

Abstract
Mutations in tumor suppressor BRCA1 lead to breast and/or ovarian cancer. Here we show that loss of BRCA1 in mice results in transcriptional derepression of the tandemly repeated satellite DNA. BRCA1 deficiency is accompanied by reduction of condensed DNA regions in the genome and loss of ubiquitylation of histone H2A at satellite repeats. BRCA1 binds to satellite DNA regions in vivo and ubiquitylates H2A in vitro. Ectopic expression of an H2A fused to ubiquitin reverses the effects of BRCA1 loss, suggesting that BRCA1 maintains heterochromatin structure via ubiquitylation of histone H2A. Satellite DNA derepression was also observed mouse and human BRCA1 deficient breast cancers. Ectopic expression of satellite DNA can phenocopy BRCA1 loss in centrosome amplification, cell cycle checkpoint defects, DNA damage and genomic instability. We propose that the role of BRCA1 in maintaining global heterochromatin integrity accounts for many of its tumor suppressor functions.
Breast cancer susceptibility gene1 (BRCA1) was identified as a hereditary cancer susceptibility gene which increased1 risk for the development of breast and ovarian cancer in BRCA1 mutation carriers to as high as 95% by age 702. The BRCA1 protein contains a RING finger domain in the N-terminus with ubiquitin E3 ligase activity and two BRCT repeats in the C-terminus3. BRCA1 is highly expressed in proliferative cells and its loss leads most prominently to genetic instability and growth arrest. These observations have implicated BRCA1 in a multitude of disparate cellular functions, including DNA damage repair, cell cycle checkpoint activation, transcriptional regulation, DNA replication, centrosome function, X inactivation, among others4. Presently no unifying mechanistic framework exists to tie the reported biochemical activities of BRCA1 to its tumor suppressor function3.
Heterochromatic centers in BRCA1 KO cells
High levels of BRCA1 expression have been found in the embryonic neuroectoderm5, the postnatal cerebellum6 and the subgranular zone of the dentate gyrus in the adult brain, which are the sites of embryonal and adult neurogenesis, respectively. We initially set out to study the contribution of BRCA1 to neural stem cell (NSC) proliferation through targeted deletion of BRCA1 in nestin-expressing NSCs (Supplementary Fig. 1a). As expected, the levels of BRCA1 protein and RNA are significantly reduced in the brains of BRCA1 KO mice (Figs.1a and Supplementary Fig. 1b). To characterize the mutant brains, we performed microarray analyses and found evidence of altered epigenetic regulation in the absence of BRCA1 as some imprinted genes are deregulated (Supplementary Table 1). Most notably, upregulation of both Igf2 and H19 expression has been verified by quantitative RT-PCR experiments using cortical RNA samples (Fig. 2a).
Figure 1. BRCA1 deficiency impairs heterochromatin structure.
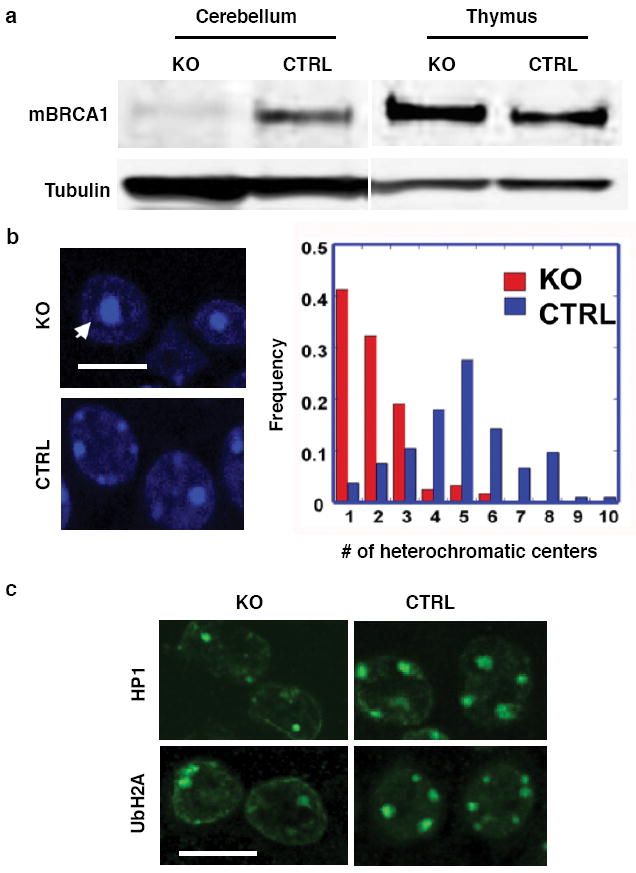
a, Expression of BRCA1 is diminished in the Nestin-Cre BRCA1 knockout brains as shown by immunoblotting. KO: samples from a P7 BRCA1 KO (Brca11+/-;Brca5-13cK+/-;nestin-Cre+) mouse. CTRL: samples from a control P7 (Brca11+/-; Brca5-13cK+/-; nestin-Cre-). BRCA1 immunoblot (upper panel), tubulin loading control (lower panel). b, Lack of BRCA1 induces changes in the nuclear morphology of P7 KO cortical cells. The numbers of strong DAPI staining nuclear foci/cell were counted and their frequencies plotted (right panel). c, Confocal microscopic images of brain sections from BRCA1 conditional KO and control mice (P7) stained with antibodies against HP1 and ubiquitin-H2A (UbH2A).
