

Abstract
Primary carnitine deficiency is caused by defective OCTN2 carnitine transporters encoded by the SLC22A5 gene. Lack of carnitine impairs fatty acid oxidation resulting in hypoketotic hypoglycemia, hepatic encephalopathy, skeletal and cardiac myopathy. Recently, asymptomatic mothers with primary carnitine deficiency were identified by low carnitine levels in their infant by newborn screening. Here we evaluate mutations in the SLC22A5 gene and carnitine transport in fibroblasts from symptomatic patients and asymptomatic women. Carnitine transport was significantly reduced in fibroblasts obtained from all patients with primary carnitine deficiency, but was significantly higher in the asymptomatic women’s than in the symptomatic patients’ fibroblasts (p<0.01). By contrast, ergothioneine transport (a selective substrate of the OCTN1 transporter, tested here as a control) was similar in cells from controls and patients with carnitine deficiency. DNA sequencing indicated an increased frequency of nonsense mutations in symptomatic patients (p<0.001). Expression of the missense mutations in CHO cells indicated that many mutations retained residual carnitine transport activity, with no difference in the average activity of missense mutations identified in symptomatic versus asymptomatic patients. These results indicate that cells from asymptomatic women have on average higher levels of residual carnitine transport activity as compared to that of symptomatic patients due to the presence of at least one missense mutation.
Keywords: carnitine deficiency, SLC22A5, OCTN2
INTRODUCTION
Primary carnitine deficiency (MIM# 212140) is an autosomal recessive disorder of the carnitine cycle that results in defective fatty acid oxidation (Roe and Ding, 2001; Scaglia and Longo, 1999). It has a frequency of about 1:40,000 newborns (Koizumi, et al., 1999; Wilcken, et al., 2003; Wilcken, et al., 2001), with extremely high prevalence in the Faroe Islands where about 5% of the population is a carrier for an abnormal allele (Lund, et al., 2007).
Primary carnitine deficiency is caused by defective activity of the OCTN2 carnitine transporter, resulting in urinary carnitine wasting, low serum carnitine levels, and decreased intracellular carnitine accumulation. Carnitine is essential for the transfer of long-chain fatty acids from the cytosol to mitochondria for subsequent β-oxidation. The lack of carnitine impairs the ability to use fat as energy source during periods of fasting or stress. This can result in an acute metabolic decompensation early in life with hypoketotic hypoglycemia, Reye syndrome, and sudden infant death, or in a more insidious presentation, later in life, with skeletal or cardiac myopathy (Longo, et al., 2006).
The gene for primary carnitine deficiency, SLC22A5 (MIM# 603377), encodes the novel organic cation transporter OCTN2 (Tamai, et al., 1998; Wu, et al., 1998). Heterogeneous mutations in the SLC22A5 gene have been identified in patients with primary carnitine deficiency (Nezu, et al., 1999; Tang, et al., 1999; Wang, et al., 1999). There are no prevalent mutations, although, in a few cases, the same mutation was found in unrelated patients (Lamhonwah, et al., 2002; Wang, et al., 2000a; Wang, et al., 2001; Wang, et al., 2000c; Wang, et al., 1999).
The OCTN2 transporter was identified by its homology to OCTN1, another organic cation transporter whose specific substrate is ergothioneine (Grundemann, et al., 2005). The OCTN1 and OCTN2 transporters are highly homologous, but have very different specificities (Amat di San Filippo, et al., 2003). In some species, OCTN1 has been reported to mediate carnitine transport (Tamai, et al., 2000), although this was not our experience with the human OCTN1 (Amat di San Filippo, et al., 2003).
The initial patients with primary carnitine deficiency presented either with an acute metabolic decompensation early in life or with severe cardiomyopathy in childhood (Wang, et al., 2000a; Wang, et al., 1999). The phenotype and age of onset was similar among the reported patients, irrespective of the type of mutation (missense or nonsense) identified. Different types of presentation have been observed within an individual family, indicating that other factors, intrinsic or environmental (likely fasting, recurrent infections, and others), were responsible for the different phenotype (Lamhonwah, et al., 2002; Wang, et al., 2001; Wang, et al., 2000c). With the advent of expanded newborn screening, infants with very low free carnitine (C0, the marker of primary carnitine deficiency) can be identified shortly after birth (Schimmenti, et al., 2007). In some cases, low carnitine in the infant is due to maternal carnitine deficiency with the infant being a carrier for this condition (El-Hattab, et al., 2010; Li, et al., 2010; Schimmenti, et al., 2007). Mothers with this condition are mostly asymptomatic, although there is anecdotal evidence that some feel better after initiation of carnitine therapy (Schimmenti, et al., 2007). It is unclear whether this apparently milder phenotype could have a genetic basis or it is simply related to lack of environmental stressors. Here we compare mutations in the SLC22A5 gene and function of the OCTN2 carnitine transporter in fibroblasts from patients who presented symptomatically and asymptomatic women with primary carnitine deficiency.
