

Summary
Protein-peptide interactions play important roles in many cellular processes, including signal transduction, trafficking, and immune recognition. Protein conformational changes upon binding, an ill-defined peptide binding surface, and the large number of peptide degrees of freedom make the prediction of protein-peptide interactions particularly challenging. To address these challenges, we perform rapid molecular dynamics simulations in order to examine the energetic and dynamic aspects of protein-peptide binding. We find that, in most cases, we recapitulate the native binding sites and native-like poses of protein-peptide complexes. Inclusion of electrostatic interactions in simulations significantly improves the prediction accuracy. Our results also highlight the importance of protein conformational flexibility, especially side-chain movement, which allows the peptide to optimize its conformation. Our findings not only demonstrate the importance of sufficient sampling of the protein and peptide conformations, but also reveal the possible effects of electrostatics and conformational flexibility on peptide recognition.
Introduction
Protein-peptide interactions play a key role in many cellular processes, such as signaling, regulation, and the formation of protein networks. Peptides are the substrates of many physiological macromolecules, including major histocompatibility complex, insulin degrading enzyme, and HIV protease. They also mediate immune recognition and the induction of immune response (Neduva et al., 2005). Protein-peptide interactions have been exploited in various biotechnological and pharmaceutical applications, such as peptide-based therapeutics (Vlieghe et al., 2010), biosensors, biomarkers (Hao et al., 2008), and functional modulators of proteins (Karanicolas and Kuhlman, 2009). Therefore, understanding the molecular mechanism of protein-peptide recognition and having the ability to predict, manipulate, and design novel protein-peptide interactions will have broad applications in the fields of biology, medicine, and pharmaceutical sciences.
High-resolution structure determination methods such as x-ray crystallography and nuclear magnetic resonance have offered atomic insight into the formation of the protein-peptide complex. Based on available structures, both hydrophobic and hydrophilic interactions, including hydrogen bonds and salt-bridges, are important for stability of the protein-peptide complex. Upon peptide binding, many receptor proteins change their conformations, known as induced fit (Koshland et al., 1958). Furthermore, peptides also experience ordering transitions upon binding to their receptors (London et al., 2010). However, the molecular mechanism of the recognition and binding events that occur between the bound and unbound states remains elusive. Computational modeling offers the opportunity to directly observe the binding event and deconstruct the determinants of protein-peptide recognition.
The modeling of protein-peptide complexes is most often approached in two steps: (i) identification of the peptide binding sites on a protein, and (ii) determination of the native pose of the peptide. A number of methods have been developed to address the first step of modeling, based on sequence (Lopez et al., 2007), structure (Brady and Stouten, 2000; Huang and Schroeder, 2006; Liang et al., 1998), or both (Capra et al., 2009). However, most structure-based methods do not consider binding-induced conformational changes of the receptor. Only a very limited number of blind docking (docking without any prior information about the binding site) studies exist for peptide binding in the literature. Autodock is a docking method commonly used for blind peptide docking; however, the length of the peptide is limited up to four residues (Hetenyi and van der Spoel, 2002). In another blind docking study, coarse-grained modeling and four-body statistical pseudo-potentials are implemented (Aita et al., 2010), however, the binding sites in the selected complexes are also usually the largest or second-largest pockets in the protein (Aita et al., 2010). However, in some cases, the peptide-unbound protein structures do not have a well-defined pocket or the binding site is not one of the largest pockets on the protein (Coleman and Sharp, 2010). In addition, it has been suggested that electrostatic interactions play an important role in the formation of the “encounter complex,” which is the meta-stable state prior to optimization of the binding pose in the formation of the final complex (Sheinerman et al., 2000; Suh et al., 2007; Tang et al., 2006). Considering the net charge variation on protein and peptide surfaces, the electrostatic contribution to peptide recognition can vary from case to case; for example, electrostatics is the major determinant in Calmodulin-peptide recognition (Andre et al., 2004), whereas it has been proposed that electrostatic interactions have no role in PDZ domain-peptide interaction (Harris et al., 2003). The questions remain as to what degree electrostatic interactions contribute to peptide recognition and how the binding site is identified without prior knowledge of peptide-binding-induced conformational changes.
The second step of the protein-peptide recognition problem is often referred to as the docking problem. Flexible docking methods considering both ligand and receptor conformational flexibilities are believed to increase the accuracy of predicting the native pose of small molecules and peptides (Anderson et al., 2001; Antes, 2010; Davis and Baker, 2009; Ding et al., 2010). However, the conformational space of peptides is significantly larger than that of small molecules, due to a larger number of rotatable bonds. As a result, most flexible docking methods developed for small molecules are not applicable in determining protein-peptide binding poses. Moreover, the modeling of protein conformational flexibility, including side-chain and/or backbone flexibility, is computationally expensive (Carlson and McCammon, 2000). Hence, a crucial step in the efficient modeling of protein-peptide interactions is to determine the optimal level of protein conformational flexibility required in order to accurately define the correct binding pose. In order to address these issues, we conduct systematic studies of peptide binding to the peptide-unbound receptor state, at various levels of receptor flexibility.
