

Abstract
Neuronal nicotinic receptors have been implicated in several diseases and disorders such as: autism, Alzheimer’s disease, Parkinson’s disease, epilepsy, and various forms of addiction. To understand the role of nicotinic receptors in these conditions, it would be beneficial to have selective molecules that target specific nicotinic receptors in vitro and in vivo. Our laboratory has previously identified novel negative allosteric modulators of human α4β2 (Hα4β2) and human α3β4 (Hα3β4) nicotinic receptors. In the following studies, the effects of novel sulfonylpiperazine analogs that act as negative allosteric modulators on both Hα4β2 nAChRs and Hα3β4 nAChRs were investigated. This work, through structure-activity relationship (SAR) studies, describes the chemical features of these molecules that are important for both potency and selectivity on Hα4β2 nAChRs.
INTRODUCTION
Neuronal nicotinic acetylcholine receptors (nAChRs) are associated with many important physiological mechanisms (i.e., cognition, arousal, pain sensation), as well as a number of neurological diseases and disorders (depression, schizophrenia, Alzheimer’s disease, Parkinson’s disease, lung cancer, Tourette’s syndrome, autism, and addiction).1,2,3,4 nAChRs are notable for their implications in the addictive properties of nicotine, the primary addictive component of tobacco products.5 nAChRs are ligand-gated ion channels composed of five protein subunits encoded by a family of related but distinct genes.6,7 Multiple nAChR subtypes have been described based on subunit (α2 – α10 and β2 – β4) composition, where the three most prominent subtype compositions in the CNS are α4β2, α3β4 and α7.8 The specific nAChR subtypes involved with most of the above mentioned physiological processes or diseases are not known, and their study is challenging due to the complex composition of some nAChRs (α4α6β2, α3α5β4, etc.).9,10 Despite the lack of understanding concerning specific subtypes of nAChRs in diseases and disorders, it is widely accepted that nicotine addiction is mediated primarily by α4β2 containing nAChRs.11,12 The discovery of novel, selective molecules that target specific subtypes of nAChRs would contribute significantly to our understanding of the role nAChR subtypes play in normal and pathophysiological states, and prove beneficial for the clinical treatment of several neuropathologies.
Worldwide, nicotine addiction is a significant problem. Smoking is the primary cause of preventable death worldwide and roughly 90% of the people who attempt to quit are unable to do so.13 Current FDA approved treatments for tobacco addiction are nicotine replacement, buproprion, and varenicline. Each of these therapies has a modest success of 20%–30% abstinence one year after quit date.14,15,16 The target site of many nAChR drug discovery programs is the orthosteric (agonist binding) site of nAChRs9,17 and few laboratories have had some success in identifying molecules that show some selectivity using this approach.18,19 However, one difficulty with this strategy is the high degree of amino acid sequence homology among nAChR α and β subunits in the ligand binding domain for acetylcholine, making it difficult to develop drugs which specifically target nAChR subtypes.9,17 Thus, the development of selective nAChR drugs directed at orthosteric sites progresses slowly. For this reason, drugs targeting “non-orthosteric” sites of nAChRs (e.g., allosteric, noncompetitive sites) is an approach now taken by many laboratories.20,21,22
Mecamylamine, a non-selective non-competitive nAChR antagonist, was shown to promote 40% abstinence at the year mark when used as an agonist-antagonist therapy in combination with the nicotine patch.23 In addition, antagonists of α4β2 nAChRs block nicotine self-administration on fixed and progressive ratio schedules in rats.20,24 These data support the use of antagonists as nicotine cessation therapies; however, to produce new therapeutic molecules it has widely been accepted that nAChR subtype selectivity must be pursued.9 Selectivity is especially important for avoiding effects on α3β4 nAChRs, as they are the prominent nAChR subtype in the peripheral nervous system.9,10 Furthermore, it has been postulated that this nAChR subtype mediates many of the adverse affects associated with therapeutics that target nAChRs.14,23 Our laboratory has identified a novel allosteric site on Hα4β2 nAChRs that is approximately 10 Å from the orthosteric site at the interface of the α and β subunits.25 This site was identified through a combination of homology modeling, blind docking, and molecular dynamics studies, and was confirmed through the use of site-directed mutagenesis.25,26 At this site, a novel class of NAMs has been shown to have relative-selectivity for Hα4β2 nAChRs (over Hα3β4 nAChRs) principally through non-conserved amino acids on the β2 subunit. Through SAR we have identified several physiochemical features that mediate relative-selectivity for Hα4β2 nAChRs. These include specific placement of aromatic residues, specific orientation of the keto group of esters or amides, and the specific length of carbon chains. Using the allosteric site that was identified through computational modeling,25,26 a small library of diverse molecules were computationally docked to identify novel chemical scaffolds that are active as nAChR NAMs. This structure-based virtual screening (SBVS) approach resulted in the identification of four novel scaffolds that show preference for Hα4β2 nAChRs and act as inhibitors of nAChRs.27 One of these SBVS hits has a sulfonylpiperazine scaffold unique among previously described nAChR NAMS. Here in these studies we present the SAR of novel sulfonyl analogs on Hα4β2 and Hα3β4 nAChRs and describe the physical and chemical features that are important for both relative-selectivity and potency on Hα4β2 nAChRs.
CHEMISTRY
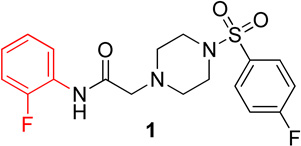
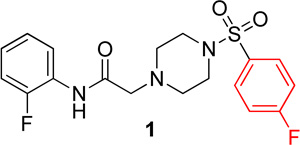
We have previously reported that SBVS has yielded active hits that target Hα4β2 nAChRs.27 A hit molecule (1) has been selected as a scaffold for the development of a focused library of compounds. Lead compound 1 was assembled through a convergent synthesis allowing for facile access to analogs. Retrosynthetically, compound 1 was formed from displacement of bromide 2 by arylsulfonyl piperazine 3 (Scheme 1). The two coupling partners were assembled from commercially available starting materials, where amide 2 resulted from acylation of o-fluoroaniline (4) with bromoacetyl bromide (5) and piperazine 3 resulted from sulfonylation of piperazine (6) with p-fluorobenzenesulfonyl chloride (7).
Scheme 1.

Retrosynthetic analysis of lead compound 1.
This synthetic plan allowed access to a small library of compounds simply by using various substituted anilines and arylsulfonyl chlorides. Analogs were thus available in two linear steps (three total) from commercially available reagents. In practice, the synthetic plan was executed as planned where o-fluoroaniline (4) was acylated with bromoacetyl bromide (5) in the presence of triethylamine to yield amide 2 (Scheme 2).28 In addition to o-fluoroaniline (4), five other aniline derivatives, 3-aminopyrazole, and o-phenylphenol were reacted in a similar manner to access a variety of α-bromoamides and an α-bromoester in unoptimized yields ranging from 26% to 96%.29
Scheme 2.

Synthesis of left hand α-bromoamides.
The piperazine fragment 3 was obtained in high yield from the reaction of p-fluorobenzenesulfonyl chloride with excess piperazine (Scheme 3).30 Four other arylsulfonyl chlorides were reacted with piperazine to obtain the arylsulfonyl piperazine right hand fragments needed to assemble analogs.
Scheme 3.

Synthesis of right hand arylsulfonyl piperazines.
With the left side bromide (2) and right side piperazine (3) in hand, the two fragments were coupled in the presence of sodium carbonate to obtain lead compound 1 (Scheme 4). The various other left and right side fragments were mixed and matched to obtain analogs 12–30 (Tables 1–5) in unoptimized yields varying from 34 to 89%.
Scheme 4.

Coupling of left side bromide with right side piperazine.
Table 1.
Series 1 SAR Studies
| Hα4β2 nAChRs | Hα3β4 nAChRs | |||
|---|---|---|---|---|
| Compound | IC50 Value (µM)a | nhb | IC50 Value (µM)a | nhb |
 |
9.3 (6.2–13.9) | −1.2 | 9.0 (6.3–12.9) | −1.2 |
 |
>100c | ~ | >100c | ~ |
 |
21.1 (15.7–28.4) | −0.9 | 29.6 (18.9–46.3) | −1.1 |
 |
13.8 (10.7–17.8) | −0.9 | 11.6 (5.5–24.5) | −1.1 |
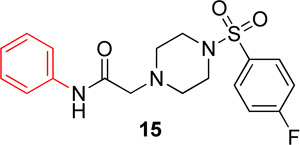 |
14.9 (11.5–19.4) | −1.4 | 18.5 (15.6–21.9) | −1.2 |
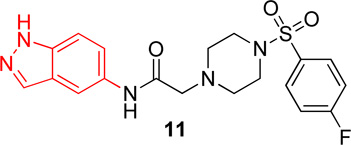 |
6.6 (3.8–11.3) | −1.0 | 23.8 (13.1–43.3) | −0.8 |
Values represent geometric means (confidence limits), n = 5–10
nh, Hill coefficient
No activity up to 100 µM
Table 5.
