

SUMMARY
Cell survival in changing environments requires appropriate regulation of gene expression, including post-transcriptional regulatory mechanisms. From reporter gene studies in glucose-starved yeast, it was proposed that translationally silenced eukaryotic mRNAs accumulate in P-bodies and can return to active translation. We present evidence contradicting the notion that reversible storage of non-translating mRNAs is a widespread and general phenomenon. First, genome-wide measurements of mRNA abundance, translation, and ribosome occupancy following glucose withdrawal show that most mRNAs are depleted from the cell coincident with their depletion from polysomes. Second, only a limited sub-population of translationally repressed transcripts, comprising fewer than 400 genes, can be reactivated for translation upon glucose re-addition in the absence of new transcription. This highly selective post-transcriptional regulation could be a mechanism for cells to minimize the energetic costs of reversing gene-regulatory decisions in rapidly changing environments by transiently preserving a pool of transcripts whose translation is rate-limiting for growth.
INTRODUCTION
Cells respond to changing environments by regulating gene expression. Regulation can occur at the level of transcription and/or post-transcriptionally during processes including pre-mRNA splicing, mRNA export, translation and mRNA decay. In some embryonic cells, gene regulation during early development is entirely post-transcriptional and involves temporally and spatially controlled translation of maternally deposited mRNAs (Johnstone and Lasko, 2001; Richter, 1991). More typically, cells employ a combination of transcriptional and post-transcriptional regulatory strategies. The logical and mechanistic relationships between transcriptional and post-transcriptional regulation are poorly understood, if indeed such relationships exist.
Various hypotheses have been proposed for the role of translational regulation in contexts where transcriptional regulatory mechanisms are also active. For example, translational activation of pre-existing mRNAs can produce new protein faster than transcriptional activation of the same genes, and may therefore be important in situations that demand rapid responses. In addition, translational mechanisms can control where proteins are produced within the cell. Furthermore, translational regulation has been suggested to act globally as an amplifier of the effects of transcriptional gene control, increasing the protein output from transcriptionally induced genes and further decreasing the protein output from transcriptionally repressed genes (Melamed et al., 2008; Preiss et al., 2003). On the other hand, translational attenuation has also been proposed to act as a global dampener of transcriptional noise in gene expression (Blake et al., 2003; Ozbudak et al., 2002; Raser and O’Shea, 2005).
We set out to determine the relationship between the programs of transcriptional and translational response to stress. We further sought to determine the biological logic behind selection of specific mRNAs for translational regulation, and the molecular differences between genes controlled at transcriptional versus translational levels. The glucose starvation response in yeast is an appropriate model system because glucose withdrawal induces widespread changes in both transcription and translation. Transcriptional changes are mediated by well-characterized signaling pathways and transcription factors (Zaman et al., 2008). Translation activity changes by an incompletely understood mechanism requiring genes that have been variously implicated in deadenylation-dependent mRNA decapping and decay, mRNA sub-cellular localization, and the formation of translationally repressed mRNPs (Ashe et al., 2000) (Brengues et al., 2005; Coller and Parker, 2004; Coller and Parker, 2005; Holmes et al., 2004; Teixeira et al., 2005). In response to glucose starvation, yeast initiate a cellular differentiation program known as haploid invasive growth, which is thought to function as a cellular foraging response (Cullen and Sprague, 2000). Because this cellular adaptive response to glucose starvation requires new protein synthesis, the ‘global’ repression of translation must either be short-lived or in fact affect only a subset of genes.
Here we used DNA microarrays to investigate changes in mRNA abundance, translation activity and ribosome occupancy during a two-hour time course following glucose withdrawal. We found that the view of ‘global’ translational repression is over-generalized. While ‘bulk’ translation was greatly reduced, hundreds of newly transcribed mRNAs associated with polysomes within ten minutes of glucose withdrawal. Functionally coherent groups of genes were co-regulated at the post-transcriptional as well as transcriptional level. Using computational approaches, we related gene-specific post-transcriptional changes to underlying mRNA properties by exploiting recent genome-wide studies of yeast mRNA characteristics including abundance, half-life, translational efficiency, poly(A) tail length and association with various RNA-binding proteins. Following a lead generated by this analysis, we examined whether all genes or only a specific sub-population of genes are capable of returning to active translation in the absence of new transcription. In contradiction of the prevailing model in the field, we found that the capacity for translational reactivation is narrowly restricted to a limited subset of mRNAs. Transient preservation of these select mRNAs, whose translation is rate-limiting for growth in rich media, could act as a buffer to minimize the fitness costs associated with “false alarms” caused by transient depletion of glucose or by noise in glucose-sensitive signaling pathways.