Molecular dynamics (MD), with its accurate description of atomic interactions, can be employed to study protein-peptide binding. However, the time scale accessible to traditional MD simulations limits their broad applications in MD-based peptide binding prediction (Shan et al., 2011). On the other hand, all-atom discrete molecular dynamics (DMD) can accurately and efficiently fold small, fast-folding proteins (Ding et al., 2008) and sample the conformational dynamics of protein complexes (Karginov et al., 2010; Proctor et al., 2011). We use replica exchange all-atom DMD simulations (Ding et al., 2008) to study protein-peptide binding in a set of ten protein-peptide systems. We perform a set of replica simulations for each system, where the receptors initially are in the unbound state, with varying levels of protein side- and main-chain conformational flexibility. In order to study the effect of long-range electrostatics on peptide binding site recognition, we conduct sets of simulations in both the presence and absence of these interactions. Our computational studies reveal the important contributions of electrostatics and conformational flexibility in protein-peptide binding. Our findings suggest that electrostatic interactions may be the driving force for the formation of an energy landscape favoring the native-like structure, independent of any conformational change of the protein. For nine out of ten complexes, we capture the native peptide binding site area, and in several cases we also recapitulate the near-native binding pose.
Results
We perform replica exchange DMD simulations of ten experimentally well-characterized protein-peptide complexes (see Table S1). No prior knowledge of the binding site location or peptide binding pose is assumed in simulations; we use the peptide-unbound structure (i.e., the apo-structure) of the receptor, and the peptide is initially positioned randomly with respect to the receptor (see Figure S1A). In order to evaluate the effect of conformational flexibility on the accurate modeling of peptide binding, we vary the level of receptor flexibility in simulations: (i) rigid receptor, where both side- and main-chain of the protein apo-structure are fixed; (ii) flexible side-chain, where the side-chains of the apo-structure are allowed to move; and (iii) flexible receptor, where we allow the side-chains to move freely but assign a bias potential to the backbone α-carbons, favoring the native apo-structure contacts. The protein backbone is therefore able to sample conformations near the apo-state.
Recapitulation of experimental binding
We first test whether our simulation methods are able to recapitulate the experimentally-observed protein-peptide complexes. In our simulations, the peptide randomly diffuses and forms both non-native and native contacts with the protein. We select for analysis only those complex structures in which the peptide and the protein are in contact, which we define as any heavy atom of the peptide being within a distance of 5.5 Å from any heavy atom of the receptor (see Figure S1B). We then perform hierarchical clustering of the peptide binding conformations using root-mean-square distances (RMSD) calculated over all heavy atoms of the peptides (see Figure S1C). Finally, we select the lowest energy poses from the highly populated clusters as the putative peptide-binding poses, and calculate the heavy-atom RMSDs of the peptide conformation with respect to the native pose (Figure S1D).
In the case of the PDZ domain-peptide complex (PDB ID: 1BFE), we observe a significant fraction of native-like populations in the flexible side-chain simulation. As illustrated in a typical trajectory starting from the unbound state (gray dots in Fig. 1A), the peptide randomly collides with the protein and forms transient complexes (scattered solid dots in Fig. 1A). Once the native binding-site is sampled (~30 ns), the peptide forms a metastable “encounter-complex” (Sheinerman et al., 2000; Suh et al., 2007; Tang et al., 2006), which allows further conformational rearrangement of the system in order to form the native-like binding complex (~40 ns; RMSD ~ 2–3 Å). In order to identify the binding poses, we collect all bound states from each of the eight replicas (Figure 1B). Without knowledge of the native binding pose, we select the putative binding ensemble of the peptide in the context of the energy landscape (Figure 1C). Here, we use MedusaScore (Yin et al., 2008) in order to evaluate the energy of binding between the peptide and the protein. MedusaScore is based on inter-atomic interactions, including van der Waals, solvation, hydrogen bonding, and electrostatic interactions. The PDZ domain-peptide complex features a well-defined funnel-like energy landscape; lower RMSD results in a more favorable binding energy. Notably, the minimum energy peptide pose in the complex has the minimum RMSD from the native pose (Figure 1C). Furthermore, we perform clustering analysis of the bound conformations. We observe that peptides are present in the native binding site if their RMSD from the native pose is lower than 10 Å. Therefore, we use 10 Å as our clustering cutoff (it is 15 Å for 1JBE in which the peptide is 13-mer). The most highly populated clusters correspond to the low free energy states. For these highly populated clusters, we select the pose with the lowest MedusaScore as the representative structure, and we compare that structure with the crystal structure. The representative structure of the most highly populated cluster of the PDZ domain-peptide complex has a RMSD of 2.5 Å from the crystal structure pose (Figure 1D). Thus, we obtain a native-like conformation of the PDZ domain-binding peptide without any knowledge of the binding site, the conformation of the peptide, or the bound-state structure of the protein.
Figure 1. Analysis of flexible side-chain simulation of PDZ-peptide complex.

(A) RMSD values of peptide conformations with respect to the crystallographic pose of the peptide for peptide-bound (black) and peptide-unbound (gray) states from a representative replica. If any atom of the peptide is within 5.5 Å of any atom of the protein in the trajectory, then that snapshot is considered as a peptide-bound conformation. (B) The backbone of PDZ domain is fixed during simulation, and we reconstruct all peptide-bound states from the simulation trajectories. The positions of the peptide in each peptide-bound frame are displayed in ribbon diagrams. The hit map of peptide interactions with the protein corresponds to the frequency with which the peptide atoms interact with the protein atoms, and these interactions range from very frequent (red) to very infrequent (blue). (C) Energy landscape with the interface energy between the peptide and protein in terms of MedusaScore. (D) The lowest energy conformation (magenta) of the peptide from the largest cluster and its experimental pose (black). See also Figure S1.