MATERIALS AND METHODS
Materials
PCR reaction buffers, deoxyribonucleotide triphosphates (dNTPs), and oligonucleotides were obtained from Idaho Technology (Salt Lake City, UT). KlenTaq1™ polymerase was obtained from AB Peptides (St Louis, MO) and TaqStart ™ antibody was obtained from BD Biosciences Clontech (Palo Alto, CA). Primers and all other conditions were as described previously (Wang, et al., 2001). To detect deletions and duplications in the SLC22A5 gene, a custom microarray for 13 genes involved in fatty acid oxidation was developed. The array was constructed on a NimbleGen 4×72K platform using unique oligomers with an average length of 53 base pairs. Probe coverage extended across all exons and introns of each gene and an additional 5kb both upstream and downstream of each genomic region. The median probe spacing was nine base pairs, providing a resolution of 100 base pairs. Self-self, within run, and between run hybridizations were performed to validate the array design. Two self-self hybridizations were performed to confirm probe validity, estimate a false positive rate, and to verify the parameters used for data analysis. The chip was validated using samples from patients with known chromosomal deletions and duplications.
Patients and Membrane Transport
All studies were approved by the IRB of the University of Utah. Patients were referred for diagnostic confirmation either after symptomatic presentation or after identification by newborn screening programs. Some of the patients reported in this study were previously individually described (Amat di San Filippo, et al., 2006; Dobrowolski, et al., 2005; Wang, et al., 2001; Wang, et al., 2000c; Wang, et al., 1999). We evaluated functional and molecular characteristics of all cell strains (n=14) from asymptomatic women available in our laboratory. An equal number of cell strains from symptomatic patients were obtained from our laboratory database. The 14 patients selected presented between 5 months and 6 years of age with cardiomyopathy (n=7), hypoglycemia (n=4) and Reye syndrome (n=3).
Fibroblasts from patients with primary carnitine deficiency were obtained by skin biopsy for diagnostic purposes. They were grown in Dulbecco-modified MEM supplemented with 15% fetal bovine serum. Chinese hamster ovary (CHO) cells were grown in Ham F12 medium supplemented with 6% fetal bovine serum. For comparison experiments, carnitine (0.5 μM) and ergothioneine (0.5 μM) transport were measured at 37 °C in adherent cells using the cluster-tray method (Wang, et al., 2000c). Nonsaturable transport was measured in the presence of 2 mM cold carnitine or ergothioneine and was subtracted from total transport to obtain saturable transport (Wang, et al., 2000c). Values are reported as means ± SE of 6 independent determinations obtained in two separate experiments. For all other experiments, conditions are reported in the text or figure legends.
Kinetic constants for ergothioneine (0.5–500 μM) transport were determined by nonlinear regression analysis according to the equation:
| (1) |
for a saturable transport system with superimposed diffusion, where [S] is the concentration of substrate, Vmax the maximal velocity of a saturable transporter, Km its Michaelis-Menten constant, and Kd the diffusion constant (Longo, et al., 1988). Non-linear parameters are expressed as means ± SE.
Data were also analyzed using a Michaelis-Menten equation (Amat Di San Filippo and Longo, 2004; Amat di San Filippo, et al., 2003) after subtraction of non-saturable ergothioneine transport (measured in the presence of 2 mM ergothioneine).
DNA Analysis and Molecular Techniques
Genomic DNA was extracted from fibroblasts or peripheral blood by standard methods. GenBank sequence AB016625.1 was used as reference for the gene, NM_003060.2 was used as the reference sequence for the cDNA. Nucleotide numbering uses the A of the ATG translation initiation start site as nucleotide +1. Frequency of missense and nonsense mutations in symptomatic and asymptomatic patients was compared using two-sided Fisher’s exact test (GraphPad).
Missense mutations were re-created in the OCTN2-EGFP expression vector by site-directed mutagenesis (Wang, et al., 2000a; Wang, et al., 2001; Wang, et al., 2000b; Wang, et al., 2000c; Wang, et al., 1999). The final clones were sequenced to confirm the presence of the mutation and the absence of PCR artifacts. Mutant cDNAs were stably transfected into CHO cells using lipofectamine and selection with G418 (0.8 mg/ml) for 14 days. Confocal microscopy was used to confirm protein production, since in our expression vector the OCTN2 transporter is tagged with the green fluorescent protein (Amat Di San Filippo and Longo, 2004; Wang, et al., 2000b).
RESULTS
Carnitine transport in human fibroblasts
Carnitine (0.5 μM) transport was significantly reduced in fibroblasts obtained from all patients with primary carnitine deficiency (Fig. 1). When data from multiple patients (Panel A) were combined, carnitine transport was significantly higher (7.4% of paired controls) in fibroblasts obtained from asymptomatic women with primary carnitine deficiency as compared to those of symptomatic patients (2.9% of paired control, p<0.01 versus asymptomatic women and controls, panel B). The significant difference in carnitine transport observed in cells from asymptomatic patients could be due to specific mutations retaining higher residual activity, a decreased prevalence of null alleles, or to a generalized increase in membrane transport activity. To distinguish among these possibilities, we evaluated the specific mutations in cells of our patients, expressed them in heterologous cells, and evaluated the activity of an unrelated, but similar membrane transporter (OCTN1).
Fig. 1.