Figure 2. BRCA1 and its ubiquitin E3 ligase activity are required for gene silencing in constitutive heterochromatin.
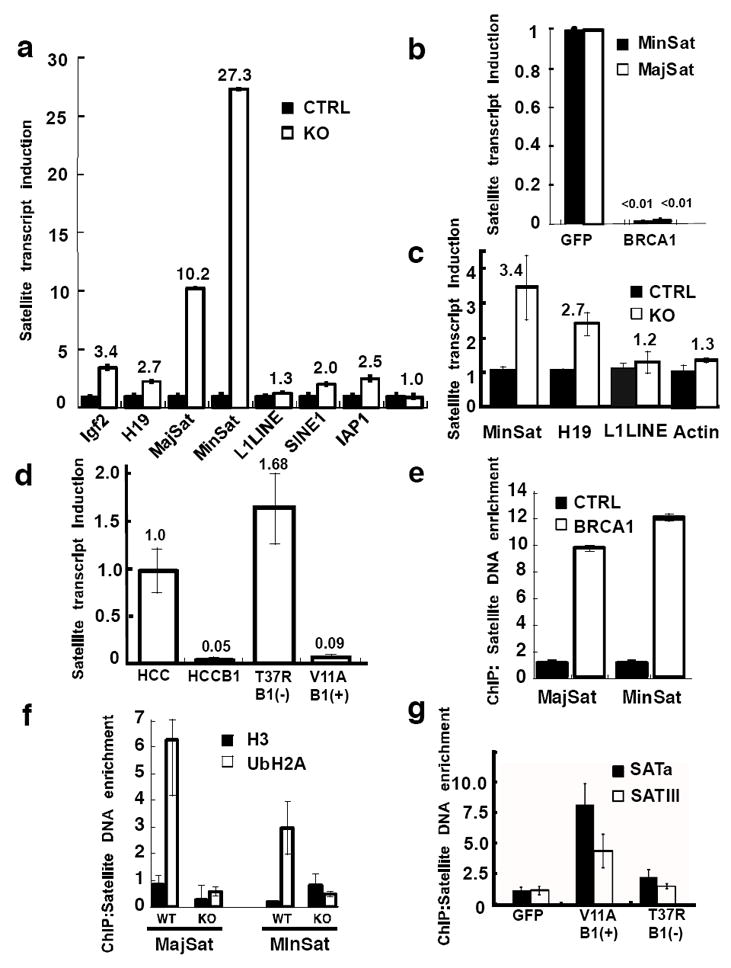
a, Quantitative RT-PCR showed that heterochromatic regions or certain imprinted genes (Igf2 & H19 but not Igf2R) were upregulated in BRCA1 KO brains. Internal controls: cyclophilin or 18SRNA. b, Rescue of the BRCA1 mediated repression in NPCs by exogenous human BRCA1 but not GFP after infection with retroviral CRE-GFP. c, Quantitative RT-PCR experiments showed that heterochromatic regions or imprinted genes were upregulated in BRCA1 KO mammary glands (6-week virgin). d, The ubiquitin E3 ligase activity of BRCA1 is required for heterochromatin silencing. HCC1937 cells were reconstituted with either wild type BRCA1 (HCCB1) or BRCA1 with a mutation in the RING domain, T37R23. P7 cerebellar ChIP experiments with antibodies against (e) mouse BRCA1 or VSVG protein as a control and (f) ubiquityl-histone H2A or histone H3 protein as a control. Enrichment of major and minor satellite DNA was measured by quantitative PCR relative to 18S control. g, ChIP experiments were performed using ubiquitinated histone H2A antibody as in (f) and HCC1937 cells reconstituted with GFP, BRCA1 (V11A) or a mutant (T37R). Error bars are shown as s.d. Each result shown is representative of three independent experiments.