Series 5 SAR Studies
| Hα4β2 nAChRs | Hα3β4 nAChRs | |||
|---|---|---|---|---|
| Compound | IC50 Value (µM)a | nhb | IC50 Value (µM)a | nhb |
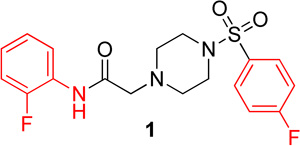 |
9.3 (6.2–13.9) | −1.2 | 9.0 (6.3–12.9) | −1.2 |
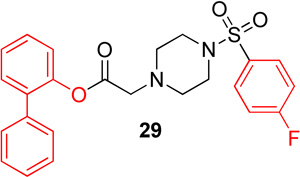 |
12.2 (6.3–23.8) | −1.1 | 23.8 (13.1–43.3) | −0.9 |
 |
8.0 (5.4–11.8) | −1.5 | 99.8 (57.2–174) | −0.6 |
 |
20.9 (12.7–34.3) | −1.2 | 23.5 (20.3–27.2) | −1.1 |
Values represent geometric means (confidence limits), n = 5–9
nh, Hill coefficient
The only analog not accessible through this convergent method was aminoindazole analog 11. The inability to cleanly acylate 5-aminoindazole (10) with bromoacetyl bromide (5) lead to a linear approach to obtain aminoindazole 11. To this end, bromoacetic acid (8) was reacted in the presence of triethylamine with p-fluorobenzenesulfonyl piperazine (3) to provide acid 9. Standard amide coupling conditions (EDCI, HOBt) were then employed to obtain analog 11 in modest 46% yield (Scheme 5).31
Scheme 5.

Linear synthesis of aminoindazole 11.
RESULTS
Series 1 SAR
Lead compound 1 produced inhibition on both Hα4β2 and Hα3β4 nAChRs with IC50 values of 9.3 µM and 9.0 µM, respectively (Table 1). The right side p-fluorobenzenesulfonyl piperazine (3) had no effect on either Hα4β2 or Hα3β4 nAChRs (data not shown). When tested for competitive or non-competitive activity on Hα4β2 and Hα3β3 nAChRs, lead compound 1 reduced the maximum efficacy for the agonist epibatidine (Figure 1). If the compounds acted at the agonist binding site, they would compete directly with the agonist, epibatidine, at its binding. This would produce a parallel shift of the concentration-response curve to the right. Compound 1 did not cause a parallel shift; but instead decreased the maximum efficacy of epibatidine. According to classical drug receptor theory, this result is indicative of non-competitive inhibition (e.g., the binding at a site other than the agonist site). Therefore, this result suggests that compound 1 modulates nAChRs from an allosteric site. As outlined in the chemistry section, analogs of 1 were synthesized. None of the compounds described here showed agonist activity on either Hα4β2 or Hα3β4 nAChRs in the calcium accumulation assay (data not shown).
Figure 1. Concentration-response effects of epibatidine in the absence and presence of Compound 1.

Calcium accumulation assays were performed as described in the methods section. The concentration-response effects of ebibatidine were investigated in the absence (■) or presence (□) of 30 µM 1 (~ IC70) by using HEK ts201 cells expressing Hα4β2 nAChRs (A) and Hα3β4 nAChRs (B). Values represent means ± SEMs (n= 4).
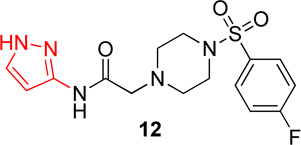
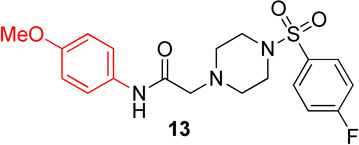
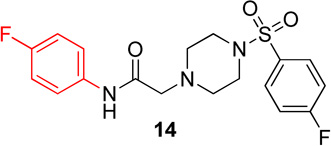
In the first series of analogs, the amide portion of lead compound 1 was modified using several different substitutions (Table 1). This study was conducted to determine the synthetic flexibility of the amide portion and to determine how these changes effect potency on nAChRs. The pyrazole analog (12) produced a significant decrease in potency for both Hα4β2 and Hα3β4 nAChRs (IC50 value, >100 µM, Table 1). The methoxy analog (13) produced a 2-fold decrease in potency for Hα4β2 nAChRs (IC50 value, 21.1 µM, Table 1) and a 3-fold decrease in potency for Hα3β4 nAChRs (IC50 value, 29.6 µM, Table 1). The p-fluorophenyl analog (14) produced no significant change in potency for either Hα4β2 or Hα3β4 nAChRs (Table 1). The benzyl analog (15) showed no significant change in potency on Hα4β2 nAChRs; but showed a 2-fold decrease in potency for Hα3β4 nAChRs (IC50 value, 18.5 µM, Table 1). The indazole analog (11) produced a slight increase in potency for Hα4β2 nAChRs (IC50 value, 6.6 µM, Table 1) and a 3-fold decrease in potency for Hα3β4 nAChRs (IC50 value, 23.8 µM, Table 1).
Series 2 SAR
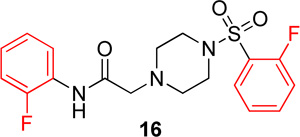
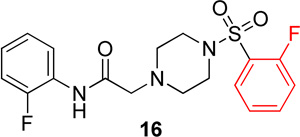
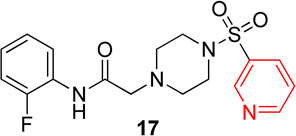
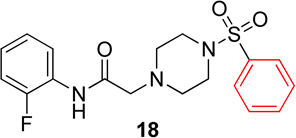
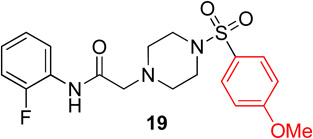
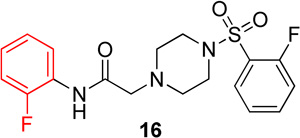
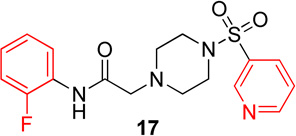
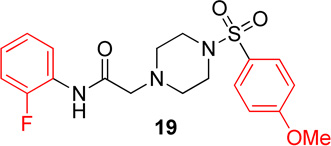
In the next series of analogs, the sulfonyl portion of scaffold 1 was modified using several different substitutions (Table 2). Similar to the Series 1 study, this was conducted to determine the synthetic flexibility of the sulfonyl portion and to determine how these changes affect potency on nAChRs. Movement of the fluorine from the para position (1) to the ortho position (16) produced no significant change in potency for Hα4β2 nAChRs; however, resulted in a significant, 12-fold decrease in potency for Hα3β4 nAChRs (IC50 value, 99.8 µM, p < 0.01, Table 2). The pyridinyl analog (17) produced a 2-fold decrease in potency for the Hα4β2 nAChR and a 5-fold decrease in potency for the Hα3β4 nAChR (IC50 values 20.3 µM and 49.6 µM respectively, Table 2). The phenyl analog (18) produced a 2-fold increase in potency for Hα4β2 and Hα3β4 nAChRs (IC50 values 4.1 and 4.6 µM respectively, Table 2). The p-methoxyphenyl analog (19) resulted in no significant change in potency for both Hα4β2 and Hα3β4 nAChRs (Table 2). In examining the inhibition curves of 1 and 16 on nAChRs, 1 produces similar hill coefficients on both Hα4β2 and Hα3β4 nAChRs (Figure 2 and Table 2). 16 produces a similar hill coefficient as 1 on Hα4β2 nAChRs (nh = −1.5); but produced a statistically significant (p<0.0005) 2-fold decreased hill coefficient on Hα3β4 nAChRs (nh = −0.6, Table 2).
Table 2.
Series 2 SAR Studies
| Hα4β2 nAChRs | Hα3β4 nAChRs | |||
|---|---|---|---|---|
| Compound | IC50 Value (µM)a | nhb | IC50 Value (µM)a | nhb |
 |
9.3 (6.2–13.9) | −1.2 | 9.0 (6.3–12.9) | −1.2 |
 |
8.0 (5.4–11.8) | −1.5 | 99.8 (57.2–174) | −0.6 |
 |
20.3 (12.6–32.6) | –1.1 | 49.6 (41.5–59.3) | −0.8 |
 |
4.1 (3.0–5.7) | −1.4 | 4.0 (3.2–6.8) | −1.3 |
 |
13.4 (9.8–18.4) | −1.3 | 15.4 (11.2–21.1) | −1.4 |
Values represent geometric means (confidence limits), n = 5–10
nh, Hill coefficient
Figure 2. Concentration-response effects of compound 1 and compound 16 on Hα4β2 and Hα3β4 nAChRs.

Calcium accumulation assays were performed as described in the methods section. The concentration-response effects of compound 1 (A) and compound 16 (B) were investigated on Hα4β2 nAChRs (■) and Hα3β4 nAChRs (□). Values are mean ± SEM (n= 6–9). IC50 values of compound 1 and compound 16 are reported together in Table 2.