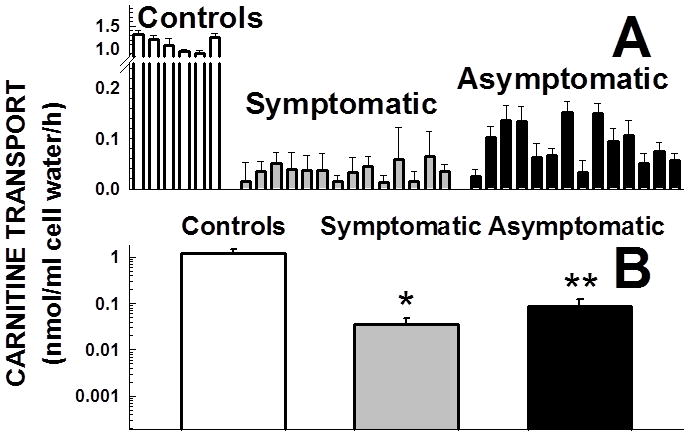
Carnitine transport by fibroblasts obtained from patients with primary carnitine deficiency. Carnitine (0.5 μM) transport was measured for 4 h at 37°C. Nonsaturable transport, measured in the presence of 2 mM cold carnitine, was subtracted from total transport to obtain saturable carnitine transport. Data are means± SE of 6 observations in two separate experiments. Panel A shows data for individual patients, panel B shows the average ± 99% confidence intervals for symptomatic patients and asymptomatic women. Note the logarithmic scale in panel B. *p<0.01 versus controls and mothers; **p<0.01 versus symptomatic patients and controls using 99% confidence intervals.
Mutational analysis of patients with primary carnitine deficiency
Table 1 lists the mutations identified in our patients with primary carnitine deficiency. In three patients (379, 417, 428), the second mutation could not be identified and no visible deletion was identified by microarrays. These patients’ cells had markedly reduced carnitine transport (Fig. 1A), consistent with primary carnitine deficiency and indicating that some mutations might escape detection. A review of the mutations identified indicated that while some of the symptomatic patients were homozygous or compound heterozygous for nonsense mutations, this was not the case for adult asymptomatic patients. Table 2 provides a summary of the types of mutations reported. Comparison of the frequency of mutations between symptomatic patients and asymptomatic women indicated an increased frequency of nonsense mutations in symptomatic patients (two sided p=0.0008 with Fisher’s exact test).
Table 1.
Mutations in the SLC22A5 carnitine transporter gene in patients with primary carnitine deficiency
| Patient | ID | Mutation 1 (Exon) | Mutation 2 (Exon) |
|---|---|---|---|
| SYMPTOMATIC PATIENTS | |||
| 1 | 1 | c.1202_1203insA, p.Y401X (7) | c.1302delG, p.G435fsX458 (8) |
| 2 | 2 | c.844C>T, p.R282X (5) | c.844C>T, p.R282X (5) |
| 3 | 23 | c.760C>T, p.R254X (4) | c.760C>T, p.R254X (4) |
| 4 | 16 | c.847T>A, p.W283R (4) | c. 847T>A, p.W283R (4) |
| 5 | 14 | c.12C>G, p.Y4X (1) | c.12C>G, p.Y4X (1) |
| 6 | 32 | c.64_66delTTC, p.ΔF22 (1) | c.64_66delTTC (1), p.ΔF22 (1) |
| 7 | 13 | c.1340A>G, p.Y447C (8) | c.1340A>G, p.Y447C (8) |
| 8 | 8 | c.56G>C, p.R19P (1) | c.254_264dup11, p.I89fsX133 (1) |
| 9 | 7 | c.505C>T, p.R169W (3) | c.1051T>C, p.W351R (6) |
| 10 | 26 | c.232delC, p.P78fsX129 (1) | c.232delC, p.P78fsX129 (1) |
| 11 | 329 | c.457delTG, p.V153fsX193 (2) | c.844C>T, p.R282X (5) |
| 12 | 330 | c.506G>A, p.R169Q (3) | c.506G>A, p.R169Q (3) |
| 13 | 19 | c.760C>T, p.R254X (4) | c.760C>T, p.R254X (4) |
| 14 | 20 | c.844C>T, p.R282X (5) | c.3G>T, p.M1I (1) |
| ASYMPTOMATIC WOMEN | |||
| 15 | 414 | c.1336G>T, p.V446F (8) | c.1412G>C, p.R471P (8) |
| 16 | 357 | c.95A>G, p.N32S (1) | c.136C>G, p.P46S(1) |
| 17 | 440 | c.136C>T, p.P46S (1) | c.695C>T, p.T232M (4) |
| 18 | 411 | c.1319C>T, p.T440M (8) | c.453G>A, p.V151V (2) |
| 19 | 428 | IVS1 -16T>A, c.394-16T>A | UNKNOWN |
| 20 | 379 | c.453G>A, p.V151V (2) | UNKNOWN |
| 21 | 400 | c.136C>T, p.P46S (1) | c.424G>T, p.A142S (2); c.1463G>A, p.R488H (9) |
| 22 | 426 | c.77G>A, p.S26N (1) | c.845G>A, p.R282Q (5) |
| 23 | 349 | c.641C>T, p.A214V (3) | c.641C>T, p.A214V (3) |
| 24 | 417 | c.839C>T, p.S280F (5) | UNKNOWN |
| 25 | 362 | c.136C>G, p.P46S (1) | c.1556_1559dupACAC, p.I521HX3 (9) |
| 26 | 363 | c.760C>T, p.R254X (4) | c.1400C>G, p.S467C (8) |
| 27 | 385 | c.1324-5GC>AT, p.A442I (8) | c.1324-5GC>AT, p.A442I (8) |
| 28 | 381 | c.43G>T, p.G15W (1) | c.248G>T, p.R83L (1) |
Patients 1–14 presented symptomatically, patients 15–28 were identified because of abnormal newborn screening results in their children. Mutations refer to the cDNA, using as reference sequence NM_003060.2. Nucleotide numbering uses the A of the ATG translation initiation start site as nucleotide +1. For intronic variations, the genomic reference sequence was AB016625.1.
