid chemokine B cell attracting chemokine-1 (CXCL13) is expressed in germinal center of ectopic lymphoid follicles within the synovium of chronic arthritis patients. J Immunol. 2001;166:650–655. doi: 10.4049/jimmunol.166.1.650. [DOI] [PubMed] [Google Scholar]
- Meagher LC, Savill JS, Baker A, Fuller RW, Haslett C. Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J Leukoc Biol. 1992;52:269–273. [PubMed] [Google Scholar]
- Abrahams VM, Cambridge G, Lydyard PM, Edwards JC. Induction of tumor necrosis factor alpha production by adhered human monocytes: a key role for Fcgamma receptor type IIIa in rheumatoid arthritis. Arthritis Rheum. 2000;43:608–616. doi: 10.1002/1529-0131(200003)43:3<608::AID-ANR18>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Al-Mughales J, Field M, Leung BP, Huang F-P, Dixon R, Sturrock RD, Wilkinson PC, Liew FY. The role of interleukin-15 in T-cell migration and activation in rheumatoid arthritis. Nat Med. 1996;2:175–182. doi: 10.1038/nm0296-175. [DOI] [PubMed] [Google Scholar]
- Mohamadzadeh M, Berard F, Essert G, Chalouni C, Pulendran B, Davoust J, Bridges G, Palucka AK, Banchereau J. Interleukin 15 skews monocyte differentiation into dendritic cells with features of Langerhans cells. J Exp Med. 2001;194:1013–1020. doi: 10.1084/jem.194.7.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Warren MK, Haskill S. Colony-stimulating factor-induced monocyte survival and differentiation into macrophages in serum-free cultures. J Immunol. 1987;139:3703–3709. [PubMed] [Google Scholar]