Heterochromatic centers are highly condensed regions of heterochromatin with repetitive tandem DNA, which can be visualized by DAPI staining. In neurons these centers are particularly prominent. We analyzed the nuclei of the mutant brains by DAPI staining and found a marked reduction in the number of heterochromatic foci in the cell, especially in the perinuclear region, when compared to control cortices (4.88 foci/cell to 1.99 foci/cell) (Student’s t-test, p<0.0005) (Fig. 1b). Moreover, the size of the heterochromatic foci appeared to be larger and diffuse in mutant brains. These observations suggest a defective heterochromatic structure in BRCA1-deficient cells. To study the nature of this defect, we stained the nuclei of cortical cells with heterochromatin protein (HP1)7, a bona fide pericentric heterochromatin associated protein, and found that HP1 localized to heterochromatic centers. The number of HP1-positive foci in mutant cortices was decreased from 2.86 foci/cell to 2.13 foci/cell (Student’s t-test, p<0.0005) (Fig. 1c, top row and Supplementary Fig. 1c, left panel). Moreover, immunoblotting of cerebellar samples showed that the protein levels of all three HP1 protein isoforms (HP1α, β and γ) were severely reduced in KO animals compared to controls (Supplementary Fig. 1d). The repressive histone H1 also displayed aberrant staining and was more diffuse in BRCA1 mutant cortices (Supplementary Fig. 2a). Other previously characterized repressive histones, such as Macro H2A1 and heterochromatin markers, such as histone H3 dimethyl Lys 9, did not appear to be affected by BRCA1 KO (Supplementary Fig. 2b). Since we have previously shown that the BRCA1 protein preferentially mono-ubiquitinates histone H2A in vitro through its N-terminal RING finger8, we investigated whether BRCA1 is involved in heterochromatin formation through histone H2A ubiquitylation. We found that the staining of mono-ubiquitylated histone H2A, which has recently been implicated in polycomb-mediated silencing9, corresponded to the heterochromatic centers described in Fig. 1b and that the staining signal was reduced in BRCA1 mutant brains (Fig.1c bottom row and Supplementary Fig.1c right panel). We verified the cell autonomous nature of our observations using cultured embryonic neural progenitor cells (NPCs) in vitro. Ten days after neuronal differentiation, the BRCA1-deficient cells displayed a significant reduction in the number of heterochromatic centers (7.85 foci/cell vs 5.00 foci/cell, Student’s t-test, p<0.0005) (Supplementary Fig. 2c). Taken together, these data strongly suggest that cells lacking BRCA1 are impaired in the organization of heterochromatin structure.
Heterochromatic silencing disruption
To ascertain that the observed heterochromatic structural deficiencies are of functional importance, we investigated the transcriptional status of known silenced genes. Defects are expected to result in derepression of normally silenced genes in heterochromatic regions10. The murine pericentric heterochromatin consists of stretches of highly repetitive DNA elements, including the major and minor satellite repeats, which make up ~3% and ~0.45% of the mouse genome respectively11. Quantitative RT-PCR experiments showed that the levels of minor and major satellite transcripts were elevated 10.2-fold and 27.3-fold, respectively, in the BRCA1 mutant cortices, indicating a loss of repression in constitutive heterochromatin (Fig. 2a). Other interspersed repetitive elements such as IAP1, SINE1 and the L1 LINE element were largely unaffected. This effect was cell autonomous as it was observed in vitro in cultured embryonic NPCs (Supplementary Fig. 3a). The defects in heterochromatic repression in BRCA1-deficient mouse cells could be reversed by re-expression of human BRCA1 using a lentiviral vector (Fig. 2b). Heterochromatin repression by BRCA1 is not unique to neural cells because deletion of BRCA1 in fibroblasts from P7 mouse ribcages also showed upregulation of both satellite transcripts (Supplementary Fig. 3b). Since BRCA1 is a tumor suppressor in breast cancer we next generated mice harboring a mammary gland specific deletion of BRCA1 and analyzed the integrity of heterochromatin by qRT-PCR (Fig. 2c). The result demonstrated that lack of BRCA1 in mammary glands from 6-week old virgin females, disrupted heterochromatic satellite DNA silencing as well as imprinted H19 epigenetic regulation, consistent with our findings in BRCA1 null brains (Fig. 2a).
The silencing of constitutive heterochromatin by BRCA1 is also not limited to mouse cells, as satellite transcripts from human HeLa cells were upregulated upon expression of an shRNA targeting BRCA1 (Supplementary Fig. 4a). Similarly, reconstitution of the human BRCA1-deficient cell line, HCC1937, with a retrovirus expressing wildtype BRCA1 repressed the expression of satellite DNA transcripts about 20-fold (Fig. 2d). To determine whether the N-terminal RING domain of BRCA1 is important for the heterochromatic silencing function, we reconstituted HCC1937 cells with either a pathogenic BRCA1 mutant devoid of ubiquitin ligase activity (T37R, known to confer increased cancer risk) or a polymorphic variant (V11A, indistinguishable from wildtype). The T37R mutant BRCA1 failed to repress satellite transcripts whereas the V11A repressed the satellite transcripts to an extent similar to wildtype BRCA1 (Fig. 2d). The lack of repressive activity was not due to reduced binding of BARD1, the natural heterodimeric partner of BRCA1, as both polymorphic and mutant immunoprecipitated BARD1 similarly (Supplementary Fig. 4b). To further verify the involvement of the E3-ligase activity of BRCA1, we assessed the effects of BARD1 knockdown on satellite DNA transcription since the heterodimerization between BRCA1 and BARD1 is required for the optimal E3 ligase activity8,12. Expression of an shRNA against BARD1 elevated the satellite DNA transcription in the BRCA1 reconstituted HCC1937 cells but not in the HCC1937 cells where BRCA1 is absent and the satellite repeat transcription is already derepressed (Supplementary Fig. 4c). These results from cultured human cells are consistent with data obtained from the BRCA1 deficient mouse brains, fibroblasts and mammary glands in which satellite DNA transcript induction was observed. Moreover, it appears that ubiquitin E3 ligase activity of BRCA1 is essential for its role in gene silencing in constitutive heterochromatin.