RESULTS
We studied wild type invasive-growth competent yeast subjected to acute glucose starvation. Cells were starved for 0, 10, 20, 30, 60, or 120 minutes before processing. For each time point, polysome profiles were generated to monitor global translation. In agreement with previous reports (Ashe et al., 2000; Kuhn et al., 2001), a collapse of the polysome region and concomitant increase in the 80S monosome peak occurred after 10 minutes of glucose withdrawal, indicating a bulk reduction in translation initiation. Previous investigations of the translational response to acute glucose withdrawal using either bulk measurements (Ashe et al., 2000; Holmes et al., 2004) or examination of a few genes (Brengues et al., 2005) suggested that little translation initiation occurs 10–20 minutes following glucose removal. In light of prior studies of glucose-stimulated transcriptional regulation, as well as genetic evidence that many of the genes induced upon glucose depletion are required for growth in the absence of glucose, we reasoned that such transcripts must at some time associate with polysomes in glucose-starved cells (Zaman et al., 2008). We extended the time course of the experiment to learn when bulk translation recovers as cells adapt to growth in low glucose concentrations. Polysomes remained low between 10 and 30 minutes post glucose withdrawal, and showed signs of partial recovery after 60 minutes, with further increases by 120 minutes (Fig. 1A).
Figure 1. Regulation of transcription and translation in glucose-starved cells.

(A) Polysome profiles of yeast starved for glucose by transfer from YPD to YP media. (B) Schematic of the microarray comparisons performed. (C) Time-resolved gene expression profiles resulting from the comparisons shown in B. Ratio values are indicated by color scale and represent the average of eight measurements (2 biological replicates × 2 technical replicates × 2 probes per gene). Rows (genes) are ordered according to the results of hierarchical clustering based on Euclidean distance with P/T values assigned twice the weight of P/P or T/T. (D) Unusual gene clusters for which polysomal mRNA abundance diverged from total mRNA abundance. (E) P/T ratios are more similar to each other than to T/T or P/T for any time point. Branch lengths represent Spearman rank correlation coefficients between data columns from C. (F) Total mRNA abundance changes parallel and exceed polysomal mRNA abundance changes after 60 minutes minus glucose. Dotted line indicates best fit by linear regression. See also Figures S1 and S2.
Changes in Polysomal mRNA Levels Closely Parallel Changes in Transcript Abundance
To examine the kinetics of gene-specific translation activity following glucose withdrawal, we isolated RNA from polysome fractions and total cell lysates, and prepared fluorescently labeled cDNA for competitive hybridization on custom microarrays. In order to determine relative changes in translation activity, mRNA abundance, and the fraction of total mRNA associated with polysomes (ribosome occupancy), we performed the following comparisons for each time point after glucose withdrawal: polysomal RNA starved with polysomal RNA mock-starved (Px/P0); total RNA starved with total RNA mock-starved (Tx/T0); and polysome starved x-min with total starved x-min (PxTx), respectively (Fig. 1B). All experiments were performed twice (biological replicates), with each RNA sample processed in duplicate (technical replicates). ‘Noisy’ genes for which mRNA abundance or polysome association varied by > 1.5-fold between biological replicate experiments in unstarved cells were omitted from further analysis (495 genes). Reproducible results were obtained for 5,590 genes.