Series 3 SAR
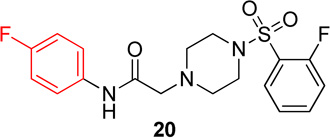
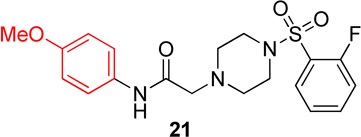
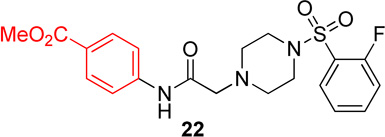
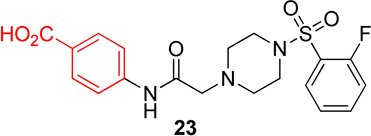
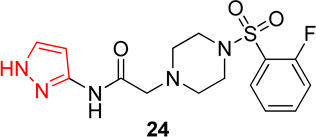
In this series, analog 16 (Table 2) was used as the basis of comparison. Replacement of the o-fluorophenyl with the p-fluorophenyl (20) resulted in no change in potency for Hα4β2 nAChRs (Table 3) and a 6-fold increase in potency for Hα3β4 nAChRs (IC50 value, 13.6 µM; Table 3). Replacement of the o-fluorophenyl with a p-methoxyphenyl (21) resulted in a 2-fold decrease in potency for Hα4β2 nAChRs (IC50 value, 17.1 µM; Table 3) and a 4-fold increase in potency for Hα3β4 nAChRs (IC50 value, 19.1 µM; Table 3). Substitution of a methyl ester at the para position (22) resulted in no significant change for Hα4β2 and an 8-fold increase in potency for Hα3β4 nAChRs (IC50 value, 11.2 µM; Table 3). Replacement of the fluorine with carboxylic acid (23) or instillation of a pyrazole heterocycle in place of the fluorophenyl (24) resulted in a loss of activity on both subtypes (Table 2).
Table 3.
Series 3 SAR Studies
| Hα4β2 nAChRs | Hα3β4 nAChRs | |||
|---|---|---|---|---|
| Compound | IC50 Value (µM)a | nhb | IC50 Value (µM)a | nhb |
 |
8.0 (5.4–11.8) | −1.5 | 99.8 (57.2–174) | −0.6 |
 |
8.0 (4.5–14.3) | −1.2 | 13.6 (8.4–22.1) | −1.1 |
 |
17.1 (14.1–20.7) | −1.0 | 19.1 (16.0–20.7) | −1.2 |
 |
12.4 (8.8–17.6) | −1.1 | 11.2 (8.6–14.7) | −1.1 |
 |
>100c | ~ | >100c | ~ |
 |
>100c | ~ | >100c | ~ |
Values represent geometric means (confidence limits), n = 6–12
nh, Hill coefficient
No activity up to 100 µM
Series 4 SAR
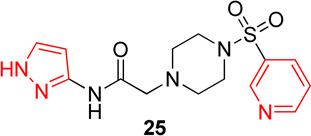
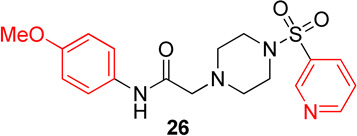
Pyridinyl, phenyl, and p-methoxyphenyl containing analogs of 1 were synthesized. The o-fluorophenyl, pyridinyl analog (17) resulted in IC50 values of 20.3 µM and 48.9 µM on Hα4β2 and Hα3β4 nAChRs, respectively (Table 4). The pyrazole, pyridinyl analog (25) resulted in IC50 values of 83.0 µM and >100 µM on Hα4β2 and Hα3β4 nAChRs, respectively (Table 4). The p-methoxyphenyl, pyridinyl analog (26) resulted in a significant decrease in potency for both Hα4β2 and Hα3β4 nAChRs (IC50 values of >100 µM and 95.0 µM respectively, Table 4). The o-fluorophenyl, phenyl analog (18) resulted in IC50 values of 4.1 µM and 4.6 µM on Hα4β2 and Hα3β4 nAChRs, respectively (Table 4). The phenyl, phenyl analog (27) resulted in IC50 values of 9.6 µM and 16.4 µM on Hα4β2 and Hα3β4 nAChRs, respectively (Table 4). The o-fluorophenyl, p-methoxyphenyl analog (19) resulted in no significant change in potency for both Hα4β2 and Hα3β4 nAChRs (Table 4). The p-fluorophenyl, p-methoxyphenyl analog (28) resulted in a 2-fold decrease in potency for both Hα4β2 and Hα3β4 nAChRs (IC50 values of 23.6 µM and 17.1 µM, respectively, Table 4)
Table 4.
Series 4 SAR Studies
| Hα4β2 nAChRs | Hα3β4 nAChRs | |||
|---|---|---|---|---|
| Compound | IC50 Value (µM)a | nhb | IC50 Value (µM)a | nhb |
 |
20.3 (12.6–32.6) | −1.1 | 48.9 (29.1–82.1) | −0.8 |
 |
83.0 (48.3–143) | −0.5 | >100c | ~ |
 |
>100c | ~ | 95.0 (74.0–122) | −1.5 |
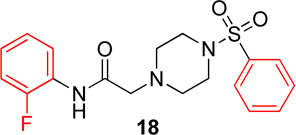 |
4.1 (3.0–5.7) | −1.4 | 4.6 (3.2–6.8) | −1.4 |
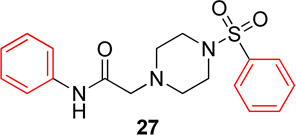 |
9.6 (6.0–15.2) | −0.9 | 16.4 (9.2–29.6) | −1.0 |
 |
13.4 (9.8–18.4) | −1.3 | 15.4 (11.2–21.1) | −1.4 |
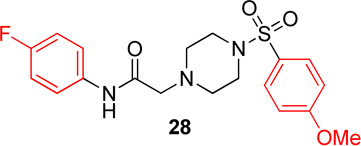 |
23.8 (20.6–27.5) | −1.0 | 17.1 (12.7–23.0) | −1.0 |
Values represent geometric means (confidence limits), n = 5–10
nh, Hill coefficient
No activity up to 100 µM
Series 5 SAR
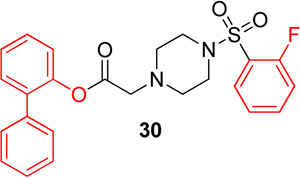
Biphenyl ester analogs were made of lead molecule 1 (29) and 16 (30). Biphenylester 29 showed no change in potency for Hα4β2 nAChRs and a 2-fold decrease in potency for Hα3β4 nAChRs (IC50 value, 23.8; Table 5) compared to 1. Biphenylester 30 showed a decrease in potency for Hα4β2 nAChRs (IC50 value, 20.9 µM, Table 5) and a 3-fold increase in potency for Hα3β4 nAChRs (IC50 value, 23.5; Table 5) in comparison to 16.
DISCUSSION AND CONCLUSION
We understand that there are a multitude of nAChR subtypes and that a study such as this, utilizing only two subtypes, cannot use the term ‘selectivity’ accurately. For this reason, the term ‘relative-selectivity’ has been used to describe the difference between only Hα4β2 and Hα3β4 nAChRs. The most significant change in series 1 was the pyrazole (12) modification, which abolished activity (up to 100 µM) for both subtypes. Movement of the fluorine led to no significant change; therefore, suggests that the fluorine is not a significant contributor to selectivity or potency for this scaffold. Substitution of the indazole (11) did not affect potency on Hα4β2 nAChRs but decreased potency on Hα3β4. This suggests that this structure or position of the scaffold is amenable to optimization for Hα4β2 nAChR selectivity.
The significant findings of series 2 were that the o-fluorophenyl and pyridinyl substitutions showed an improvement in the relative-selectivity for Hα4β2 nAChRs (9-fold and 3-fold, respectively). This result suggests that this position of the scaffold may improve the range of selectivity with further modification. Moving the fluorine from the para (1) to the ortho (16) positions may cause rotation of the ring due to proximity to the sulfonyl oxygen groups.
Series 3 data suggest that these substitutions have little effect on the potency of these molecules on Hα4β2 nAChRs (with the exception of pyrazole 24); yet every change showed a decrease in relative-selectivity for Hα4β2 nAChRs. The results of the comparison between analogs 20 and 16 (Table 3) contradict earlier results (e.g., the comparison between analogs 14 and 1, Table 1). The first comparison showed that the fluorine position, ortho or para, had no effect on potency for Hα3β4 nAChRs (Table 1). However, we noticed that movement of the fluorine (20 and 16) makes a significant difference on potency for Hα3β4 nAChRs (Table 3). This suggests that this portion of the molecule is interacting at a different position in the ligand binding domain depending on the fluorine position on the phenyl of the sulfonyl portion. Compound 12 (Series 1) and compound 30 both contained pyrazole moieties and resulted in a loss of activity on both nAChR subtypes. This suggests that more hydrophilic substitutions decrease binding and/or efficacy on nAChRs.
In pyridinyl analogs of series 4, the methoxybenzyl substitution to the amide portion (26) produced a significant change in potency on both subtypes (Table 4). This is surprising considering similar substitutions had little effect on potency (13 in Table 1 and 21 in Table 3). These data also show that other aromatics, such as a pyrazole, drastically reduce potency on nAChRs (25). In both the phenyl and p-methoxyphenyl analogs, molecules with fluorine substitutions at the ortho position (18 and 19) showed improved Hα4β2 nAChR potency compared to molecules that lacked a fluorine substitution (27) or had a fluorine substitution at the para position (28). This suggests that the ortho position is preferred for Hα4β2 nAChR potency.
Previously published data25 showed that the incorporation of biphenyl structures is important for selectivity of molecules targeting Hα4β2 nAChRs. Therefore, biphenyl analogs of 1 and 16 were made in series 5. It was hypothesized that the ester carbonyl and fluorinated phenyl groups would act in a similar way as the ester and phenylpropyl of KAB-18.25 However, these features found in this novel scaffold lack the flexibility of the phenylpropyl in KAB-18-like molecules and therefore may have a different binding mode within the binding site.