Table 2.
Mutations in the SLC22A5 gene in patients with primary carnitine deficiency identified symptomatically or because of abnormal newborn screening results in their child (asymptomatic)
| NONSENSE/FRAMESHIFT | MISSENSE/IN FRAME DELETION | SPLICING/OTHERS | TOTAL | |
|---|---|---|---|---|
| SYMPTOMATIC | 16 | 12 | 0 | 28 |
| ASYMPTOMATIC | 2 | 20 | 6 | 28 |
| TOTAL | 18 | 32 | 6 | 56 |
Statistical analysis of missense and nonsense mutations indicates that nonsense mutations are more prevalent in the symptomatic group (p=0.0008 with Fisher’s exact test for two-tailed p value).
The missense mutations identified in symptomatic patients and asymptomatic women with primary carnitine deficiency were inserted in the OCTN2 cDNA and stably expressed in CHO cells. Fig. 2 summarizes the results obtained. All mutations significantly impaired carnitine transport (panel A). When the values of carnitine transport activity were grouped for type of patients, no significant difference were observed in the average transport activity measured in cells expressing mutations identified in symptomatic patients versus asymptomatic women (panel B). Therefore, the increased residual carnitine transport measured in fibroblasts from asymptomatic women was likely due to an increased frequency of missense mutations (Table 2) rather than the specific type of missense mutation (Fig. 2A).
Fig. 2.
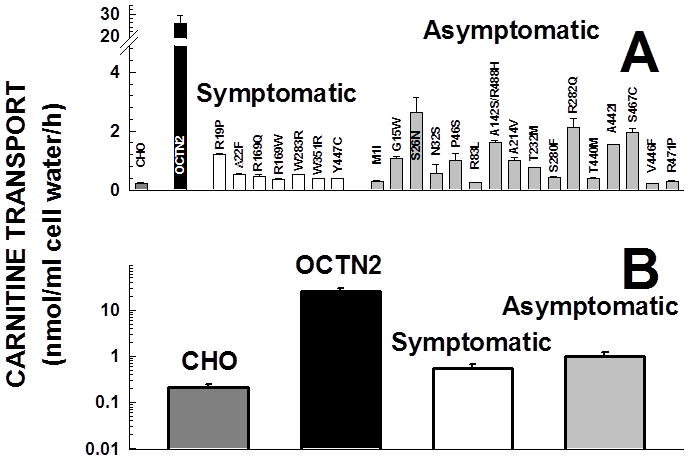
Carnitine transport by Chinese Hamster Ovary (CHO) cells expressing normal and mutant carnitine transporters. CHO cells were stably transfected with normal and mutant OCTN2 cDNA cloned in the pEGFP mammalian expression vector. After selection for resistance to G418 (0.8 mg/ml), expression of the trans-gene was verified by fluorescent microscopy. Carnitine (0.5 μM) transport was measured for 1 h (to measure initial entry rates) and corrected for nonsaturable uptake (measured in the presence of 2 mM cold carnitine). Points are averages ± SE of 6 samples. Panel A shows data for individual missense mutations, panel B shows the average for mutations identified in symptomatic patients and mothers. Note the logarithmic scale in panel B. In both groups, carnitine transport was significantly (p<0.01 using ANOVA) reduced as compared to wild type OCTN2, but there was no significant difference between the group of missense mutations identified in symptomatic patients versus asymptomatic women using ANOVA.
Ergothioneine transport in human fibroblasts and CHO cells
Ergothioneine is the physiological substrate of the OCTN1 transporter (Grundemann, et al., 2005), a transporter highly homologous to OCTN2 that maps to the same chromosomal region. To compare ergothioneine transport between cells of symptomatic and adult patients with primary carnitine deficiency, we had first to determine appropriate conditions to estimate initial entry rates of the compound in adherent cells. Fig. 3 shows the time course of ergothioneine uptake in human fibroblasts (Fig. 3). Ergothioneine transport was also evaluated in the presence of saturating concentrations of ergothioneine (2 mM) to obtain saturable ergothioneine transport. In human fibroblasts, there was a linear increase of saturable ergothioneine transport up to 4 h of incubation (Panels A, B). Nonsaturable ergothioneine transport, measured in the presence of 2 mM cold ergothioneine, reached a maximum after 2 h of incubation and then remained stable (Fig. 3). This suggested that, as in the case of carnitine, in the initial phases of the uptake process, a portion of ergothioneine might bind to cellular components. Once this process is saturated, only active transport is observed. In fact, when nonsaturable uptake was subtracted from total uptake, the saturable component was perfectly (R2=0.999) linear up to 4 h (Fig. 3B). Therefore, transport times of up to 4 h could be used to assess the initial rate of saturable ergothioneine entry into these cells.
Fig. 3.