Glucose withdrawal led to changes in the relative mRNA abundance of hundreds of genes within 10 minutes, consistent with prior studies of carbon source-mediated regulation of yeast transcription (Zaman et al., 2008). These changes persisted and were amplified over the course of 120 minutes of starvation. Our data do not distinguish between transcriptional induction/repression and mRNA stabilization/destabilization as the mechanism responsible for changing total mRNA abundance. Both mechanisms likely contribute. Many of the genes that showed relatively increased mRNA levels following glucose withdrawal are known targets of glucose regulated transcription factors (Fig. S1). For most genes, changes in their relative mRNA abundance in polysomes (P/P) mirrored changes in overall mRNA levels (T/T) (Fig. 1C). Notably, for the more than 1,000 genes whose relative mRNA abundance increased by 2-fold or more after glucose withdrawal, relative polysomal abundance similarly increased. Furthermore, most induced genes did not show a noticeable lag between the increase of mRNA in the total RNA pool and appearance in the polysomal fraction. Thus, despite the reduction in the rate of ‘bulk’ protein synthesis, translation initiation occurred on transcriptionally up-regulated mRNAs as early as 10 minutes following glucose withdrawal. Reduction in polysomal mRNA closely paralleled reduction in total mRNA levels for almost all down-regulated genes. These data contradict the model that glucose withdrawal leads to widespread sequestration of mRNAs in stable translationally repressed mRNPs (Brengues and Parker, 2007; Brengues et al., 2005; Hoyle et al., 2007; Teixeira et al., 2005).
Unsupervised hierarchical clustering revealed small groups of genes that appear to be regulated primarily at the post-transcriptional level: cluster I genes showed relatively reduced polysomal mRNA levels despite increased total mRNA abundance due to low ribosome occupancy; conversely, cluster II genes had relatively unaffected polysomal mRNA levels despite reduced total mRNA abundance due to high ribosome occupancy (Fig. 1D). Nevertheless, there was strong overall agreement between the relative increase or decrease of an mRNA in the cell at any time point after glucose withdrawal (T/T) and the change in polysome association at that time point (P/P). In contrast, ribosome occupancy (P/T) at any time point was more highly correlated with ribosome occupancy at other times than with either P/P or T/T at the same time point after glucose withdrawal (Fig. 1E), suggesting that it is largely an intrinsic property of each mRNA, although some starvation-induced changes in P/T were observed. At the global scale, fold-changes in polysomal mRNA levels were somewhat compressed compared to changes in total mRNA levels (Fig. 1F, S2). Thus, our data do not indicate widespread ‘potentiation’, whereby changes in total mRNA levels are amplified by homodirectional changes in translation efficiency, as was suggested in studies of yeast subject to rapamycin treatment or heat shock (Preiss et al., 2003). Stress specificity of ‘potentiation’ was previously noticed in comparison of the translational responses to amino acid starvation and butanol stress (Smirnova et al., 2005).
Relationships Between Changes in Transcript Levels and Ribosome Occupancy
The complex groupings of genes produced by combining the analysis of transcriptional and post-transcriptional regulatory behavior using unsupervised hierarchical clustering did not readily reveal the logic underlying the relationship between the two modes of regulation. To investigate this relationship more directly, changes in total mRNA levels (T/T) and changes in ribosome occupancy (P/T) were analyzed separately using k-means clustering to identify groups of genes displaying similar behavior for each mode of regulation. Experimenting with various group numbers (k = 2–20) revealed that k = 7 for the T/T comparisons and k = 3 for the P/T comparisons gave robust solutions reflecting a reasonable compromise between preserving the complexity of the data and simplifying the subsequent analysis (Fig. S3). Clustering genes by their ribosome occupancy (P/T) produced simple divisions into groups with high, low, and neutral P/T ratios (Fig. 2A).
Figure 2. Ribosome occupancy and mRNA abundance are divergently regulated.

(A) The seven groups of genes identified by k-means clustering of T/T ratios include different proportions of genes with high, low, or neutral P/T ratios. Data and color scale are as in Figure 1C, with rows (genes) re-ordered to highlight the differences in ribosome occupancy (P/T) among genes with similar glucose-withdrawal induced changes in total mRNA abundance (T/T). P/P ratios are displayed for comparison but were not considered during clustering. Selected GO categories enriched in specific P/T groups are indicated. (“mito.” – mitochondrial; “RBG” – ribosome biogenesis; “RPG” – ribosomal protein gene) (B) P/T ratios are not equally distributed among T/T groups. Asterisks (*) indicate significant deviations from the distributions predicted by chance (Fisher’s exact test 2-tailed p-value, * < 0.05; ** < 0.005; *** < 0.0005). See also Figure S3.