In conclusion, the SAR of sulfonylpiperizine analogs on Hα4β2 and Hα3β4 nAChRs has been described here. Compound 16 showed the highest relative-selectivity for Hα4β2 nAChRs (12-fold, Table 3) while 18 showed the highest potency (Table 4) among the compounds described here. The SAR of these compounds has identified that the position of fluorine substitution on the sulfonyl portion (ortho vs. para) has a significant effect on relative-selectivity for Hα4β2 nAChRs. In these analogs, relative-selectivity for Hα4β2 nAChRs is associated with o-fluorophenyl (16) while p-fluorobenzenes show no preference for Hα3β4 or Hα4β2 nAChRs. Additionally, the ortho substitution of halogens in the amide portion has shown improvement in potency for Hα4β2 nAChRs. In the future discovery of novel Hα4β2 nAChR antagonists, it would be informative to incorporate both of these features in the design of new NAMs to improve both potency and selectivity. Compounds 11 and 16, which have modifications to the amide and sulfonyl positions, respectively, both led to an increase in selectivity for Hα4β2 nAChRs. Further studies may include making both modifications in a single molecule to improve selectivity for Hα4β2 nAChRs.
The structural diversity of these new analogs provides additional insight into the physiochemical features that are important for antagonism of nAChRs at allosteric sites. As mentioned before, the discovery of selective molecules targeting nAChRs has been slow. Studies like these contribute to the discovery and development of selective compounds that can be used as novel therapeutics for nAChR related diseases and disorders.
Experimental Section
Materials
Calcium 5NW dye was obtained from Molecular Devices (Sunnyvale, CA). Dulbecco’s Modified Eagle Medium (DMEM), penicillin, streptomycin and L-glutamine were obtained from Invitrogen Corporation (Grand Island, NY). Epibatidine was purchased from Sigma-Aldrich (St. Louis, MO). All other reagents were purchased from Fisher Scientific (Pittsburg, PA). For pharmacological evaluation, all compounds were initially dissolved in 100% DMSO (0.01 M stocks). Stock solutions of compounds at concentrations less than or equal to 100 µM were made in HBK buffer.
Calcium Accumulation Assays
A procedure previously reported by our laboratory25,32,33 was used with minor modifications. For the calcium accumulation assays, HEK ts201 cells stably expressing either Hα4β2 nAChRs or Hα3β4 nAChRs (obtained from Professor Jon Lindstrom, University of Pennsylvania, Philadelphia, PA) were used. Cells were plated at a density of 2.0 – 2.3 × 105 cells per well in clear 96-well culture plates previously coated with poly-l-ornithine. On the day of the experiment (typically ~24 h later), cells were washed (100 µl) with HEPES-buffered Krebs (HBK) solution,25,32,33 and incubated (protected from light) for 1 hour at 24 °C with 50% Calcium 5 NW dye (Molecular Devices). The plates were then placed into a fluid handling integrated fluorescence plate reader (FlexStation, Molecular Devices, Sunnyvale, CA) and fluorescence was read at excitation of 485 nm and emission of 525 nm from the bottom of the plate, and changes in fluorescence were monitored at ~1.5 second intervals. Inhibition curves were obtained in the concentration-response studies using 6 concentrations within a range of 0.1 to 100 µM for each compound reported here. Results are reported as IC50 values (see Tables 1–5). Results were expressed as a percentage of control-1µM epibatidine group. Due to solubility problems, compound concentrations greater than 100 µM were not used in our concentration-response studies. Therefore, compounds that showed no inhibition up to the highest concentration have been labeled with IC50 values of ‘>100’ (See Tables 1–4). The DMSO concentration at this compound concentration was 1% and had no effect on basal or agonist-induced increases in fluorescence intensity.
Calculations and Statistics
Functional data were calculated from the number of observations (n) performed in triplicate. Curve fitting was performed by Prism software (GraphPad, San Diego, CA) using the equation for a single-site sigmoidal dose-response curve with a variable slope. IC50 values are expressed as geometric means (95% confidence limits). When statistical significance was calculated, the t-test was used. Where data were labeled ‘not significant’ or when ‘no significant difference’ was reported, then p>0.05.
General Chemistry Methods
1H (500 MHz or 400 MHz) and 13C (125 MHz or 100 MHz) NMR spectra were recorded on a Bruker DRX-500 or Bruker DPX-400 spectrometer in CDCl3 using CHCl3 (1H δ 7.26) and CDCl3 (13C δ 77.0) or DMSO-d6 using DMSO (1H δ 2.49) and DMSO-d6 (13C δ 39.51) as internal standards. High resolution mass spectra were recorded on a Bruker MicrOTOF ESI spectrometer provided by OBIC. Elemental analyses were carried out by Galbraith Laboratories, Inc. (Knoxville, TN). All reactions were conducted in either oven-dried (120 °C) glassware or flame-dried glassware, under an N2 atmosphere when necessary. Tetrahydrofuran (THF) was distilled from benzophenone ketyl. Triethylamine and CH2Cl2 were distilled from calcium hydride prior to use. All other chemicals were used as received. All compounds were > 95% purity as determined by elemental analysis (C, H, N), unless otherwise noted.
Synthesis
Preparation of N-aryl-2-bromoacetamides General Procedure A
Bromoacetyl bromide (10 mmol, 1 equiv) was added dropwise over 5 min to a solution of amine or alcohol (10 mmol, 1 equiv) and triethylamine (11 mmol, 1.1 equiv) in dichloromethane (50 mL, 0.2 M) at 0 °C. The reaction mixture was stirred at 0 °C for 20 min to 1 h, diluted with dichloromethane (50 mL), washed with saturated NH4Cl (3 × 30 mL), dried (Na2SO4), and concentrated in vacuo to afford crude product. The crude product was purified by flash column chromatography, crystallization, or trituration to yield pure N-aryl-2-bromoacetamide (26% to 96%).
Preparation of arylsulfonylpiperazines General Procedure B
Arylsulfonyl chloride (10 mmol, 1 equiv) was added in one portion to a solution of piperazine (60 mmol, 6 equiv) in CH2Cl2 (100 mL, 0.1 M) at 0 °C. The reaction mixture was stirred at 0 °C for 30 min, diluted with CH2Cl2 (200 mL), quenched by the addition of saturated NaHCO3(aq) (50 mL), washed with brine (50 mL), dried (Na2SO4), and concentrated in vacuo to provide crude product. The crude product was used directly, or purified by crystallization or trituration to yield pure arylsulfonylpiperazines (75% to quant).
Preparation of N-aryl-2-((arylsulfonyl)piperazinyl)acetamides General Procedure C
Sodium carbonate (0.4 mmol, 2 equiv) or triethylamine (0.4 mmol, 2 equiv) was added to a solution of N-aryl-2-bromoacetamide (0.2 mmol, 1 equiv) and arylsulfonyl piperazine (0.2 mmol, 1 equiv) in THF (1 mL, 0.2 M) at 23 °C. The reaction mixture was allowed to stir for 16 h, diluted with CH2Cl2 (5 mL), filtered, and concentrated in vacuo to afford crude product. The crude product was purified by flash column chromatography to yield pure N-aryl-2-((arylsulfonyl)piperazinyl)acetamides (48% to 86%).
2-bromo-N-(2-fluorophenyl)acetamide (2)
Following general procedure A, the crude product was precipitated (Et2O/Hexanes) to give pure 2 as a white solid (26%): 1H NMR (CDCl3, 400 MHz) δ = 8.40 (br s, 1H), 8.24 (t, J = 7.8 Hz, 1H), 7.04–7.19 (m, 3H), 4.04 (s, 2H); 13C NMR (CDCl3, 101 MHz) δ = 163.6, 154.1, 151.6, 125.7, 125.6, 125.5, 125.4, 124.7, 121.7, 115.2, 115.0, 29.4; IR (neat) λmax 3313, 1669, 1620, 1330, 1105, 758; HRMS (ESI) m/z calcd for C8H7BrFNNaO: 253.9587; found: 253.9591.
1-([4-fluorophenyl]sulfonyl)piperazine (3)
Following general procedure B, the crude product was crystallized (EtOAc/Hexanes) to give pure 3 as a white solid (93%): 1H NMR (CDCl3, 500 MHz) δ = 7.70–7.79 (m, 2H), 7.16–7.24 (m, 2H), 2.93–3.00 (m, 4H), 2.87–2.93 (m, 4H), 1.52 (s, NH); 13C NMR (CDCl3, 126 MHz) δ = 166.3, 164.3, 131.8, 131.8, 130.5, 130.5, 116.4, 116.2, 46.9, 45.4; IR (neat) λmax 2762, 1592, 1495,1341, 1170, 541, 539; Calcd. for C10H13FN2O2S: C(49.17%), H(5.36%), N(11.47%); Found: C(49.04%), H(5.45%), N(11.30%); HRMS (ESI) m/z calcd for C10H14FN2O2S1 : 245.0755; found: 245.0758.
N-(2-fluorophenyl)-2-(4-((4-fluorophenyl)sulfonyl)piperazin-1-yl)acetamide, (1)
Following general procedure C, the crude product was purified by flash column chromatography (silica, 4:6 EtOAc/Hexanes) to provide 1 as a white solid (59%): 1H NMR (CDCl3, 400 MHz) δ = 9.08 (br s, 1H), 8.25 (t, J = 8.0 Hz, 1H), 7.73–7.82 (m, 2H), 7.18–7.28 (m, 2H), 7.04–7.13 (m, 1H), 6.93–7.04 (m, 2H), 3.16 (s, 2H), 3.11 (br s, 4H), 2.69 (t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 101 MHz) δ = 167.5, 166.7, 164.1, 153.7, 151.2, 131.9, 131.9, 130.5, 130.4, 125.9, 125.8, 124.7, 124.7, 124.6, 124.5, 121.5, 116.6, 116.4, 114.9, 114.7, 61.6, 52.5, 46.1; IR (neat) λmax 3504, 2831, 1699, 1531, 1173, 951, 735, 548; Calcd. for C18H19F2N3O3S: C(54.67%), H(4.84%), N(10.63%); Found: C(54.72%), H(4.94%), N(10.44%); HRMS (ESI) m/z calcd for C18H20F2N3O3S: 396.1188; found: 396.1173.