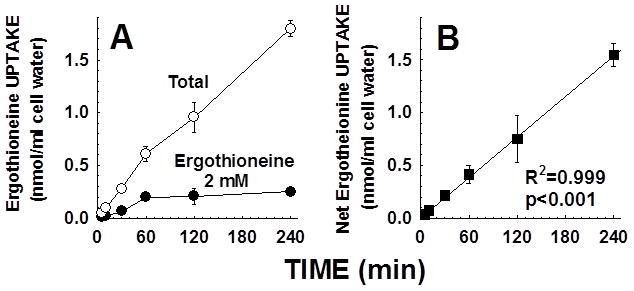
Time-course of ergothioneine transport in normal human fibroblasts. A. Ergothioneine (0.5 μM) transport was measured for the time indicated at 37°C in the absence (open circles) and presence of 2mM cold ergothioneine (nonsaturable transport, filled circles). B. Nonsaturable ergothioneine transport was subtracted from total transport to obtain saturable (net) ergothioneine transport. Data are means ± SE of 3observations. The line in panel B is a linear regression passing for the origin with the parameters indicated.
Fig. 4 shows the kinetic constants for ergothioneine (0.5–500μM) transport in normal human fibroblasts. Total ergothioneine uptake (panel A) could be explained by the operation of a single saturable transporter and a linear component, formally indistinguishable from diffusion or binding to cellular components. Fitting of transport data to a kinetic model composed of a single saturable transporters and a linear component (Equation 1 in methods) generated the constants reported in Fig. 4. If nonspecific transport measured in the presence of 2 mM unlabeled substrate was subtracted from total uptake, kinetic analysis according to a Michaelis-Menten equation confirmed Km values around 45μM for human fibroblasts. The values of Km value measured using total ergothioneine transport or subtraction of saturating concentrations of substrate were not significantly different (using 95% confidence intervals). These values are in the same range of those seen in other cell systems overexpressing the OCTN1 transporter after subtraction of endogenous transport activity (21±5 μM in HEK-293 cells (Grundemann, et al., 2005), 4.6±0.7 μM in for the rat OCTN1 expressed in HEK293 cells (Nakamura, et al., 2008), 26±9 μM for PC12 cells (Nakamura, et al., 2008)). These results indicate that the conditions for measuring OCTN1 activity in human fibroblasts were appropriate and comparable to those of other studies.
Fig. 4.
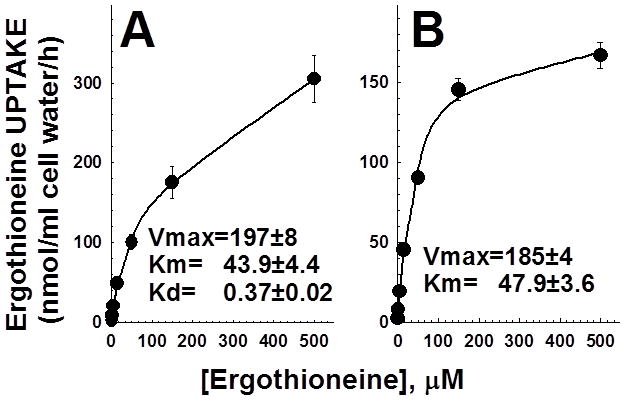
Kinetic constants for ergothioneine transport in human fibroblasts. Ergothioneine (0.5–500 μM) uptake was measured for 60 min at 37°C in normal human fibroblasts. Points are averages of triplicates ± SD. A. Total ergothioneine transport was fitted to equation 1 (see methods) for a single saturable transporter with superimposed diffusion. Lines show the best fit to total ergothioneine uptake. B. Ergothioneine transport was corrected for that measured in the presence of 2 mM cold ergothioneine to correct for diffusion. Data were fitted to a Michaelis-Menten equation. Parameters are shown in the figure as averages ± SE. Units for the parameters are: Vmax= nmol/ml cell water/h; Km=μM; Kd=h−1.
Ergothioneine transport by fibroblasts from patients with primary carnitine deficiency
Fig. 5 summarizes OCTN1 transport activity, assessed as saturable ergothioneine transport, in fibroblasts from normal controls and patients with primary carnitine deficiency. Ergothioneine transport (OCTN1 activity) was similar in cells from controls and patients with carnitine deficiency, with no significant difference between the group of symptomatic patients and asymptomatic women (p>0.05 using ANOVA).
Fig. 5.
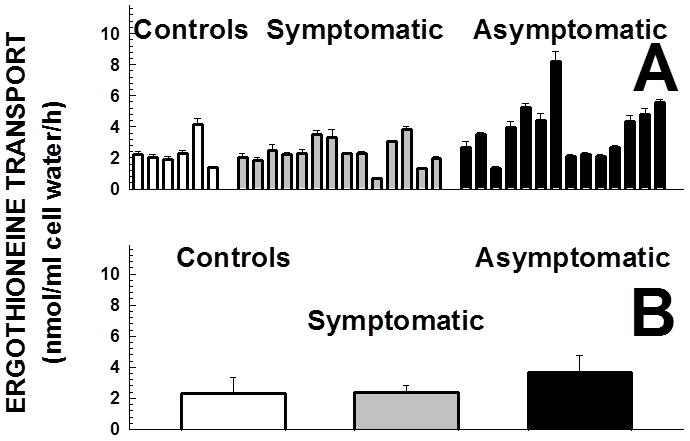
Ergothioneine transport by fibroblasts obtained from patients with primary carnitine deficiency. Ergothioneine (0.5 μM) transport was measured for 4 h at 37°C. Nonsaturable transport, measured in the presence of 2 mM cold ergothioneine, was subtracted from total transport to obtain saturable ergothioneine transport. Data are means ± SE of 6 observations from two separate experiments. Panel A shows data for individual patients, panel B shows the average for symptomatic patients and asymptomatic women. In panel B, there were no significant differences among groups using ANOVA.