In molecular dynamics simulations, the most-populated cluster corresponds to the lowest free energy state, which is not always the state with the lowest potential energy. In proteins with more than one potential binding site, the energy landscapes demonstrate different trends from those of proteins with only one binding site (Figures 1, 2). If multiple binding sites are identified during simulations, clustering analysis is necessary to determine the lowest free energy binding state. In the case of Keap1-peptide complex (PDB ID: 1X2J), the minimum energy pose from the most populated cluster is associated with the native-like conformation, but it does not correspond to the global minimum energy, whereas the lowest energy pose from the entire trajectory (from the second most-populated cluster) suggests a different binding site (Figure 2). Using our clustering analysis, we are able to obtain a conformation similar to the native pose of the peptide in the Keap1-peptide complex.
Figure 2. Analysis of flexible side-chain simulation of Keap1-peptide complex.

Two binding sites exist for this peptide, as exhibited by two low-energy clusters in the energy landscape. The purple ribbon is the lowest energy peptide pose from the most populated cluster, whereas the black ribbon is the experimentally-determined pose. The global minimum energy corresponds to the red conformation; however, that state is less populated than the purple conformation. For the results of all complexes, see also Figure S2 and Figure S3.
We perform similar analysis on six additional protein-peptide complexes. For each complex, we report the clustering results (see Table S2). Except the case 2ZGC, we identify the native binding site from within the first two most-populated clusters (Table 1, see Figure S2). In addition, we test two more cases of longer peptides that form secondary structure in the bound form (PDB IDs: 1RWZ, 1JBE). For these two cases, we can recapitulate the binding sites of the protein and the helical structure of the peptides in the bound conformation (see Figure S3). Our ability to identify the native binding site of the peptide from an arbitrary initial position highlights the sampling efficiency of DMD simulations and the accuracy of our all-atom force field.
Table 1. RMSD (Å) values and cluster population percentages in the presence of electrostatic interactions.
Heavy atom RMSD values are given on the first line of each row, and population sizes in terms of percentage are given on the second line, for each protein-peptide complex (see also Table S1). Backbone RMSD (Å) is given in the parenthesis for the flexible side chain simulations. We report data for only the largest two clusters. The number of clusters is dependent upon the cluster population distribution; we report the clusters having a significant number of samples in Table S2. The bolded values belong to conformations having RMSD lower than 10 Å.
| PDB ID | Fixed receptor | Flexible side-chain | Flexible receptor | |||
|---|---|---|---|---|---|---|
| 1st cluster | 2nd cluster | 1st cluster | 2nd cluster | 1st cluster | 2nd cluster | |
| 1BFE | 8.80 | 9.39 | 2.51(1.0) | 11.31 | 3.19 | |
| 36.5% | 5.2% | 70.8% | 12.9% | 49.2% | -- | |
|
| ||||||
| 1DDW | 5.74 | 9.74 | 7.14(6.49) | 7.01 | 10.04 | 5.67 |
| 26.8% | 21.8% | 34.9% | 15.5% | 12.7% | 8.5% | |
|
| ||||||
| 1CL5 | 11.56 | 22.07 | 8.27(7.17) | 26.21 | 6.57 | 4.85 |
| 16.5% | 5.0% | 19.8% | 5.4% | 14.6% | 14.4% | |
|
| ||||||
| 2HPJ | 4.93 | 3.26(3.25) | 4.46 | |||
| 71.2% | -- | 51.9% | -- | 64.9% | -- | |
|
| ||||||
| 1X2J* | 10.25 | 26.53 | 34.49 | 10.5 (8.02) | 9.36 | 36.26 |
| 57.4% | 4.5% | 24.7% | 17.0% | 22.6% | 11.7% | |
|
| ||||||
| 1SRL | 8.31 | 22.74 | 7.73(5.48) | 20.14 | 6.98 | 10.12 |
| 34.9% | 10.8% | 11.6% | 10.6% | 12.1% | 7.9% | |
|
| ||||||
| 2ID8 | 6.69 | 24.22 | 9.59(9.03) | 33.50 | 11.87 | |
| 31.7% | 18.8% | 35.7% | 5.0% | 54.2% | -- | |
|
| ||||||
| 2ZGC | 37.59 | 35.36 | 24.04(23.7) | 22.51 | 21.82 | 26.73 |
| 18.2% | 9.4% | 27.3% | 6.7% | 10.8% | 5.5% | |
| 1RWZ | 7.21 | 20.9 | 6.43(5.77) | 33.9 | n/a | n/a |
| 26.2% | 14.6% | 29.4% | 11.9% | n/a | n/a | |
| 1JBE | 12.91 | 23.1 | 12.94(10.5) | 29.16 | n/a | n/a |
| 71.5% | 3.8% | 44.2% | 25.9% | n/a | n/a | |
The peptide in this complex is a nonamer, therefore RMSD value of the predictions are expectedly higher.
Electrostatic interactions may be necessary for the identification of the native peptide-binding site
To test the effect of electrostatics on protein-peptide recognition, we also perform simulations without electrostatic interactions (Table 2). Here, we use the Debye-Hückel approximation to model screened electrostatic interactions between charged residues (Methods). We find a significant improvement in the prediction of the binding site and native pose of peptides with the addition of electrostatics to the force field (Tables 1 and 2). In the absence of electrostatics, we observe decoy-binding poses that correspond to lower energies than that of the native pose. With the addition of electrostatics, the number of favorable decoys decreases and the size of the native-like population increases. For example, in most simulations we observe that with the addition of electrostatics, the native-like state becomes the most populated state, as opposed to the second most populated state when electrostatics is not included. However, we do not observe a significant difference in the selected binding pose between simulations with and without electrostatics in the cases of PDZ domain and Serine proteinase K (PDB ID: 2ID8). Our observed nil effect of electrostatic interactions in the special case of peptide recognition by PDZ domain is consistent with experimental observation (Harris et al., 2003). We conclude that, for the majority of protein-peptide complexes, long-range electrostatic interactions play an important role in protein-peptide recognition in simulations by guiding the peptide toward the binding site.