2-(4-((4-fluorophenyl)sulfonyl)piperazin-1-yl)acetic acid (9)
Following general procedure C, using triethylamine, the crude product was purified by crystallization (CH3OH) to provide 9 as a white crystal (30%): 1H NMR (DMSO-d6, 500 MHz) δ = 7.80 (dd, J = 8.4, 5.2 Hz, 2H), 7.47 (br t, J = 8.7 Hz, 2H), 3.13 (s, 2H), 2.88 (br s, 4H), 2.58 (br s, 4H); 13C NMR (DMSO-d6, 126 MHz) δ = 171.2, 165.7, 163.7, 131.2, 131.1, 130.7, 130.6, 116.7, 116.5, 57.8, 50.8, 45.8; IR (neat) λmax 3470, 1634, 1355, 1173, 935, 733, 548; HRMS (ESI) m/z calcd for C12H16FN2O4S: 303.0809; found: 303.0794.
2-(4-((4-fluorophenyl)sulfonyl)piperazin-1-yl)-N-(1H-indazol-5-yl)acetamide, (11)
EDCI (66.9 mg, 0.349 mmol) was added to a solution of 9 (105.4 mg, 0.349 mmol), 5-aminoindazole (51.1 mg, 0.384 mmol), and HOBt (47.2 mg, 0349 mmol) in DMF (1.7 mL) at 0 °C. The reaction mixture was stirred 16 h while warming to 23 °C, diluted with CH2Cl2 (10 mL), washed with H2O (3 × 5 mL), brine (5 mL), dried (Na2SO4), and concentrated in vacuo to produce crude brown solid. The crude material was purified by flash column chromatography (silica, 8:2 EtOAc/Hexanes) to yield 11 as a white solid (67.7 mg, 46%): 1H NMR (DMSO-d6, 400 MHz) δ = 12.93 (br s, NH), 9.60 (s, NH), 7.98 (s, 1H), 8.03 (s, 1H), 7.83 (dd, J = 8.7, 5.3 Hz, 2H), 7.50 (t, J = 8.7 Hz, 2H), 7.34–7.46 (m, 2H), 3.14 (s, 2H), 3.00 (br s, 4H), 2.60 (apparent t, J = 4.3 Hz, 4H); 13C NMR (DMSO-d6, 126 MHz) δ = 167.7, 165.7, 163.7, 137.0, 133.3, 131.5, 131.5, 131.4, 130.7, 130.6, 122.6, 120.9, 116.7, 116.5, 110.5, 109.9, 61.0, 51.6, 45.7; IR (neat) λmax 3628, 1682, 1171, 948, 735, 548; Calcd. for C19H20FN5O3S: C(54.67%), H(4.83%), N(16.78%); Found: C(32.55%), H(2.98%), N(9.52%); HRMS (ESI) m/z calcd for C19H20FN5NaO3S: 440.1163; found: 440.1148.
2-(4-((4-fluorophenyl)sulfonyl)piperazin-1-yl)-N-(1H-pyrazol-3-yl)acetamide, (12)
Following general procedure C, crude product was purified by flash column chromatography (silica, EtOAc) to provide 12 as a white solid (47%): 1H NMR (CDCl3, 500 MHz) δ = 10.50 (br s, NH) 9.15 (s, NH), 7.67–7.82 (m, 2H), 7.40 (d, J = 2.4 Hz, 1H), 7.15–7.30 (m, 2H), 6.64 (d, J = 2.4 Hz, 1H), 3.14 (s, 2H), 3.05 (br s, 4H), 2.65 (br t, J = 4.7 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.5, 166.5, 164.4, 146.1, 131.4, 131.4, 130.5, 130.5, 130.1, 116.7, 116.6, 96.7, 61.3, 52.6, 45.9; IR (neat) λmax 3613, 3308, 2938, 2834, 1668, 1592, 1169, 954, 836, 732, 547; Calcd. for C15H18FN5O3S: C(49.04%), H(4.94%), N(19.06%); Found: C(48.07%), H(5.11%), N(17.77%); HRMS (ESI) m/z calcd for C15H19FN5O3S: 368.1187; found: 368.1170.
2-(4-((4-fluorophenyl)sulfonyl)piperazin-1-yl)-N-(4-methoxyphenyl)acetamide, (13)
Following general procedure C, crude product was purified by flash column chromatography (silica, 1:1 EtOAc/hexanes) to provide 13 as a white solid (79%): 1H NMR (CDCl3, 400 MHz) δ = 8.53 (s, 1H), 7.73–7.82 (m, 2H), 7.30–7.39 (m, 2H), 7.20–7.28 (m, 2H), 6.77–6.85 (m, 2H), 3.75 (s, 3H), 3.10 (s, 2H), 3.08 (br s, 4H), 2.66 (br t, J = 4.8 Hz, 4H); 13C NMR (CDCl3, 101 MHz) δ = 167.2, 166.6, 164.1, 156.5, 131.8, 131.8, 130.5, 130.4, 130.4, 121.5, 116.7, 116.4, 114.2, 61.6, 55.5, 52.6, 45.9; IR (neat) λmax 2840, 1916, 1683, 1515, 1172, 952, 839, 735, 548; Calcd. for C19H22FN3O4S: C(56.01%), H(5.44%), N(10.31%); Found: C(56.29%), H(5.64%), N(10.22%); HRMS (ESI) m/z calcd for C19H23FN3O4S: 408.1388; found: 408.1383.
N-(4-fluorophenyl)-2-(4-((4-fluorophenyl)sulfonyl)-piperazin-1-yl)acetamide, (14)
Following general procedure C, crude product was purified by flash column chromatography (silica, 1:1 EtOAc/hexanes) to provide pure 14 as a white solid (86%): 1H NMR (CDCl3, 500 MHz) δ = 8.63 (br s, 1H), 7.69–7.85 (m, 2H), 7.37–7.47 (m, 2H), 7.25 (apparent t, J = 8.5 Hz, 2H), 6.92–7.01 (m, 2H), 3.13 (s, 2H), 3.11 (br s, 4H), 2.69 (t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.4, 166.5, 164.4, 160.5, 158.5, 133.4, 133.3, 132.0, 132.0, 130.6, 130.5, 121.5, 121.4, 116.7, 116.5, 115.8, 115.7, 61.7, 52.7, 46.0; IR (neat) λmax 2982, 1666, 1523, 1351, 1172, 947, 834, 740, 549; Calcd. for C18H19F2N3O3S: C(54.67%), H(4.84%), N(10.63%); Found: C(55.00%), H(5.21%), N(10.47%); HRMS (ESI) m/z calcd for C18H20F2N3O3S: 396.1188; found: 396.1177.
2-(4-((4-fluorophenyl)sulfonyl)piperazin-1-yl)-N-phenylacetamide, (15)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4:6 EtOAc/hexanes) to provide 15 as a white solid (43%): 1H NMR (CDCl3, 500 MHz) δ = 8.65 (br s, 1H), 7.74–7.83 (m, 2H), 7.43–7.50 (m, 2H), 7.19–7.33 (m, 4H), 7.03–7.14 (m, 1H), 3.13 (s, 2H), 3.10 (br s, 4H), 2.68 (t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.4, 166.4, 164.4, 137.3, 132.0, 131.9, 130.6, 130.5, 129.1, 124.5, 119.7, 116.7, 116.5, 61.8, 52.6, 46.0; IR (neat) λmax 3270, 3059, 2944, 1677, 1524, 1173, 946, 737, 548; Calcd. for C18H20FN3O3S: C(57.28%), H(5.34%), N(11.13%); Found: C(57.39%), H(5.54%), N(11.00%); HRMS (ESI) m/z calcd for C18H21FN3O3S: 378.1282; found 378.1270.
N-(2-fluorophenyl)-2-(4-((2-fluorophenyl)sulfonyl)-piperazin-1-yl)acetamide, (16)
Following general procedure C, crude product was purified by flash column chromatography (silica, 3:7 EtOAc/hexanes) to provide 16 as a white solid (53%): 1H NMR (CDCl3, 400 MHz) δ = 9.20 (br s, NH), 8.30 (t, J = 8.1 Hz, 1H), 7.80–7.92 (m, 1H), 7.54–7.69 (m, 1H), 7.27–7.35 (m, 1H), 7.23–7.29 (m, 1H), 7.08–7.17 (m, 1H), 6.91–7.08 (m, 2H), 3.26–3.42 (m, 4H), 3.20 (s, 2H), 2.65–2.77 (m, 4H); 13C NMR (CDCl3, 101 MHz) δ = 167.6, 160.3, 157.8, 153.7, 151.3, 135.4, 135.3, 131.3, 125.9, 125.8, 125.4, 125.3, 124.7, 124.7, 124.7, 124.6, 124.6, 124.5, 121.5, 117.6, 117.4, 114.9, 114.7, 61.8, 52.9, 45.8, 45.8; IR (neat) λmax 3308, 2857, 1700, 1185, 766, 736, 586; Calcd. for C18H19F2N3O3S: C(54.67%), H(4.84%), N(10.63%); Found: C(54.82%), H(4.77%), N(10.49%); HRMS (ESI) m/z calcd for C18H20F2N3O3S: 396.1188; found 396.1168.