DISCUSSION
Primary carnitine deficiency is an autosomal recessive disorder that impairs fatty acid oxidation. It can present in children of different ages with hypoketotic hypoglycemia, cardiomyopathy and/or skeletal myopathy. This disease is suspected based on reduced levels of carnitine in plasma and confirmed by measurement of carnitine transport in the patient’s fibroblasts. Newborn screening programs can identify patients with primary carnitine deficiency finding low levels of free carnitine (C0) at birth. Low levels of other acylcarnitines (C3, C16, C18) are also usually detected. Low levels of free carnitine are also seen in infants of mothers with primary and secondary (glutaric acidemia type 1, medium chain acylCoA dehydrogenase deficiency, cystic fibrosis) carnitine deficiency. Mothers with primary carnitine deficiency are usually asymptomatic, but are at risk of sudden death from arrhythmia (Schimmenti, et al., 2007). It is unclear why these women remain asymptomatic while other patients with the same disease can present early in life.
Here we show that cells from these asymptomatic women had on average more than twice residual carnitine transport activity as compared to cells from patients presenting symptomatically (Fig. 1). DNA sequencing indicated that none of the asymptomatic women was homozygous or compound heterozygous for two null alleles (Table 1). The presence of two null alleles for the carnitine transporter has not been reported in any of the asymptomatic women or adult patients described to date (El-Hattab, et al., 2010; Lee, et al., 2010; Li, et al., 2010; Schimmenti, et al., 2007; Spiekerkoetter, et al., 2003) whereas it has been observed in several patients who presented symptomatically (Lamhonwah, et al., 2002; Wang, et al., 2001; Wang, et al., 2000c; Wang, et al., 1999). The analysis of our patients indicates that the frequency of nonsense mutations is significantly increased in patients presenting symptomatically as compared to asymptomatic women (Table 2). This is the likely explanation for the increased residual carnitine transport observed in fibroblasts from asymptomatic women (Fig. 1). In fact, the residual activity of missense mutations identified in the two groups of patients was similar when missense mutations were expressed in CHO cells (Fig. 2). While no asymptomatic woman was homozygous or compound heterozygous for two null alleles in the carnitine transporter gene, some of the mutations identified in this group were previously described in symptomatic patients. This is the case for all null alleles and for some of the missense mutations (p.N32S, p.R83L, p.T440M, p.V446F, the combination p.A142S/p.R488H) (Amat di San Filippo, et al., 2006; Dobrowolski, et al., 2005; Lamhonwah, et al., 2002; Mayatepek, et al., 2000). However, none of the symptomatic women was compound heterozygote for missense mutations previously identified in symptomatic patients or one of these and a null allele. A specific mutation, p.P46S, has only been identified in asymptomatic women. This mutation reduces, but does not abolish carnitine transport and affects glycosylation and maturation to the plasma membrane of the OCTN2 carnitine transporter (Filippo, et al., 2011). It is possible that this and other similar missense mutations are protective against early clinical manifestations in primary carnitine deficiency. While this is the general case, there are exceptions to this rule. For example, a symptomatic infant and an asymptomatic father with the same identical mutational (p.R471H) were reported within a single-family (Spiekerkoetter, et al., 2003). Also, from our own data, carnitine transport in cells from some symptomatic individuals is higher than that of cells from some asymptomatic women (Fig. 1). Clearly, in addition to the nature of the specific mutation identified in our study, environmental factors or other genes still play an important role in triggering symptoms in some patients.
Other membrane transporters possibly recognizing carnitine could have had a compensatory increased activity in cells from asymptomatic women and explain lack of symptoms. The OCTN1 and OCTN2 transporters are highly homologous, and, in some species, OCTN1 mediates carnitine transport (Tamai, et al., 2000). OCTN1 transports ergothioneine, a natural antioxidant and a scavenger of hydroxyl radicals produced by several bacteria and fungi (Grundemann, et al., 2005). Human fibroblasts possess the OCTN1 transporter and transport ergothioneine in a saturable manner (Figs. 3 and 4). The kinetic constants for ergothioneine transport by human fibroblasts were in the same order of magnitude of those observed for the OCTN1 transporter when expressed in other cells (Grundemann, et al., 2005; Nakamura, et al., 2008). There were no significant differences in ergothioneine transport between control cells and cells from patients with primary carnitine deficiency (Fig. 5). In addition, ergothioneine transport was not significantly different between fibroblasts of symptomatic patients as compared to asymptomatic women (Fig. 5), indicating that asymptomatic patients did not have a generalized increase in the activity of other membrane transporters that could compensate for decreased carnitine transport. Therefore, the increased residual carnitine transport activity of the OCTN2 transporter (Fig. 1) due to a decreased prevalence of nonsense mutations (Table 1 and 2) is likely associated with the asymptomatic status of these women.
Previous studies found no association between genotype and phenotype in primary carnitine deficiency (Lamhonwah, et al., 2002; Wang, et al., 2001; Wang, et al., 2000c; Wang, et al., 1999). In these studies, however, correlation was searched with type and timing of clinical presentation in children. Here, a new phenotype (patients relatively asymptomatic of adult age) correlated with milder mutations. This phenotype was not known at the time the disease was initially described.