Table 2. RMSD (Å) values and cluster population percentages in the absence of electrostatic interactions.
The electrostatic interactions in the force field are removed, and we perform simulations with conformational constraints similar to those in Table 1. Heavy atom RMSD values are given on the first line of each row, and population sizes in terms of percentage are given on the second line, for each protein-peptide complex. We report data for only the largest two clusters. The number of clusters is dependent upon the cluster population distribution; we report the clusters having a significant number of samples (see Table S2). The bolded values are the conformations having RMSD lower than 10 Å.
| PDB ID | Fixed receptor
|
Flexible side-chain
|
Flexible receptor
|
|||
|---|---|---|---|---|---|---|
| 1st cluster | 2nd cluster | 1st cluster | 2nd cluster | 1st cluster | 2nd cluster | |
| 1BFE | 12.34 | 18.12 | 1.51 | 6.40 | 2.96 | |
| 29.4% | 5.4% | 70.2% | -- | 9.0% | 6.7% | |
|
| ||||||
| 1DDW | 5.74 | 9.74 | 6.25 | 6.25 | 7.51 | 25.76 |
| 26.8% | 21.8% | 34.1% | 18.5% | 26.9% | 13.2% | |
|
| ||||||
| 1CL5 | 11.66 | 21.99 | 11.25 | 6.80 | 7.84 | 6.40 |
| 24.2% | 12.1% | 11.9% | 11.1% | 20% | 7.6% | |
|
| ||||||
| 2HPJ | 25.51 | 25.75 | 18.51 | 4.70 | 17.08 | 24.31 |
| 23.3% | 13.7% | 25.5% | 16.9% | 17.7% | 9.3% | |
|
| ||||||
| 1X2J | 10.72 | 34.01 | 9.73 | 39.93 | 7.41 | 39.37 |
| 30.5% | 17.1% | 19.3% | 14.4% | 16.9% | 11.2% | |
|
| ||||||
| 1SRL | 15.59 | 9.78 | 20.85 | 20.96 | 14.86 | 19.76 |
| 14.1% | 10.3% | 6.7% | 4.8% | 3.6% | 3.4% | |
|
| ||||||
| 2ID8 | 6.69 | 24.34 | 9.93 | 21.24 | 10.28 | |
| 31.7% | 18.8% | 20.2% | 5.2% | 72.3% | -- | |
|
| ||||||
| 2ZGC | 31.18 | 19.80 | 33.79 | 22.25 | 37.31 | 24.55 |
| 8.7% | 7.3% | 35.8% | 3.7% | 8.9% | 4.0% | |
Modeling of protein side-chain flexibility is necessary for accurate peptide binding pose prediction
To investigate the effect of protein conformational dynamics on protein-peptide recognition, we compare binding simulations with increasing levels of receptor conformational flexibility: fixed receptor, flexible side-chain, and flexible receptor constraints (Table 1). In the fixed receptor simulations, we correctly identify the binding sites of all cases except for the PUB domain of PNGase (PDB ID: 2HPJ), the Src SH3 domain (PDB ID: 1SRL), and Granzyme M (PDB ID: 2ZGC). The accuracy of the predictions is significantly increased in these cases if we implement a protocol featuring increased flexibility of the protein receptor (flexible side-chain or flexible receptor). Interestingly, there is no significant difference in the accuracy of binding site prediction between the flexible side-chain and flexible receptor models. However, the inclusion of backbone flexibility in the flexible receptor simulations significantly increases the computational time; including only side-chain flexibility is sufficient to predict the peptide-binding pose. Therefore, we find that flexible side-chain fixed backbone simulations with electrostatic interactions have the most promising results for peptide binding determination, considering the compromise of decreased RMSD of the predicted binding poses from the native pose (compared to fixed receptor) and the decreased computational time required for sampling (compared to flexible receptor).
Discussion
Based on the results of our simulations (Tables 1, 2, S2), we propose a two-step peptide binding mechanism. The binding process includes random collisions of the peptide with various regions of the protein surface. If the peptide encounters a site with which it has favorable interactions, thermodynamically it will remain in this site to form the meta-stable “encounter complex” (Sheinerman et al., 2000; Suh et al., 2007; Tang et al., 2006), which allows the system to find an energetically optimal conformation. In terms of finding the binding site, our results suggest that electrostatic interactions may play an important role in many cases, considering the fact that the majority of peptides contain charged residues. Even in peptides with no charged residues, the amino and carboxyl termini are always charged, making the peptide highly polar. Therefore, it is not surprising that even in fixed receptor simulations, the addition of electrostatic interactions significantly improves the prediction of the peptide-binding site on the receptor (Tables 1,2). This observation suggests that long-range electrostatic interactions guide the peptide toward the peptide-binding surface site, which does not require the formation of a complementary receptor surface. In the absence of electrostatics, the energy well corresponding to the native-like pose is broad and has a higher energy than that of the decoy pose (Figure 3). However, when we include electrostatics in the force field, we observe a lower energy well for the native-like states (Figure 4). Finally, the addition of conformational flexibility provides additional definition to the energy landscape, as well as narrows and lowers the energy well (Figure 4). Therefore, in simulations, both electrostatics and flexibility of the protein receptor are necessary for forming the energetic landscape of peptide binding.