N-(2-fluorophenyl)-2-(4-(pyridin-3-ylsulfonyl)piperazin-1-yl)acetamide, (17)
Following general procedure C, crude product was purified by flash column chromatography (silica, 8:2 EtOAc/hexanes) to provide 17 as a white solid (66%): 1H NMR (CDCl3, 500 MHz) δ = 9.06 (br s, 1H), 9.00 (dd, J = 2.4, 0.8 Hz, 1H), 8.86 (dd, J = 5.0, 1.6 Hz, 1H), 8.21–8.30 (m, 1H), 8.03–8.10 (m, 1H), 7.52 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 7.07–7.12 (m, 1H), 6.96–7.05 (m, 2H), 3.18 (s, 6H), 2.72 (t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.4, 153.7, 153.4, 151.5, 148.5, 135.4, 132.8, 125.8, 125.7, 124.7, 124.7, 124.6, 124.6, 123.9, 121.5, 114.9, 114.8, 61.7, 52.5, 46.0; IR (neat) λmax 3311, 2830, 1694, 1456, 1176, 953, 756, 583; Calcd. for C17H19FN4O3S: C(53.96%), H(5.06%), N(14.81%); Found: C(54.21%), H(5.22%), N(14.67%); HRMS (ESI) m/z calcd for C17H20FN4O3S: 379.1235; found: 379.1236.
N-(2-fluorophenyl)-2-(4-(phenylsulfonyl)piperazin-1-yl)acetamide, (18)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4:6 EtOAc/hexanes) to provide 18 as a white solid (79%): 1H NMR (CDCl3, 500 MHz) δ = 9.09 (br s, NH), 8.26 (t, J = 8.0 Hz, 1H), 7.74–7.80 (m, 2H), 7.60–7.67 (m, 1H), 7.50–7.60 (m, 2H), 7.06–7.15 (m, 1H), 6.91–7.06 (m, 2H), 3.16 (s, 2H), 3.13 (br s, 4H), 2.69 (br t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.6, 153.4, 151.5, 135.9, 133.1, 129.2, 127.7, 125.9, 125.8, 124.7, 124.7, 124.6, 124.5, 121.5, 114.9, 114.7, 61.7, 52.6, 46.1; IR (neat) λmax 3302, 2829, 1918, 1693, 1172, 952, 743, 568; Calcd. for C18H20FN3O3S: C(57.28%), H(5.34%), N(11.13%); Found: C(57.61%), H(5.51%), N(11.16%); HRMS (ESI) m/z calcd for C18H21FN3O3S: 378.1282; found: 378.1272.
N-(2-fluorophenyl)-2-(4-((4-methoxyphenyl)sulfonyl)-piperazin-1-yl)acetamide, (19)
Following general procedure C, crude product was purified by flash column chromatography (silica, 1:1 EtOAc/hexanes) to provide 19 as a white solid (77%): 1H NMR (CDCl3, 500 MHz) δ = 9.07 (br s, 1H), 8.23 (t, J = 7.9 Hz, 1H), 7.61–7.70 (m, 2H), 7.01–7.09 (m, 1H), 6.95–7.01 (m, 4H), 3.85 (s, 4H), 3.13 (s, 2H), 3.06 (br s, 4H), 2.65 (br t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.6, 163.2, 153.4, 151.4, 129.8, 127.2, 125.8, 125.7, 124.5, 124.5, 124.4, 124.4, 121.5, 114.8, 114.6, 114.3, 61.5, 55.6, 52.4, 46.0; IR (neat) λmax 2843, 1953, 1699, 1164, 949, 735, 559; Calcd. for C19H22FN3O4S: C(56.01%), H(5.44%), N(10.31%); Found: C(56.20%), H(5.64%), N(10.17%); HRMS (ESI) m/z calcd for C19H23FN3O4S: 408.1388; found: 408.1390.
N-(4-fluorophenyl)-2-(4-((2-fluorophenyl)sulfonyl)-piperazin-1-yl)acetamide, (20)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4:6 EtOAc/hexanes) to provide 20 as a white solid (86%): 1H NMR (CDCl3, 500 MHz) δ = 8.73 (br s, 1H), 7.75–7.87 (m, 1H), 7.54–7.64 (m, 1H), 7.39–7.47 (m, 2H), 7.26–7.31 (m, 1H), 7.17–7.25 (m, 1H), 6.90–7.00 (m, 2H), 3.28 (br s, 4H), 3.12 (s, 2H), 2.48–2.70 (br t, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.4, 160.3, 160.0, 158.4, 157.9, 135.4, 135.3, 133.4, 131.2, 125.3, 125.1, 124.7, 124.6, 121.4, 121.4, 117.5, 117.3, 115.7, 115.5, 61.6, 52.8, 45.6, 45.6; IR (neat) λmax 3403, 2839, 1690, 1177, 950, 836, 739, 584; Calcd. for C18H19F2N3O3S: C(54.67%), H(4.84%), N(10.63%); Found: C(54.79%), H(5.17%), N(10.64%); HRMS (ESI) m/z calcd for C18H20F2N3O3S: 396.1188; found: 396.1183.
2-(4-((2-fluorophenyl)sulfonyl)piperazin-1-yl)-N-(4-methoxyphenyl)acetamide, (21)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4:6 EtOAc/hexanes) to provide 21 as a white solid (89%): 1H NMR (CDCl3, 500 MHz) δ = 8.61 (br s, NH), 7.74–7.82 (m, 1H), 7.49–7.59 (m, 1H), 7.31–7.38 (m, 2H), 7.22–7.28 (m, 1H), 7.19 (t, J = 9.3 Hz, 1H), 6.69–6.82 (m, 2H), 3.70 (s, 3H), 3.23 (br s, 4H), 3.07 (s, 2H), 2.48–2.65 (br t, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.2, 159.9, 157.8, 156.4, 135.3, 135.2, 131.1, 130.4, 125.1, 125.0, 124.6, 124.5, 121.4, 117.4, 117.2, 114.0, 61.5, 55.4, 52.7, 45.6; IR (neat) λmax 3313, 2939, 1686, 1598, 1514, 1174, 955, 828, 734; Calcd. for C19H22FN3O4S: C(56.01%), H(5.44%), N(10.31%); Found: C(55.53%), H(5.70%), N(10.07%); HRMS (ESI) m/z calcd for C19H23FN3O4S: 408.1388; found: 408.1390.
methyl 4-(2-(4-((2-fluorophenyl)sulfonyl)piperazin-1-yl)acetamido)benzoate, (22)
Following general procedure C, crude product was purified by flash column chromatography (silica, 1:1 EtOAc/hexanes) to provide 22 as a white solid (80%): 1H NMR (CDCl3, 500 MHz) δ = 8.95 (br s, NH), 7.97 (d, J = 8.5 Hz, 2H), 7.85 (t, J = 6.8 Hz, 1H), 7.59–7.69 (m, 1H), 7.57 (d, J = 8.5 Hz, 2H), 7.31 (t, J = 7.6 Hz, 1H), 7.15–7.28 (m, 1H), 3.88 (s, 3H), 3.32 (br s, 4H), 3.17 (s, 2H), 2.57–2.76 (m, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.9, 166.5, 160.1, 158.0, 141.4, 135.4, 135.4, 131.3, 130.9, 125.9, 125.5, 125.4, 124.7, 124.7, 118.8, 117.6, 117.4, 61.9, 53.0, 52.1, 45.7; IR (neat) λmax 3282, 1702, 1598, 1281, 1175, 954, 733, 584; Calcd. for C20H22FN3O5S: C(55.16%), H(5.09%), N(9.65%); Found: C(55.15%), H(5.31%), N(9.43%); HRMS (ESI) m/z calcd for C20H23FN3O5S: 436.1337; found: 436.1350.
4-(2-(4-((2-fluorophenyl)sulfonyl)piperazin-1-yl)acetamido)benzoic acid, (23)
Following general procedure C, crude product was purified by flash column chromatography (silica, 3% MeOH/EtOAc) to provide 23 a white solid (44%): 1H NMR (DMSO-d6,500 MHz) δ = 12.66 (br s, NH), 9.91 (s, NH), 7.87 (d, J = 8.7 Hz, 2H), 7.74–7.83 (m, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.46–7.53 (m, 1H), 7.44 (t, J = 7.6 Hz, 1H), 3.19 (s, 2H), 3.14 (br s, 4H), 2.60 (br s, 4H); 13C NMR (DMSO-d6, 126 MHz) δ = 168.5, 166.9, 159.3, 157.3, 142.5, 136.1, 136.0, 130.9, 130.2, 125.5, 125.2, 125.2, 124.0, 123.9, 118.9, 117.7, 117.5, 61.0, 51.8, 45.4; IR (neat) λmax 2924, 2854, 1707, 1600, 1174, 952, 733, 584; Calcd. for C19H20FN3O5S: C(54.15%), H(4.78%), N(9.97%); Found: C(54.52%), H(5.20%), N(9.40%); HRMS (ESI) m/z calcd for C19H21FN3O5S: 422.1180; found: 422.1162.