Previous studies have shown that ever heterozygotes for primary carnitine deficiency can have left ventricular hypertrophy (Koizumi, et al., 1999) or other cardiac symptoms that improve with carnitine supplementation (Sarafoglou, et al., 2010). On the other hand, no increased rate of mutations in the carnitine transporter gene was found in an unselected populations of adults with cardiomyopathy (Amat di San Filippo, et al., 2008). Here we find that the pathological significance of markedly decreased carnitine transport activity (consistent with compound heterozygosity in the carnitine transporter gene) in cells from asymptomatic women is still unclear. Some of these mothers have cardiac involvement, such as cardiomyopathy (Lee, et al., 2010) or cardiac arrhythmia with long QT syndrome, that can result in cardiac arrest and syncope (Schimmenti, et al., 2007) preventable by carnitine supplementation (De Biase, et al., 2011). Other women have easy fatigability that seems the most common symptom in this group of women (De Biase, et al., 2011). Retrospective and prospective studies of this patient population is necessary to determine the real clinical significance and the need for therapy for attenuated forms of primary carnitine deficiency caused by milder mutations in the SLCA22A5 gene.
Acknowledgments
Supported by grant R01 DK 53824 from the National Institutes of Health.
References
- Amat Di San Filippo C, Longo N. Tyrosine Residues Affecting Sodium Stimulation of Carnitine Transport in the OCTN2 Carnitine/Organic Cation Transporter. J Biol Chem. 2004;279:7247–53. doi: 10.1074/jbc.M309171200. [DOI] [PubMed] [Google Scholar]
- Amat di San Filippo C, Pasquali M, Longo N. Pharmacological rescue of carnitine transport in primary carnitine deficiency. Hum Mutat. 2006;27:513–23. doi: 10.1002/humu.20314. [DOI] [PubMed] [Google Scholar]
- Amat di San Filippo C, Taylor MR, Mestroni L, Botto LD, Longo N. Cardiomyopathy and carnitine deficiency. Mol Genet Metab. 2008;94:162–6. doi: 10.1016/j.ymgme.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat di San Filippo C, Wang Y, Longo N. Functional domains in the carnitine transporter OCTN2, defective in primary carnitine deficiency. J Biol Chem. 2003;278:47776–84. doi: 10.1074/jbc.M307911200. [DOI] [PubMed] [Google Scholar]
- De Biase I, Champaigne N, Schroer R, Pollard L, Longo N, Wood T. Primary carnitine deficiency presents atypically with long QT syndrome: A case report. Journal Inherited Metabolic Diseases. 2011 doi: 10.1007/8904_2011_52. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski SF, McKinney JT, Amat di San Filippo C, Giak Sim K, Wilcken B, Longo N. Validation of dye-binding/high-resolution thermal denaturation for the identification of mutations in the SLC22A5 gene. Hum Mutat. 2005;25:306–313. doi: 10.1002/humu.20137. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Li FY, Shen J, Powell BR, Bawle EV, Adams DJ, Wahl E, Kobori JA, Graham B, Scaglia F, et al. Maternal systemic primary carnitine deficiency uncovered by newborn screening: clinical, biochemical, and molecular aspects. Genet Med. 2010;12:19–24. doi: 10.1097/GIM.0b013e3181c5e6f7. [DOI] [PubMed] [Google Scholar]
- Filippo CA, Ardon O, Longo N. Glycosylation of the OCTN2 carnitine transporter: study of natural mutations identified in patients with primary carnitine deficiency. Biochim Biophys Acta. 2011;1812:312–20. doi: 10.1016/j.bbadis.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundemann D, Harlfinger S, Golz S, Geerts A, Lazar A, Berkels R, Jung N, Rubbert A, Schomig E. Discovery of the ergothioneine transporter. Proc Natl Acad Sci U S A. 2005;102:5256–61. doi: 10.1073/pnas.0408624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi A, Nozaki J, Ohura T, Kayo T, Wada Y, Nezu J, Ohashi R, Tamai I, Shoji Y, Takada G, et al. Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum Mol Genet. 1999;8:2247–54. doi: 10.1093/hmg/8.12.2247. [DOI] [PubMed] [Google Scholar]
- Lamhonwah AM, Olpin SE, Pollitt RJ, Vianey-Saban C, Divry P, Guffon N, Besley GT, Onizuka R, De Meirleir LJ, Cvitanovic-Sojat L, et al. Novel OCTN2 mutations: no genotype-phenotype correlations: early carnitine therapy prevents cardiomyopathy. American journal of medical genetics. 2002;111:271–84. doi: 10.1002/ajmg.10585. [DOI] [PubMed] [Google Scholar]
- Lee NC, Tang NL, Chien YH, Chen CA, Lin SJ, Chiu PC, Huang AC, Hwu WL. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol Genet Metab. 2010;100:46–50. doi: 10.1016/j.ymgme.2009.12.015. [DOI] [PubMed] [Google Scholar]
- Li FY, El-Hattab AW, Bawle EV, Boles RG, Schmitt ES, Scaglia F, Wong LJ. Molecular spectrum of SLC22A5 (OCTN2) gene mutations detected in 143 subjects evaluated for systemic carnitine deficiency. Hum Mutat. 2010;31:E1632–51. doi: 10.1002/humu.21311. [DOI] [PubMed] [Google Scholar]
- Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C:77–85. doi: 10.1002/ajmg.c.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo N, Griffin LD, Elsas LJ. Influx and efflux of 3-O-methyl-D-glucose by cultured human fibroblasts. Am J Physiol. 1988;254:C628–33. doi: 10.1152/ajpcell.1988.254.5.C628. [DOI] [PubMed] [Google Scholar]