Figure 3. Proposed model for the structural and dynamic determinants of peptide recognition.
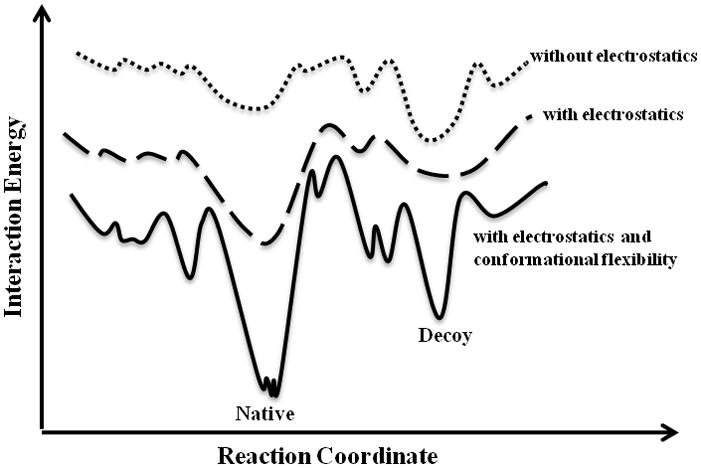
The dotted line represents binding without electrostatic interactions. The dashed line represents binding with electrostatic interactions. In the presence of electrostatics, the number of decoy states decreases, whereas the native-like funnel becomes more populated. The solid line represents the binding with both electrostatic interactions and conformational flexibility. Here, the native-like funnel experiences more sampling and a decrease in its energy.
Figure 4. MedusaDock-refined experimental and predicted conformations.

Using MedusaDock, we improve the prediction accuracy of (A) 1DDV, (B) 1PRM, (C) 1X2R, (D) 2FNX, (E) 2PQ2 complexes. The selected conformations from the simulations with flexible side-chain constraints in the presence of electrostatics are employed as initial conformations for docking optimization. Shown are the native binding pose (black) and the predicted binding pose before (magenta) and after (blue) docking refinement.
According to our results, peptides are able to find the binding site in many cases with a fixed receptor. This finding is consistent with a recent study (London et al., 2010) that systematically compared the bound and unbound forms of protein structures upon peptide binding. London et al. (London et al., 2010) found that, in 86% of cases, the protein does not significantly change its conformation upon peptide binding. The peptide binds to the protein by minimizing the conformational change of the protein, while maximizing the enthalpy gained by hydrogen bonds and packing. Thus, the peptide, rather than the protein, undergoes induced fit, since it adapts its conformation to the binding site of the protein. This phenomenon is different from small-molecule binding, where proteins adopt their conformations upon ligand binding (Mobley and Dill, 2009), because small molecules are relatively rigid in comparison to peptides. However, there may be some exceptional cases where a large conformational change occurs upon peptide binding. In the case of PCNA-FEN-1-peptide complex (see Figure S3A), the C-terminal flexible loop of PCNA forms β-strands with the N-terminus of the peptide upon binding, resulting in an average RMSD of 3.5 Å with respect to the unbound conformation, while the C-terminus of the peptide forms a helical secondary structure. This peptide-binding-induced conformational change in the protein is suggested as the structural basis for the allosteric control of enzyme activities in DNA mismatch repair (Chapados et al., 2004). In our simulations, we are able to predict the correct binding site for the peptide and its helical secondary structure (1RXZ), (see Figure S3A-D), but the prediction of the ligand-binding-induced protein backbone changes remains a major challenge. As an additional analysis, we also calculate the size of pockets/cavities on the proteins using the CASTp server (Dundas et al., 2006) to check whether the peptide always binds to the largest pockets. According to these results (see Table S5), the binding site is located in the largest pocket only in 1CL5. The binding sites of 2PQ2 and 2ZGC are the second-largest pockets on their surfaces, the binding sites of 1DDV and 2HPJ are in the fourth-largest pockets, and the binding sites of 1BFE, 1SRL, 1X2J are not located in any of the five largest pockets.
The same protein can populate multiple binding modes (Birdsall et al., 1989; Ma et al., 2002). Conversely, a ligand can bind to a target with multiple conformations, due to symmetries in the ligand or receptor protein (Mobley and Dill, 2009). For example, similar amino acids on the two termini of a peptide can result in a flipped conformation relative to the x-ray structure. Although observing multiple binding modes is rare (Constantine et al., 2008; Lazaridis et al., 2002; Montfort et al., 1990), some studies showed multiple-mode binding without a symmetry effect (Jayachandran et al., 2006; Lazaridis et al., 2002; Oostenbrink and van Gunsteren, 2004); for instance, we observe multiple binding modes in our simulations of Keap1-peptide complex (Figure 2). In the case of the Granzyme M-peptide complex, we cannot recapitulate the crystal structure binding site, but instead the peptide binds to a completely different region of the protein surface. Examination of the peptide-bound structure shows that there is a large conformational change in the binding site of Granzyme M upon binding of the peptide (Wu et al., 2009). However, the possibility remains that the identified binding site is an alternative to the crystallographic site for Granzyme M (Wu et al., 2009).