BRCA1 and heterochromatin relationship
To gain a mechanistic insight into the role of BRCA1 in maintaining heterochromatin structure, we first investigated if BRCA1 directly associates with the constitutive heterochromatic region in vivo. Chromatin immunoprecipitation (ChIP) experiments showed that BRCA1 was indeed enriched on major and minor satellite DNA (Fig. 2e) in mouse cells and on various α satellite sequences in HeLa cells (Supplementary Fig. 4d).
We next tested whether BRCA1 influenced histone ubiquitylation in vivo. The results of ChIP experiments showed that ubiquitylated histone H2A was enriched in both major and minor satellite regions when compared with a histone H3 antibody (Fig. 2f). However, this satellite region enrichment was abrogated in the absence of BRCA1, indicating that BRCA1 integrity is essential for the accumulation of ubiquityl-histone H2A at satellite repeats in vivo. Our data also show that ubiquityl-histone H2A is enriched at the satellite regions of the mouse genome to a greater extent than at other interspersed repeats such as LINE1 (data not shown) suggestive of a distinct heterochromatin. If BRCA1 is indeed the ubiquitin ligase for pericentric heterochromatic histone H2A, it would imply that the integrity of the RING finger is required for the accumulation of ubiquitylated histone H2A at satellite repeats, as we have shown that the same region is critical for transcriptional silencing in the heterochromatin (Fig. 2d). To test this hypothesis, ChIP experiments were performed using HCC1937 cells reconstituted with BRCA1-T37R, BRCA1-V11A, or GFP (Fig. 2g). The T37R mutant BRCA1 failed to significantly enrich ubiquitylated histone H2A at satellite regions, in a manner similar to GFP control, while the polymorphic BRCA1-V11A showed enrichment of ubiquitylated histone H2A within the same regions. A distinct mutant I26A which has been reported to not destabilize the BARD1 interaction but has no ubiquityl ligase activity13 fails to complement the loss of BRCA1 (Supplementary Fig. 5). The ubiquityl ligase function of BRCA1 appears to be specific as deletion of Ring1B or knockdown of RNF8 fail to appreciably derepress satellite transcription (Supplementary Fig. 6). These findings provide further evidence that the BRCA1 ubiquitin ligase activity is essential for the maintenance of the ubiquityl histone H2A mark within constitutive heterochromatic regions not only in mouse tissue but also human cells derived from breast cancers.
Ubiquitylated histone H2A rescues BRCA1
To assess the contribution of ubiquityl-histone H2A to the function of BRCA1 in heterochromatic silencing we generated a lentiviral vector construct expressing an H2A-Ub fusion protein that mimics the natural ubiquityl histone H2A (Fig. 3a). This was accomplished by fusing a single ubiquitin moiety to the C-terminus of histone H2A in lieu of the modification of lysine 119. Subnuclear fractionation of cells expressing this fusion protein shows that the H2A-Ub protein and mutants can be incorporated into cellular chromatin (Supplementary Fig. 7). BRCA1 deficient HCC1937 cells were transduced with lentivirus expressing the H2A-Ub fusion protein. Quantification of the levels of satellite DNA transcription showed that the H2A-Ub expression can re-establish silencing of the satellite repeats (mcBox, SATa and SATIII) within heterochromatin in BRCA1-deficient human cells, suggesting that mono-ubiquityl-H2A is likely to be the main target of BRCA1 ubiquitin E3 ligase that mediates silencing at constitutive heterochromatin (Fig. 3b). Ectopic expression of H2A-Ub (Supplementary Fig. 8) in murine NPCs restored the levels of satellite DNA silencing to more than 90% of endogenous wildtype silencing (Fig. 3c). Mutations within the ubiquitin domain of the fusion protein reduced the ability to restore silencing of the H2A-Ub fusion protein (Supplementary Fig. 9a). H2A-Ub expression in wild type cells had no significant effect on the satellite DNA transcription. To inquire whether the ubiquitylation of histone H2A affected previously well-documented BRCA1 functions, we tested if the restored satellite DNA silencing in BRCA1 null cells had any of the expected consequences. It has been reported that primary BRCA1 null cells display a severe growth defect and p53-activated apoptosis and that p53 inactivation largely rescues the growth defect14. By performing BrdU incorporation assays, we found that ectopic expression of H2A-Ub led to a restoration of over 75% of proliferation in BRCA1 deficient cells, only slightly lower than a p53 knockdown (Fig. 3d). Again, in this case expression of H2A-Ub bypassed the requirement for BRCA1. It is worthy to note that although p53 is able to rescue the proliferative defect of BRCA1 deficient cells, it does not rescue the formation of heterochromatic centers, or the derepression of satellite transcripts (Supplementary Fig. 10). We also evaluated the ability of H2A-Ub to suppress apoptosis. Staining for an apoptotic marker, active caspase3, showed a similar result (Fig. 3e) to what was seen for BrdU incorporation or transcriptional silencing. Thus H2A-Ub can suppress the apoptosis induced by the loss of BRCA1. Taken together, ectopic expression of H2A-Ub not only restored silencing but also rescued the proliferative defect and apoptosis of BRCA1 deficient cells. This rescue of the proliferative capacity of H2A-Ub is specific for the ubiquitylation modification, as mutations within the ubiquitin domain can abrogate the effect (Supplementary Figs. 9b and 9c). Homologous recombination, another reported cellular process in which BRCA1 has been implicated, can also be rescued by ectopic H2A-Ub expression in the absence of BRCA1, whereas the same protein with a triple point mutation within the ubiquitin domain is unable to complement (Supplementary Fig. 11). Thus, mono-ubiquityl-H2A is likely the principal target of BRCA1 for maintaining heterochromatin integrity in vivo.