- Lund AM, Joensen F, Hougaard DM, Jensen LK, Christensen E, Christensen M, Norgaard-Petersen B, Schwartz M, Skovby F. Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. Journal of inherited metabolic disease. 2007;30:341–9. doi: 10.1007/s10545-007-0527-9. [DOI] [PubMed] [Google Scholar]
- Mayatepek E, Nezu J, Tamai I, Oku A, Katsura M, Shimane M, Tsuji A. Two novel missense mutations of the OCTN2 gene (W283R and V446F) in a patient with primary systemic carnitine deficiency. Hum Mutat. 2000;15:118. doi: 10.1002/(SICI)1098-1004(200001)15:1<118::AID-HUMU28>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Yoshida K, Yabuuchi H, Maeda T, Tamai I. Functional characterization of ergothioneine transport by rat organic cation/carnitine transporter Octn1 (slc22a4) Biol Pharm Bull. 2008;31:1580–4. doi: 10.1248/bpb.31.1580. [DOI] [PubMed] [Google Scholar]
- Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, Nikaido H, Sai Y, Koizumi A, Shoji Y, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21:91–4. doi: 10.1038/5030. [DOI] [PubMed] [Google Scholar]
- Roe C, Ding J. Mitochondrial fatty acid oxidation disorders. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 2297–2326. [Google Scholar]
- Sarafoglou K, Tridgell AH, Bentler K, Redlinger-Grosse K, Berry SA, Schimmenti LA. Cardiac conduction improvement in two heterozygotes for primary carnitine deficiency on L-carnitine supplementation. Clin Genet. 2010;78:191–4. doi: 10.1111/j.1399-0004.2009.01368.x. [DOI] [PubMed] [Google Scholar]
- Scaglia F, Longo N. Primary and secondary alterations of neonatal carnitine metabolism. Semin Perinatol. 1999;23:152–61. doi: 10.1016/s0146-0005(99)80047-0. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, Crombez EA, Schwahn BC, Heese BA, Wood TC, Schroer RJ, Bentler K, Cederbaum S, Sarafoglou K, McCann M, et al. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol Genet Metab. 2007;90:441–5. doi: 10.1016/j.ymgme.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Huener G, Baykal T, Demirkol M, Duran M, Wanders R, Nezu J, Mayatepek E. Silent and symptomatic primary carnitine deficiency within the same family due to identical mutations in the organic cation/carnitine transporter OCTN2. Journal of inherited metabolic disease. 2003;26:613–5. doi: 10.1023/a:1025968502527. [DOI] [PubMed] [Google Scholar]
- Tamai I, Ohashi R, Nezu J, Yabuuchi H, Oku A, Shimane M, Sai Y, Tsuji A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273:20378–82. doi: 10.1074/jbc.273.32.20378. [DOI] [PubMed] [Google Scholar]
- Tamai I, Ohashi R, Nezu JI, Sai Y, Kobayashi D, Oku A, Shimane M, Tsuji A. Molecular and functional characterization of organic cation/carnitine transporter family in mice. J Biol Chem. 2000;275:40064–72. doi: 10.1074/jbc.M005340200. [DOI] [PubMed] [Google Scholar]
- Tang NL, Ganapathy V, Wu X, Hui J, Seth P, Yuen PM, Wanders RJ, Fok TF, Hjelm NM. Mutations of OCTN2, an organic cation/carnitine transporter, lead to deficient cellular carnitine uptake in primary carnitine deficiency. Hum Mol Genet. 1999;8:655–60. doi: 10.1093/hmg/8.4.655. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kelly MA, Cowan TM, Longo N. A missense mutation in the OCTN2 gene associated with residual carnitine transport activity. Hum Mutat. 2000a;15:238–45. doi: 10.1002/(SICI)1098-1004(200003)15:3<238::AID-HUMU4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Korman SH, Ye J, Gargus JJ, Gutman A, Taroni F, Garavaglia B, Longo N. Phenotype and genotype variation in primary carnitine deficiency. Genet Med. 2001;3:387–92. doi: 10.1097/00125817-200111000-00002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Meadows TA, Longo N. Abnormal sodium stimulation of carnitine transport in primary carnitine deficiency. J Biol Chem. 2000b;275:20782–6. doi: 10.1074/jbc.M000194200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Taroni F, Garavaglia B, Longo N. Functional analysis of mutations in the OCTN2 transporter causing primary carnitine deficiency: lack of genotype-phenotype correlation. Hum Mutat. 2000c;16:401–7. doi: 10.1002/1098-1004(200011)16:5<401::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ye J, Ganapathy V, Longo N. Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc Natl Acad Sci U S A. 1999;96:2356–60. doi: 10.1073/pnas.96.5.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348:2304–12. doi: 10.1056/NEJMoa025225. [DOI] [PubMed] [Google Scholar]
- Wilcken B, Wiley V, Sim KG, Carpenter K. Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J Pediatr. 2001;138:581–4. doi: 10.1067/mpd.2001.111813. [DOI] [PubMed] [Google Scholar]
- Wu X, Prasad PD, Leibach FH, Ganapathy V. cDNA sequence, transport function, and genomic organization of human OCTN2, a new member of the organic cation transporter family. Biochem Biophys Res Commun. 1998;246:589–95. doi: 10.1006/bbrc.1998.8669. [DOI] [PubMed] [Google Scholar]