We also address the question of whether we can improve prediction accuracy by performing additional sampling in the vicinity of the binding site. We initiate sampling using the receptor conformation from the simulation using the flexible side-chain model with electrostatic interactions. We constrain the peptide near the binding site and perform replica exchange simulations with two types of flexible receptor models: (i) flexible side-chain and (ii) flexible receptor. We do not observe a significant increase in prediction accuracies; the sampling used in the initial simulations is already sufficient to identify the binding site and near-native pose of the peptide (see Table S3). In addition, to improve the prediction accuracy, we perform molecular docking using MedusaDock (Ding et al., 2010) for the seven cases in which we were able to obtain the native binding site (Table 3, Figure 4). Interestingly, for the complexes in which we predict native-like poses with DMD simulation alone (PDB ID: 1BFE and 2HPJ), we do not observe a decrease in RMSD values after refining with MedusaDock. Only in the case of Phospholipase A2 (PDB ID: 1CL5) does the MedusaDock refinement result in a significantly improved binding pose, with RMSD decreased from 8.3 Å to 3.7 Å. In the other four cases, we observe only minor improvement in prediction, with the top five MedusaDock predicted poses having slightly lower RMSDs than those obtained with simulation alone (Table 3, Figure 4). To test whether another peptide docking method will improve the optimization of the peptide pose, we perform a similar procedure with the Flexpepdock server (Raveh et al., 2010) and PepSite (Petsalaki et al., 2009). Flexpepdock (Raveh et al., 2010) is a freely available peptide docking protocol that is proposed to refine peptide binding poses. According to FlexPepDock results, compared to the initial poses, we do not observe significant improvement in terms of the RMSD values (Table 3). The Pepsite algorithm is knowledge-based, and incorporates information from known protein-peptide complexes based on spatial position specific scoring matrices (S-PSSMs) to identify the binding preference of amino acids onto protein surfaces (Petsalaki et al., 2009). The surface of the protein is then scanned using this matrix to find potential binding sites for the peptide. Conversely, our algorithm, replica exchange DMD, is a physical method that does not rely on any protein-peptide complex structural information. The Pepsite server provides predictions of potential binding sites for each individual residue in the peptide and the top nine conformations of the peptide. For 1BFE, 1CL5, 2HPJ, 2ID8, 2ZGC, 1RWZ, and 1JBE the server cannot correctly predict the binding sites of any residues (Table S5). In 1DDW, only the location of one proline residue is predicted correctly at the third highest rank. For 2ZGC, the binding site of Lysine is predicted at the 7th rank. We conclude from these results that, at least for certain targets, existing protein-peptide complex information is not sufficient to provide an adequate knowledge base for evaluation. In addition, the accurate prediction of native-like peptide binding poses, especially for long peptides, remains a challenging task.
Table 3. RMSD (Å) with respect to native pose before and after molecular docking.
The predicted conformations from the simulations with flexible side-chain constraints are used as initial conformations for docking calculations. We report the lowest RMSD values from the top five lowest energy conformations and their rank in predicted models predicted by MedusaDock and FlexPepDock. We also compare our results with castP and PEPSITE server (see Table S5).
| PDB ID | Initial | MedusaDock | FlexPepDock |
|---|---|---|---|
| 1BFE | 2.51 | 2.85 (2) | 2.49 (4) |
| 1DDW | 7.14 | 5.98 (5) | 6.83 (3) |
| 1CL5 | 8.27 | 3.73 (4) | 7.42 (1) |
| 2HPJ | 3.26 | 5.05 (2) | 3.02 (1) |
| 1X2J | 10.51 | 8.33 (4) | 9.56 (4) |
| 1SRL | 7.73 | 5.71 (4) | 6.83 (3) |
| 2ID8 | 9.59 | 6.70 (1) | 9.80 (3) |
Conclusion
The prediction of peptide binding poses is one of the most challenging problems in computational structural biology, due to the large number of peptide degrees of freedom. Here, we have developed a protein-peptide docking procedure that allows us to identify the peptide-binding region of proteins, as well as a near-native pose of the peptides. The direct observation of peptide binding in simulations reveals a possible two-step protein-peptide recognition mechanism. The initial step, the route of the peptide to the binding site to form the meta-stable “encounter complex”, is suggested to be guided by electrostatics. Electrostatic interactions determine the formation of a funnel-like energy landscape directed toward the native binding-site. In most cases, recognition of the binding site on the receptor surface does not depend on whether or not the protein is in the binding-competent state. The second step corresponds to the docking of the peptide on the protein surface, which requires conformational change of the receptor in order to reach the native-like binding pose. Our benchmark study suggests that the flexible receptor side-chain model is the optimal method to identify the peptide binding site and to search for the near-native binding pose; however, the fixed receptor approach may be sufficient to identify the approximate peptide binding site. The proposed method both aids in the understanding of the protein-peptide interaction mechanism, and can also be used for various biotechnological purposes, including the design of peptide-based drugs and protein-peptide interfaces.
Methods
We provide a flowchart of our procedure in the supplementary material (Figure S1D).
Dataset
We select proteins that have both holo (co-crystallized with a peptide) and apo (crystallized without a peptide) structures available (Table S1). Our data set includes PDZ domain (PDB ID: 1BFE), homer evh1 (PDB ID: 1DDW), Src SH3 domain (PDB ID: 1SRL), Keap1 (PDB ID: 1X2J), Phospholipase A2 (PDB ID: 1CL5), p97/PNGase (PDB ID: 2HPJ), serine proteinase K (PDB ID: 2ID8), Granzyme M (PDB ID: 2ZGC), PGNC (PDB ID: 1RXZ), CheY (PDB ID: 1JBE). We place the peptide at a randomly selected position around the unbound state of the protein.
All-atom replica exchange DMD
Discrete molecular dynamics (DMD) is an event-driven molecular dynamics simulation engine in which inter-atomic interactions are approximated by square well potentials (Dokholyan et al., 1998). We model proteins using the united atom representation, where all heavy atoms and polar hydrogen atoms are explicitly modeled (Ding et al., 2008). We model van der Waals interactions using the Lennard-Jones potential, and solvation interactions using the Lazaridis-Karplus solvation effect (Lazaridis and Karplus, 1999). All of these continuous functions are discretized by multi-step square well functions.