Figure 3. Ubiquitinated histone H2A is directly involved in BRCA1 mediated heterochromatic silencing.
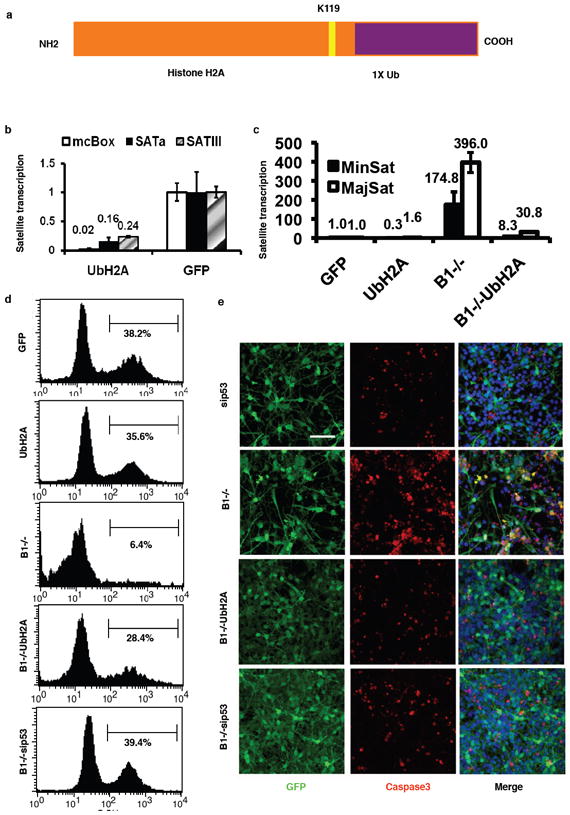
a, A diagram showing the artificial ubiquitin-H2A expression vector. b, HCC1937 cells were infected with a lentiviral vector expressing ubiquitylated histone H2A mimic fusion protein or GFP as a control and the levels of alpha satellite variants (mcBox, SATa and SATIII) transcription were measured by qRT-PCR. c, Ectopic expression of ubiquitylated histone H2A re-established heterochromatin silencing in BRCA1 null NPCs. NPCs were infected with GFP alone, ubH2A, CRE-GFP alone and CRE-GFP coinfected with UbH2A. The levels of satellite DNA transcription were measured by qRT-PCR. d, NPCs showed increased proliferation upon expression of ubiquitylated histone H2A in BRCA1 null NPCs. Two days after infection (2 dpi), cells were labeled with BrdU prior to fixation, immunostaining and FACS analysis. Result shown is representative of 3 independent experiments. e, NPCs showed reduced apoptosis upon expression of ubiquitylated histone H2A in BRCA1 null NPCs. NPCs were cultured and infected as in (d). Cells were fixed, immunostained with anti-activated caspase 3 antibody and subjected to confocal imaging. Error bars are shown as s.d. Scale bar: 100 μm
Heterochromatin and BRCA1 tumor suppression
We next investigated the significance of heterochromatin function in BRCA1 mediated tumor suppression. Mouse breast tumors (Fig. 4a) were harvested from animals where BRCA1 was deleted in the mammary gland (also shown in Fig. 2c). Analyses by qRT-PCR showed disrupted satellite DNA silencing in BRCA1 deficient mouse mammary tumors, but not in tissues where BRCA1 is intact (Fig. 4b). Furthermore, we analyzed satellite transcripts in human breast tumors derived from eight individual BRCA1 mutation carriers and found significant derepression of the expression of alpha satellite repeats: CFXr and SATa (two of human alpha satellite DNA sequences; Fig. 4c). The differences are apparent, even without normalizing for any other factors (e.g. the percentage of non-tumor cell contamination), in comparison with a cohort of normal breast biopsies.
Figure 4. Derepression of satellite DNA transcription occurs in BRCA1-deficient breast cancers.