2-(4-((2-fluorophenyl)sulfonyl)piperazin-1-yl)-N-(1H-pyrazol-3-yl)acetamide, (24)
Following general procedure C, crude product was purified by flash column chromatography (silica, EtOAc) to provide 24 as a white solid (48%): 1H NMR (CDCl3, 400 MHz) δ = 10.57 (br s, NH), 9.23 (s, NH), 7.75–7.86 (m, 1H), 7.54–7.66 (m, 1H), 7.41 (d, J = 2.3 Hz, 1H), 7.26–7.33 (m, 1H), 7.18–7.25 (m, 1H), 6.63 (d, J = 2.3 Hz, 1H), 3.23 (br s, 4H), 3.14 (s, 2H), 2.64 (br t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 101 MHz) δ = 167.5, 160.3, 157.7, 146.1, 135.5, 135.4, 131.3, 130.2, 124.8, 124.7, 124.7, 117.7, 117.5, 96.6, 61.4, 52.8, 45.7; IR (neat) λmax 3212, 1668, 1567, 1176, 735, 583; Calcd. for C15H18FN5O3S: C(49.04%), H(4.94%), N(19.06%); Found: C(46.55%), H(4.95%), N(17.40%); HRMS (ESI) m/z calcd for C15H19FN5O3S: 368.1187; found: 368.1173.
N-(1H-pyrazol-3-yl)-2-(4-(pyridin-3-ylsulfonyl)piperazin-1-yl)acetamide, (25)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4% MeOH/EtOAc) to provide 25 as a white solid (34%): 1H NMR (CDCl3, 500 MHz) δ = 9.12 (s, NH), 8.99 (d, J = 1.9 Hz, 1H), 8.87 (dd, J = 4.7, 1.6 Hz, 1H), 8.05 (dt, J = 8.1, 1.9 Hz, 1H), 7.49–7.59 (m, 1H), 7.44 (d, J = 2.5 Hz, 1H), 6.67 (d, J = 2.5 Hz, 1H), 3.16 (s, 2H), 3.14 (br s, 4H), 2.69 (br t, J = 4.7 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.3, 153.7, 148.5, 146.2, 135.5, 132.6, 130.2, 124.0, 96.8, 61.4, 52.6, 45.9; IR (neat) λmax 3608, 3240, 2947, 2829, 1683, 1176, 951, 757, 583; Calcd. for C14H18N6O3S: C(47.99%), H(5.18%), N(23.98%); Found: C(47.61%), H(5.54%), N(22.48%); HRMS (ESI) m/z calcd for C14H19N6O3S: 351.1234; found: 351.1216.
N-(4-methoxyphenyl)-2-(4-(pyridin-3-ylsulfonyl)-piperazin-1-yl)acetamide, (26)
Following general procedure C, crude product was purified by flash column chromatography (silica, 9:1 EtOAc/hexanes) to provide 26 as a white solid (59%): 1H NMR (CDCl3, 500MHz) δ = 8.98 (d, J = 2.2 Hz, 1H), 8.84 (dd, J = 4.8, 1.1 Hz, 1H), 8.50 (s, NH), 8.04 (dd, J = 8.2, 1.6 Hz, 1H), 7.50 (dd, J = 7.9, 4.8 Hz, 1H), 7.34 (d, J = 9.0 Hz, 2H), 6.80 (d, J = 9.0 Hz, 2H), 3.74 (s, 3H), 3.14 (br s, 4H), 3.11 (s, 2H), 2.68 (br t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.0, 156.6, 153.7, 148.5, 135.4, 132.8, 130.4, 123.9, 121.5, 114.2, 61.6, 55.5, 52.5, 45.9; IR (neat) λmax 3320, 2834, 1679, 1514, 1175, 951, 756, 583; Calcd. for C18H22N4O4S: C(55.37%), H(5.68%), N(14.35%); Found: C(55.50%), H(5.95%), N(13.85%); HRMS (ESI) m/z calcd for C18H23N4O4S: 391.1435; found: 391.1439.
N-phenyl-2-(4-(phenylsulfonyl)piperazin-1-yl)acetamide, (27)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4:6 EtOAc/hexanes) to provide 27 as a white solid (70%): 1H NMR (CDCl3, 500 MHz) δ = 8.66 (br s, NH), 7.77 (d, J = 7.6 Hz, 2H), 7.64 (t, J = 7.6 Hz, 1H), 7.57 (t, J = 7.6 Hz, 2H), 7.45 (d, J = 7.6 Hz, 2H), 7.28 (t, J = 7.6 Hz, 2H), 7.08 (t, J = 7.6 Hz, 1H), 3.11 (s, 6H), 2.67 (br t, J = 4.7 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 167.4, 137.3, 135.8, 133.1, 129.3, 129.0, 127.8, 124.5, 119.6, 61.7, 52.6, 45.9; IR (neat) λmax 3283, 2848, 1674, 1523, 1176, 945, 745, 694, 580; Calcd. for C18H21N3O3S: C(60.15%), H(5.89%), N(11.69%); Found: C(60.23%), H(6.08%), N(11.80%); HRMS (ESI) m/z calcd for C18H22N3O3S: 360.1376; found: 360.1361.
N-(4-fluorophenyl)-2-(4-((4-methoxyphenyl)sulfonyl)-piperazin-1-yl)acetamide, (28)
Following general procedure C, crude product was purified by flash column chromatography (silica, 1:1 EtOAc/hexanes) to provide 28 as a white solid (77%): 1H NMR (CDCl3, 500 MHz) δ = 8.76 (br s, 1H), 7.61–7.72 (m, J = 8.8 Hz, 2H), 7.41 (dd, J = 8.7, 4.6 Hz, 2H), 6.97–7.03 (m, J = 8.8 Hz, 2H), 6.94 (t, J = 8.7 Hz, 2H), 3.85 (s, 3H), 3.14 (br s, 2H), 3.07 (br s, 4H), 2.69 (br s, 4H); 13C NMR (CDCl3, 101 MHz) δ = 167.4, 163.3, 160.6, 158.2, 133.4, 133.4, 129.9, 127.2, 121.5, 121.4, 115.8, 115.5, 114.4, 77.3, 61.5, 55.7, 52.6, 45.9; IR (neat) λmax 3312, 2842, 1686, 1510, 1163, 949, 835, 735; Calcd. for C19H22FN3O4S: C(56.01%), H(5.44%), N(10.31%); Found: C(57.30%), H(5.73%), N(10.32%); HRMS (ESI) m/z calcd for C19H23FN3O4S: 408.1388; found: 408.1394.
N-([1,1'-biphenyl]-2-yl)-2-(4-((4-fluorophenyl)sulfonyl)-piperazin-1-yl)acetamide, (29)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4:6 EtOAc/hexanes) to provide 29 as a white solid (83%): 1H NMR (CDCl3, 500 MHz) δ = 7.68–7.80 (m, 2H), 7.28–7.41 (m, 8H), 7.15–7.23 (m, 2H), 7.09 (dd, J = 7.7, 1.1 Hz, 1H), 3.22 (s, 2H), 2.97 (br s, 4H), 2.44 (br t, J = 4.9 Hz, 4H); 13C NMR (CDCl3, 126 MHz) δ = 168.3, 166.3, 164.3, 147.5, 137.6, 135.1, 131.5, 131.5, 130.9, 130.6, 130.5, 129.0, 128.7, 128.4, 127.6, 126.6, 122.5, 116.4, 116.3, 58.6, 51.5, 45.8; IR (neat) λmax 3448, 1771, 1592, 1155, 1139, 737, 548; Calcd. for C24H23FN2O4S: C(63.42%), H(5.10%), N(6.16%); Found: C(60.69%), H(5.16%), N(5.97%); HRMS (ESI) m/z calcd for C24H24FN2O4S: 455.1435; found 455.1444.
N-([1,1'-biphenyl]-2-yl)-2-(4-((2-fluorophenyl)sulfonyl)-piperazin-1-yl)acetamide, (30)
Following general procedure C, crude product was purified by flash column chromatography (silica, 4:6 EtOAc/hexanes) to provide 30 as a white solid (84%): 1H NMR (CDCl3, 500 MHz) δ = 7.77–7.83 (m, 1H), 7.51–7.60 (m, 1H), 7.28–7.41 (m, 8H), 7.23–7.28 (m, 1H), 7.19 (td, J = 9.2, 0.8 Hz, 1H), 7.06–7.12 (m, 1H), 3.24 (s, 2H), 3.14 (br t, 4H), 2.43 (br t, 4H); 13C NMR (CDCl3, 126 MHz) δ = 168.3, 160.1, 158.1, 147.5, 137.6, 135.2, 135.2, 135.1, 131.4, 130.9, 129.0, 128.7, 128.3, 127.6, 126.6, 124.7, 124.6, 124.5, 124.5, 122.5, 117.5, 117.3, 58.7, 51.8, 45.6; IR (neat) λmax 3370, 2860, 1770, 1599, 1130, 739, 584; Calcd. for C24H23FN2O4S: C(63.42%), H(5.10%), N(6.16%); Found: C(61.95%), H(5.20%), N(5.98%); HRMS (ESI) m/z calcd for C24H24FN2O4S: 455.1435; found: 455.1433.