In addition to the previous version of the all-atom DMD force field (Ding et al., 2008), we also incorporate electrostatic interactions between charged residues, including basic and acidic residues (Ding et al., 2010). We assign integer charges to the central atoms of charged groups: CZ for Arg, NZ for Lys, CG for Asp, and CD for Glu. We use the Debye-Hückel approximation to model the screened charge-charge interactions. The Debye length is set at 10 Å by assuming a monovalent electrolyte concentration of 0.1 mM. We use 80 as the relative permittivity of water in order to compute the screened charge-charge interaction potential. We discretize the continuous electrostatic interaction potential with an interaction range of 30 Å, where the screened potential approaches zero.
We employ the replica-exchange sampling scheme (Okamoto, 2004; Zhou et al., 2001) to overcome energy barriers while maintaining conformational sampling corresponding to the relevant free energy surface. In replica exchange computing, multiple simulations or replicas of the same system are performed in parallel at different temperatures. The individual simulations are coupled through Monte Carlo-based exchanges of simulation temperatures between replicas at periodic time intervals. We perform simulation replicas with temperatures ranging from 0.50 kcal/(mol•kB) (approximately 250 K) to 0.75 kcal/(mol•kB) (approximately 375 K), with an increment of 0.035 kcal/(mol•kB) (approximately 17.5 K). The length of each simulation is 106 time units, corresponding to approximately 50 ns. In addition, wall clock and CPU hours for simulations are provided in Table S4.
Molecular Docking
For refinement, we use MedusaDock (Ding et al., 2010) which is a flexible docking method that allows simultaneous modeling of both ligand and receptor flexibility with a set of discrete rotamers. We employ as initial structures the predicted poses from the flexible side-chain simulations with electrostatics. For all cases, the heavy-atom RMSD values from the experimentally-determine conformation decrease significantly, approaching the native-like pose (Table 3, Figure 3).
Supplementary Material
Highlights.
Direct observation of protein-peptide binding in molecular dynamics simulations
Electrostatic interactions guide protein-peptide recognition
Direct observation of induced-fit phenomenon in peptides
Novel method for protein-peptide docking
Acknowledgments
We thank Rachel L. Redler and Irem Dagliyan for critical reading of the manuscript. This work is supported by the National Institute of Health Grant R01GM080742 and the ARRA supplement 3R01GM080742-03S1 (to N.V.D.), National Institute of Health Predoctoral Fellowship F31AG039266-01 from the National Institute on Aging (to E.A.P.), and by the UNC Research Council (to F.D.). Calculations were performed on the high-performance computing cluster at the University of North Carolina at Chapel Hill.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aita T, Nishigaki K, Husimi Y. Toward the fast blind docking of a peptide to a target protein by using a four-body statistical pseudo-potential. Comput Biol Chem. 2010;34:53–62. doi: 10.1016/j.compbiolchem.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Anderson AC, O’Neil RH, Surti TS, Stroud RM. Approaches to solving the rigid receptor problem by identifying a minimal set of flexible residues during ligand docking. Chem Biol. 2001;8:445–457. doi: 10.1016/s1074-5521(01)00023-0. [DOI] [PubMed] [Google Scholar]
- Andre I, Kesvatera T, Jonsson B, Akerfeldt KS, Linse S. The role of electrostatic interactions in calmodulin-peptide complex formation. Biophys J. 2004;87:1929–1938. doi: 10.1529/biophysj.104.040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antes I. DynaDock: A new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins. 2010;78:1084–1104. doi: 10.1002/prot.22629. [DOI] [PubMed] [Google Scholar]
- Birdsall B, Feeney J, Tendler SJ, Hammond SJ, Roberts GC. Dihydrofolate reductase: multiple conformations and alternative modes of substrate binding. Biochemistry. 1989;28:2297–2305. doi: 10.1021/bi00431a048. [DOI] [PubMed] [Google Scholar]
- Brady GP, Jr, Stouten PF. Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des. 2000;14:383–401. doi: 10.1023/a:1008124202956. [DOI] [PubMed] [Google Scholar]
- Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput Biol. 2009;5:e1000585. doi: 10.1371/journal.pcbi.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson HA, McCammon JA. Accommodating protein flexibility in computational drug design. Mol Pharmacol. 2000;57:213–218. [PubMed] [Google Scholar]
- Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- Coleman RG, Sharp KA. Protein pockets: inventory, shape, and comparison. J Chem Inf Model. 2010;50:589–603. doi: 10.1021/ci900397t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantine KL, Mueller L, Metzler WJ, McDonnell PA, Todderud G, Goldfarb V, Fan Y, Newitt JA, Kiefer SE, Gao M, et al. Multiple and single binding modes of fragment-like kinase inhibitors revealed by molecular modeling, residue type-selective protonation, and nuclear overhauser effects. J Med Chem. 2008;51:6225–6229. doi: 10.1021/jm800747w. [DOI] [PubMed] [Google Scholar]
- Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Ding F, Tsao D, Nie H, Dokholyan NV. Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure. 2008;16:1010–1018. doi: 10.1016/j.str.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Yin S, Dokholyan NV. Rapid flexible docking using a stochastic rotamer library of ligands. J Chem Inf Model. 2010;50:1623–1632. doi: 10.1021/ci100218t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Discrete molecular dynamics studies of the folding of a protein-like model. Fold Des. 1998;3:577–587. doi: 10.1016/S1359-0278(98)00072-8. [DOI] [PubMed] [Google Scholar]
- Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Serohijos AW, Newton G, Tassone G, Wang Z, Sgroi DC, Dokholyan NV, Basilion JP. Identification and rational redesign of peptide ligands to CRIP1, a novel biomarker for cancers. PLoS Comput Biol. 2008;4:e1000138. doi: 10.1371/journal.pcbi.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BZ, Lau FW, Fujii N, Guy RK, Lim WA. Role of electrostatic interactions in PDZ domain ligand recognition. Biochemistry. 2003;42:2797–2805. doi: 10.1021/bi027061p. [DOI] [PubMed] [Google Scholar]
- Hetenyi C, van der Spoel D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002;11:1729–1737. doi: 10.1110/ps.0202302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Schroeder M. LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct Biol. 2006;6:19. doi: 10.1186/1472-6807-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayachandran G, Shirts MR, Park S, Pande VS. Parallelized-over-parts computation of absolute binding free energy with docking and molecular dynamics. J Chem Phys. 2006;125:084901. doi: 10.1063/1.2221680. [DOI] [PubMed] [Google Scholar]
- Karanicolas J, Kuhlman B. Computational design of affinity and specificity at protein-protein interfaces. Curr Opin Struct Biol. 2009;19:458–463. doi: 10.1016/j.sbi.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM. Engineered allosteric activation of kinases in living cells. Nat Biotechnol. 2010;28:743–747. doi: 10.1038/nbt.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE, Jr, Ray WJ, Jr, Erwin MJ. Protein structure and enzyme action. Fed Proc. 1958;17:1145–1150. [PubMed] [Google Scholar]
- Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Lazaridis T, Masunov A, Gandolfo F. Contributions to the binding free energy of ligands to avidin and streptavidin. Proteins. 2002;47:194–208. doi: 10.1002/prot.10086. [DOI] [PubMed] [Google Scholar]
- Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure. 2010;18:188–199. doi: 10.1016/j.str.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Lopez G, Valencia A, Tress ML. firestar--prediction of functionally important residues using structural templates and alignment reliability. Nucleic Acids Res. 2007;35:W573–577. doi: 10.1093/nar/gkm297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley DL, Dill KA. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montfort WR, Perry KM, Fauman EB, Finer-Moore JS, Maley GF, Hardy L, Maley F, Stroud RM. Structure, multiple site binding, and segmental accommodation in thymidylate synthase on binding dUMP and an anti-folate. Biochemistry. 1990;29:6964–6977. doi: 10.1021/bi00482a004. [DOI] [PubMed] [Google Scholar]
- Neduva V, Linding R, Su-Angrand I, Stark A, de Masi F, Gibson TJ, Lewis J, Serrano L, Russell RB. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 2005;3:e405. doi: 10.1371/journal.pbio.0030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y. Generalized-ensemble algorithms: enhanced sampling techniques for Monte Carlo and molecular dynamics simulations. J Mol Graph Model. 2004;22:425–439. doi: 10.1016/j.jmgm.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C, van Gunsteren WF. Free energies of binding of polychlorinated biphenyls to the estrogen receptor from a single simulation. Proteins. 2004;54:237–246. doi: 10.1002/prot.10558. [DOI] [PubMed] [Google Scholar]
- Petsalaki E, Stark A, Garcia-Urdiales E, Russell RB. Accurate prediction of peptide binding sites on protein surfaces. PLoS Comput Biol. 2009;5:e1000335. doi: 10.1371/journal.pcbi.1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor EA, Ding F, Dokholyan NV. Structural and Thermodynamic Effects of Post-translational Modifications in Mutant and Wild Type Cu, Zn Superoxide Dismutase. J Mol Biol. 2011;408:555–567. doi: 10.1016/j.jmb.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh B, London N, Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins. 2010;78:2029–2040. doi: 10.1002/prot.22716. [DOI] [PubMed] [Google Scholar]
- Shan Y, Kim ET, Eastwood MP, Dror RO, Seeliger MA, Shaw DE. How Does a Drug Molecule Find Its Target Binding Site? J Am Chem Soc. 2011 doi: 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein-protein interactions. Curr Opin Struct Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Suh JY, Tang C, Clore GM. Role of electrostatic interactions in transient encounter complexes in protein-protein association investigated by paramagnetic relaxation enhancement. J Am Chem Soc. 2007;129:12954–12955. doi: 10.1021/ja0760978. [DOI] [PubMed] [Google Scholar]
- Tang C, Iwahara J, Clore GM. Visualization of transient encounter complexes in protein-protein association. Nature. 2006;444:383–386. doi: 10.1038/nature05201. [DOI] [PubMed] [Google Scholar]
- Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Wu L, Wang L, Hua G, Liu K, Yang X, Zhai Y, Bartlam M, Sun F, Fan Z. Structural basis for proteolytic specificity of the human apoptosis-inducing granzyme M. J Immunol. 2009;183:421–429. doi: 10.4049/jimmunol.0803088. [DOI] [PubMed] [Google Scholar]
- Yin S, Biedermannova L, Vondrasek J, Dokholyan NV. MedusaScore: an accurate force field-based scoring function for virtual drug screening. J Chem Inf Model. 2008;48:1656–1662. doi: 10.1021/ci8001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Berne BJ, Germain R. The free energy landscape for beta hairpin folding in explicit water. Proc Natl Acad Sci U S A. 2001;98:14931–14936. doi: 10.1073/pnas.201543998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.