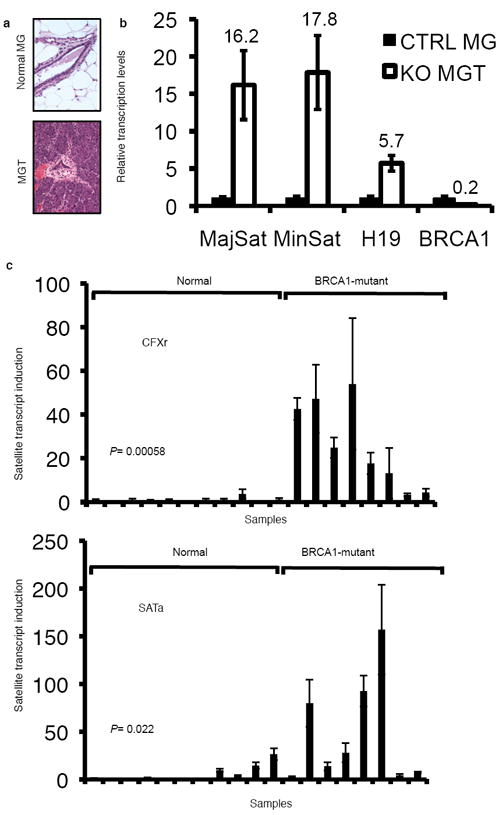
a, Haematoxylin/eosin staining of BRCA1KO mouse breast cancer. Because BRCA1 KO female mice develop mammary tumors very rarely, these mice were crossed into a p53 heterozygous background for accelerated tumorigenesis. MG, mammary gland from normal mouse littermates. MGT, mammary tumors from BRCA1KO mouse (Brca11+/-;Brca5-13cK+/-;MMTV-Cre+;p53+/-) developed at 6 months. b, Quantitative RT-PCR experiments showed that heterochromatic regions and imprinted genes were upregulated in mouse BRCA1 KO breast cancer in comparison to that of wildtype mammary glands from littermates (internal control 18SRNA). The result shown is an aggregate of 5 mouse independent sample pairs. c, Quantitative RT-PCR experiments showed the satellite DNA transcripts, CFXr and SATa, significantly derepressed (Student’s 2-tailed t-test) in human BRCA1-mutant breast tumors (n=8) in comparison with that of normal breast tissues (n=11) . Ct values of each sample were normalized with GAPDH. Error bars are shown as s.d.
Satellite transcript induced genomic instability
To investigate whether the observed satellite DNA derepression in mouse and human breast cancers is either an epiphenomenon or of etiological significance for the genesis of cancer, we ectopically expressed satellite DNA from a transduced lentiviral vector (Fig. 5a) in cultured primary human mammary epithelial cells (HMECs). These cells increased abnormal mitotic figures, including bridged and lagging chromosomes and disorganized metaphases shown by DAPI staining (Figs. 5b and supplementary Fig.12). Immunofluorescence staining of pericentrin and tubulin revealed amplified centrosomes, as previously described in BRCA1 null cells (Fig. 5c). Cells overexpressing satellite RNA also displayed numerous foci of γH2Ax (Fig. 5d), a well-established marker of DNA double strand breaks. This mirrors the phenotype of BRCA1 deficient cells. Flow cytometric analyses showed an impaired mitotic spindle checkpoint when cells overexpressing satellite DNA were treated with nocodazole (Fig. 5e), which has also been reported as a BRCA1 loss phenotype15. Just as expected, ectopic expression of satellite RNA was able to create a deficiency in homologous recombination similar in magnitude to the knockdown of BRCA1 (Supplementary Fig.13). Importantly, the cellular defects associated with satellite RNA overexpression are generally considered leading causes for genomic instability.
Figure 5. Ectopic expression of satellite DNA leads to genomic instability in human mammary epithelial cells.
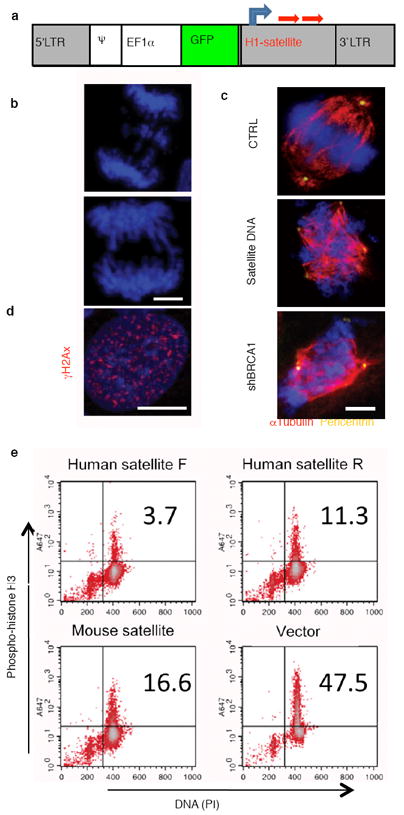
a, A diagram of the lenti-viral vector expressing the human or mouse satellite DNA sequence (red arrows) under the H1 promoter (gray arrow). b, c, d, Overexpression of satellite RNA induced mitotic catastrophe (b), centrosome amplification (c), histone γH2Ax phosphorylation (d), in primary HMECs. e, Defective mitotic checkpoints induced by satellite DNA overexpression. 48h after transduction of a lentivirus expressing human satellite DNA sequence with forward direction (human satellite F), or with reverse direction (human satellite R), or mouse satellite DNA sequence, or an empty vector, U-2OS cells were blocked with thymidine, followed by treatment of nocodazole. The cells were stained with phosho-histone H3 antibody and PI prior to flow cytometric analysis. The shown experiment is representative of 3 independent replicates.