Supplementary Material
ACKOWLEDGEMENTS
All stably-transfected human cell lines were kindly provided by Dr. Jon M. Lindstrom, Department of Neuroscience School of Medicine, University of Pennsylvania, Philadelphia, PA. This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant DA029433]. Financial support for BJH is from the National Institutes of Health National Institute on Drug Abuse Diversity Supplement. Financial support for REP is from an American Chemical Society Predoctoral Fellowship in Medicinal Chemistry. We thank OBIC for funding for a Bruker MicrOTOF MS. We would like to thank Karl Werbovetz, Ph.D. and Rostislav Likhotvorik for their help preparing this manuscript.
List of nonstandard abbreviations
- NAM
negative allosteric modulator
- nAChRs
neuronal nicotinic acetylcholine receptors
- HBK
HEPES-buffered Krebs
- SAR
structure activity relationship
- nh
Hill coefficient
- SBVS
structure-based virtual screening
Footnotes
Supporting Information Available: Experimental procedures and characterization of precursors of compounds 12–30. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.LLoyd GK, Williams M. Neuronal nicotinic acetylcholine receptors as novel drug targets. J. Pharmacol. Exp. Ther. 2000;292:461–467. [PubMed] [Google Scholar]
- 2.Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors. Nature Rev. Cancer. 2009;9:195–205. doi: 10.1038/nrc2590. [DOI] [PubMed] [Google Scholar]
- 3.Improgo MRD, Scofield MD, Tapper AR, Gardner PD. The nicotinic acetylcholine receptor CHRNA5/A3/B4 gene cluster: dual role in nicotine addiction and lung cancer. Prog. Neurobiol. 2010;92:212–226. doi: 10.1016/j.pneurobio.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gotti C, Riganti L, Vailati S, Clementi F. Brain neuronal nicotinic receptors as new targets for drug discovery. Curr. Pharma. Des. 2006;12:407–428. doi: 10.2174/138161206775474486. [DOI] [PubMed] [Google Scholar]
- 5.Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nat. Neurosci. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- 6.Cooper E, Couturier S, Ballivet M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature. 1991;350:235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- 7.Anand R, Conroy WG, Schoepfer R, Whiting P, Lindstrom J. Neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes have a pentameric quaternary structure. J. Biol. Chem. 1991;266:11192–11198. [PubMed] [Google Scholar]
- 8.Lukas RJ, Changeux J-P, le Novère N, Albuquerque EX, Balfour JK, Berg DK, Bertrand D, Chiappinelli VA, Clarke PBS, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol. Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- 9.Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J. Med. Chem. 2005;48:4705–4745. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- 10.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 12.Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux J-P. Acetylcholine receptors containing the β2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 13.Knight C, Howard P, Baker CL, Marton JP. The cost-effectiveness of an extended course (12 + 12 weeks) of varenicline compared with other available smoking cessation strategies in the United States: an extension and update to the BENESCO Model. Value Health. 2009;13:209–214. doi: 10.1111/j.1524-4733.2009.00672.x. [DOI] [PubMed] [Google Scholar]
- 14.Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at α4β2 and a full agonist at α7 neuronal nicotinic receptors. Mol. Pharmacol. 2006;70:801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- 15.Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, Sands SB, Schaeffer E, Schulz DW, Tingley FD, Williams KE. Pharmacological profile of the α4β2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacol. 2007;52:985–994. doi: 10.1016/j.neuropharm.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 16.Fiore MC, McCarthy DE, Jackson TC, Zehner ME, Jorenby DE, Mielke M, Smith SS, Guiliani TA, Baker TB. Integrating smoking cessation treatment into primary care: an effectiveness study. Prev. Med. 2004;38:412–420. doi: 10.1016/j.ypmed.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Taly A, Corringer P-J, Guedin D, Lestage P, Changeux J-P. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nature Rev. Drug Disc. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 18.Grady SR, Drenan RM, Breining SR, Yohannes D, Wageman CR, Fedorov NB, McKinney S, Whiteaker P, Bencherif M, Lester HA, Marks MJ. Structural differences determine the relative selectivity of nicotinic compounds for native α4β2*-, α6β2*-, α3β4*-, and α7-nicoine acetylcholine receptors. Neuropharmacol. 2010;58:1054–1066. doi: 10.1016/j.neuropharm.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.López-Hernández GY, Thinschmidt JS, Zheng G, Zhang Z, Crooks PA, Dwoskin LP, Papke RL. Selective inhibition of acetylcholine-evoked responses of α7 neuronal nicotinic acetylcholine receptors by novel tris- and tetrakis-azaaromatic quaternary ammonium antagonists. Mol. Pharmacol. 2009;76:652–666. doi: 10.1124/mol.109.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshimura RF, Hogenkamp DJ, Li WY, Tran MB, Belluzzi JD, Whittemore ER, Leslie FM, Gee KW. Negative allosteric modulation of nicotinic acetylcholine receptors blocks nicotine self-administration in rats. J. Pharmacol. Exp. Therap. 2007;323:907–915. doi: 10.1124/jpet.107.128751. [DOI] [PubMed] [Google Scholar]
- 21.Weltzin MM, Schulte MK. Pharmacological characterization of the allosteric modulator desformylflustrabromine and its interaction with α4β2 neuronal nicotinic acetylcholine receptor orthosteric ligands. J. Pharmacol. Exp. Ther. 2010;334:917–926. doi: 10.1124/jpet.110.167684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen SB, Taylor P. Galanthamine and non-competitive inhibitor binding to ACh-binding protein: evidence for a binding site on non-α-subunit interfaces of heteromeric neuronal nicotinic receptors. J. Mol. Biol. 2007;369:895–901. doi: 10.1016/j.jmb.2007.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rose JE, Behm FM, Westman AC, Evin ED, Tein RM, Ripka GV. Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clin. Pharmacol. Ther. 1994;56:86–99. doi: 10.1038/clpt.1994.105. [DOI] [PubMed] [Google Scholar]
- 24.Watkins SS, Epping-Jordan MP, Koob GF, Markou A. Blockade of nicotine self-administration with nicotinic antagonists in rats. Pharmacol. Biochem. Behav. 1999;62:143–751. doi: 10.1016/s0091-3057(98)00226-3. [DOI] [PubMed] [Google Scholar]
- 25.Henderson BJ, Pavlovicz RE, Allen JD, Gonzalez-Cestari TF, Orac CM, Bonnel AB, Zhu MX, Boyd RT, Li C, Bergmeier SC, McKay DB. Negative allosteric modulators that target human α4β2 neuronal nicotinic receptors. J. Pharmacol. Exp. Therap. 2010;334:761–774. doi: 10.1124/jpet.110.168211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlovicz RE, Henderson BJ, Bonnel AB, Boyd RT, McKay DB, Li C. Identification of a novel negative allosteric site on human α4β2 and α3β4 neuronal nicotinic acetylcholine receptors. PloS. ONE. 2011;6(9):e24949. doi: 10.1371/journal.pone.0024949. PLoS ONE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahasenan KV, Pavlovicz RE, Henderson BJ, González-Cestari TF, Yi B, McKay DB, Li C. Discovery of novel α4β2 neuronal nicotinic receptor modulators through structure-based virtual screening. ACS Med. Chem. Lett. 2011 doi: 10.1021/ml2001714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Wu B, Kuhen KL, Bursulaya B, Nguyen TN, Nguyen DG, He Y. Synthesis and biological evaluations of sulfanyltriazoles as novel HIB-1 non-nuleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2006;16:4174–4177. doi: 10.1016/j.bmcl.2006.05.096. [DOI] [PubMed] [Google Scholar]
- 29.Xie H, Ng D, Savinov SN, Dey B, Kwong PD, Wyatt R, Smith AB, Hendrickson WA. Structure-activity relationships in the binding of chemically derivatized CD4 to gp120 from human immunodeficiency virus. J. Med. Chem. 2007;50:4898–4908. doi: 10.1021/jm070564e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Castro Barbosa ML, de Aluquerue Melo GM, da Silva YKC, de Oliveira Lopes R, de Souza ET, de Queiroz AC, Smaniotto S, Alexandre-Moreira MS, Barreiro EJ, Lima L, Moreira D. Synthesis and pharmacological evaluation of N-phenyl-acetamide sulfonamides designed as novel non-hepatotoxic analgesic candidates. Eur. J. Med. Chem. 2009;44:3612–3620. doi: 10.1016/j.ejmech.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 31.Carpino LA, El-Haham A. The diisopropylcarbodiimide/1-hydroxy-7-azabenzotriazole system: segment coupling and stepwise peptide assembly. Tetrahedron. 1999;55:6813–6830. [Google Scholar]
- 32.McKay DB, Cheng C, González-Cestari TF, McKay SB, El-Hajj R, Bryant DL, Swaan PW, Arason KM, Pulipaka AB, Orac CM, Bergmeier SC. Analogs of methyllycaconitine as novel noncompetitive inhibitors of nicotinic receptors: pharmacological characterization, computational modeling, and pharmacophore development. Mol. Pharmacol. 2007;71:1288–1297. doi: 10.1124/mol.106.033233. [DOI] [PubMed] [Google Scholar]
- 33.González-Cestari TF, Henderson BJ, Pavlovicz RE, McKay SB, El Hajj RA, Pulipaka AB, Orac CM, Reed DD, Boyd RT, Zhu MX, Li C, Bergmeier SC, McKay DB. Effect of novel negative allosteric modulators of neuronal nicotinic receptors on cells expressing native and recombinant nicotinic receptors: implications for drug discovery. J. Pharmacol. Exp. Ther. 2009;328:504–515. doi: 10.1124/jpet.108.144576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.