Discussion
The present work shows that BRCA1 deficiency impairs the integrity of constitutive heterochromatin which leads to the disruption of gene silencing at the tandemly repeated DNA regions, probably through the loss of ubiquitination of histone H2A. BRCA1/BARD1 proteins preferentially mono-ubiquitylate H2A at high stoichiometry8, which comprises 5-15% of the total cellular nucleosomal histone H2A16. BRCA1 accumulates H2A-Ub at satellite DNA regions, which encompass ~3.45% of the mouse genome11 (Fig. 2f) and itself localizes there by ChIP experiments (Fig. 2e). The BRCA1 RING domain is essential for both satellite DNA binding and silencing. BARD1 is also required for repression of satellite repeats and loss of BARD1 is of no consequence if BRCA1 is already absent (Supplementary Fig. 4c). Although the exact function is not well understood, the emerging notion is that mono-ubiquitinated histone H2A is most frequently associated with transcriptionally repressive states9,17,18. Other ubiquityl ligases: Ring1A/B, of the polycomb repressive complex 1 (PRC1), and RNF8, a DNA repair ligase19,20 ubiquitylate H2A but do not contribute to the silencing of satellite repeats (Supplementary Fig. 6). Similarly, BAP1, a known BRCA1 binding protein, has been shown to antagonize the polycomb complex by deubiquitinating histone H2A21,22. Since a mimic of natural ubiquityl-H2A can rescue the most prominent BRCA1 deficiency defects, it suggests that ubiquitylation of histone H2A is likely to be the primary substrate of BRCA1 (Figs. 3 and Supplementary Fig. 9).
It is worth noting that overexpression of the satellite transcripts in the presence of wildtype BRCA1 partially phenocopies BRCA1 loss (Fig. 5), including centrosome amplification and γH2Ax foci formation. These observations suggest constitutive heterochromatin formation prevents DNA damage or a DNA damage response. The function of satellite transcripts is largely unknown. However, it has been known that the integrity of centromeric heterochromatin requires an unknown RNA component7 that could conceivably be satellite transcripts.
Tumor suppression by BRCA1 was presumed to occur via homologous recombination3. Recent work showed that cells expressing a mutant BRCA1 devoid of E3 ligase activity do not exhibit defects in homologous recombination13. Thus, the defect in genomic instability in BRCA1 null cells seems not to require this function although an intact RING domain is required for recovery from gamma irradiation23. Nevertheless, mutations that compromise the ubiquityl ligase activity of BRCA1 are cancer predisposing and constitute hotspots of mutational occurrences in patients23. Our work shows that satellite DNA repression indeed requires an intact BRCA1 RING domain, consistent with satellite DNA derepression observed in BRCA1 tumors (Fig. 2d). Indeed satellite transcripts in clinical BRCA1 patient tumors are upregulated (Fig. 4c). Since satellite transcripts when expressed ectopically can induce centrosome amplification, cell cycle check point defect and the DNA damage response, it fulfills the criteria to be a main effector of BRCA1 mediated tumorigenesis. Although it is presently unclear how these processes are elicited at a mechanistic level it is tempting to speculate that histone H2A ubiquitylation pathways are involved. Alternatively, the derepression of satellite might be elicited by some indirect “global effect” in trans that would not require H2A-Ub to be at the derepressed site. It is conceivable that induction of satellite transcripts could be developed into a marker of loss of heterozygosity for clinical diagnostic application. Recent work has shown that a variety of epithelial cancers overexpress satellite repeats that encompass up to 50% of cellular transcripts24. Although the pathological significance of this finding is unknown, our work suggests that these could contribute to the evolution of the cancer cell through the induction of genomic instability (Supplementary Fig. 14).
Methods Summary
Cell culture, virus infection, quantitative RT-PCR and ChIP experiments. Primary neural progenitor cells were isolated from adult mice containing one BRCA1 null allele and one floxed BRCA1 allele as described25. Embryonic neural progenitors were isolated and cultured as described26. Cultured cells were infected with retroviruses with an MOI of 5 for 12-h and harvested at the time indicated in corresponding figure legends. Cells were harvested with TRIZOL for RNA extraction. Reverse transcription was carried out using SuperScript III First-strand Synthesis System (Invitrogen). The quantitation of PCR products was analyzed with SYBR Green using ABI PRISM 7700 Sequence Detection system software (Applied Biosystems). Crosslinking and ChIP were performed as a modified version of a protocol described by the Clevers laboratory27 and supplementary information.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Material
Acknowledgments
The authors thank Barbara Miller for assistance in culturing NPC, Eugene Ke for discussion and analysis of Affymetrix data, Dr. Amiel Yanai for the BRCA1 targeting shRNA construct, Dr. Caroline Lilley for assistance with Western blotting, and Dr. Zhongsheng You for assistance with the LI-COR Odyssey Infrared Imaging System. We thank Dr. Anton Berns for his sustained interest in this work and providing mutant mice and materials and Dr. Miguel Vidal for providing mouse embryo fibroblasts containing conditional deletion allele of Ring 1B. QZ was supported by the California Breast Cancer Research Program and Ruth L. Kirschtein National Research Service Award. GMP was supported by a fellowship of the California Institute of Regenerative Medicine. HS is a recipient of ASPET-Merck fellowship. IMV is an American Cancer Society Professor of Molecular Biology, and holds the Irwin and Joan Jacobs Chair in Exemplary Life Science. This work was supported in part by grants from the NIH, Ipsen/Biomeasure, Sanofi Aventis, and the H.N. and Frances C. Berger Foundation. FHG is supported by NIH NS52842, NS50217, and the Lookout Fund.
Footnotes
The authors declare no competing financial interests.
QZ generated and QZ, NT and GMP maintained all the KO animals. GMP and QZ made the initial heterochromatin observation. QZ and AMH performed confocal microscopy experiments. GMP, QZ and NT performed ChIP experiments. RNA isolationand microarray experiments were performed by QZ, GMPand NT. Microdissection of murine brains were performed by GMP and AMH under the guidance of FHG. GMP designed the H2A-ubiquitin fusion experiments that were performed by QZ, GMP and NT. Satellite RNA experiments were designed by GMP and QZ and performed by QZ, GMP and NT. HS established the embryonic neural stem cell isolation and culture. PN obtained, isolated and curated the clinical patient samples, which were analyzed by QZ, GMP and NT. All other experiments were performed by QZ, GMP and NT. All experiments and experimental design was performed under the supervision of IMV. GMP, QZ and IMV wrote the manuscript.
References
- 1.Tutt A, Ashworth A. The relationship between the roles of BRCA1 genes in DNA repair and cancer predisposition. Trends in Molec Med. 2002;8:571–576. doi: 10.1016/s1471-4914(02)02434-6. [DOI] [PubMed] [Google Scholar]
- 2.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 3.Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010;11(2):138–148. doi: 10.1038/nrm2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pageau GJ, Hall LL, Ganesan S, Livingston DM, Lawrence JB. The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer. 2007;7(8):628–633. doi: 10.1038/nrc2172. [DOI] [PubMed] [Google Scholar]
- 5.Lane TF, et al. Expression of Brca1 is associated with terminal differentiation of ectodermally and mesodermally derived tissues in mice. Genes Dev. 1995;9:2712–2722. doi: 10.1101/gad.9.21.2712. [DOI] [PubMed] [Google Scholar]
- 6.Korhonen L, Brannvall K, Skoglosa Y, Lindholm D. Tumor suppressor gene BRCA-1 is expressed by embryonic and adult neural stem cells and involved in cell proliferation. J Neurosci Res. 2003;71(6):769–776. doi: 10.1002/jnr.10546. [DOI] [PubMed] [Google Scholar]
- 7.Maison C, Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat Rev Mol Cell Biol. 2004;5(4):296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- 8.Xia Y, Pao GM, Chen HW, Verma IM, Hunter T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem. 2003;278(7):5255–5263. doi: 10.1074/jbc.M204591200. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431(7010):873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 10.Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112(5):725–736. doi: 10.1016/s0092-8674(03)00123-5. [DOI] [PubMed] [Google Scholar]
- 11.Martens JH, et al. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. Embo J. 2005;24(4):800–812. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashizume R, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276:14537–14540. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 13.Reid LJ, et al. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc Natl Acad Sci U S A. 2008;105(52):20876–20881. doi: 10.1073/pnas.0811203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X, et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001;28(3):266–271. doi: 10.1038/90108. [DOI] [PubMed] [Google Scholar]
- 15.Sankaran S, Starita LM, Groen AC, Ko MJ, Parvin JD. Centrosomal microtubule nucleation activity is inhibited by BRCA1-dependent ubiquitination. Mol Cell Biol. 2005;25(19):8656–8668. doi: 10.1128/MCB.25.19.8656-8668.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui SI, Seon BK, Sandberg AA. Disappearance of a structural chromatin protein A24 in mitosis: implications for molecular basis of chromatin condensation. Proc Natl Acad Sci U S A. 1979;76(12):6386–6390. doi: 10.1073/pnas.76.12.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kallin EM, et al. Genome-wide uH2A localization analysis highlights Bmi1-dependent deposition of the mark at repressed genes. PLoS Genet. 2009;5(6):e1000506. doi: 10.1371/journal.pgen.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou W, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29(1):69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doil C, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136(3):435–446. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 20.Stewart GS, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136(3):420–434. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 21.Scheuermann JC, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen DE, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16(9):1097–1112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- 23.Ruffner H, Joazeiro CAP, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: Loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci (USA) 2001;98(9):5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ting DT, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011;331(6017):593–596. doi: 10.1126/science.1200801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19(19):8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakashima K, et al. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 1999;284(5413):479–482. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- 27.van Es JH, et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 2005;7(4):381–386. doi: 10.1038/ncb1240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.