Although we anticipated that kinetic analysis of the total mRNA changes in response to glucose starvation would reveal temporal distinctions between genes, the groups of co-regulated genes identified by k-means clustering at k = 7 differed primarily by the magnitude of relative increase/decrease rather than by the timing of the maximal changes in gene expression (Fig. 2A). Clustering with values of k > 12 did reveal kinetically distinct patterns of mRNA accumulation that are consistent with current understanding of transcriptional responses to glucose withdrawal (Fig. S3). For example, genes subject to repression by Mig1 in the presence of glucose (e.g. SUC2 and CAT8) were de-repressed within 10 minutes following glucose withdrawal, whereas known targets of the Cat8 transcription factor (e.g. PCK1, ICL1, and FBP1) accumulated mRNA strongly only after 120 minutes (Fig. S1). Groups of genes clustered based on starvation-induced changes in total relative mRNA levels ranged from strongly induced (119 genes, median induction at t = 60 min of 19.7-fold) to strongly repressed (311 genes, median repression at t = 60 min of 11.9-fold). The use of multiple time points as well as both biological and technical replicates allowed the confident identification of genes displaying modest yet consistent changes in mRNA levels. The most weakly induced category includes more than 100 genes encoding mitochondrial proteins known to be important for growth in the absence of glucose, highlighting the potential biological significance of coordinated small changes in gene expression.
Changes in mRNA relative abundance and ribosome occupancy were not independent of one another (Χ2 = 179, p < 0.0001 for T/T vs. P/T). Starvation-induced genes were more likely to show low ribosome occupancy after glucose withdrawal, and repressed genes were more likely to show high ribosome occupancy (Fig. 2B). Despite the statistical interdependence of changes in total mRNA levels and ribosome occupancy, for each group of genes having similarly induced/repressed total mRNA levels there were many genes displaying each of the possible post-transcriptional regulatory behaviors.
Functionally Distinct Groups of Genes Are Co-Regulated at the Post-Transcriptional Level
To investigate the possibility that differences in post-transcriptional regulatory behavior are biologically significant, the function of genes in each category was examined by gene ontology (GO) analysis. Notably, the GO terms that were significantly enriched (p < 0.01 with Bonferroni correction for multiple hypothesis testing) for the seven mRNA abundance-based groups (highly induced, moderately induced, weakly induced, strongly repressed, moderately repressed, weakly repressed, and unchanged) segregated within these groups along post-transcriptional regulatory divisions. A complete list of significant GO terms for each of the 21 regulatory groups (7 T/T × 3 P/T) is provided in Supplemental Table 1. Rarely were GO categories split between multiple post-transcriptional (P/T) regulatory groups, and where such a split occurred, the GO category spanned two out of three most similar groups – ‘high’ and ‘neutral’ or ‘low’ and ‘neutral’, not ‘high’ and ‘low’. This suggests that the post-transcriptional behavior of a gene is related to its biological function in the starved cell.
The post-transcriptional partitioning of functionally related genes may derive from mechanistic similarities in their gene expression pathways. For example, the nuclear-encoded mitochondrial protein genes showed consistently low ribosome occupancy despite the fact that these genes are transcriptionally up-regulated in response to glucose withdrawal and encode proteins required for the cellular adaptation to low glucose conditions. mRNAs of some nuclear-encoded mitochondrial proteins are translated on cytosolic ribosomes that associate with mitochondria (Marc et al., 2002; Sylvestre et al., 2003). Subcellular localization of these mRNAs is driven by cis-acting elements in their 3′UTRs, and requires both the trans-acting RNA-binding protein Puf3 and the translocase of the mitochondrial outer membrane complex for full localization (Eliyahu et al., 2010; Saint-Georges et al., 2008). The apparent lag between the appearance of these transcriptionally induced mRNAs in the cell and their association with polysomes in our experiments could be explained by translational silencing of messages in transit from the nucleus to the periphery of mitochondria. In light of this explanation for the low P/T ratios of mRNAs encoding mitochondrial proteins, it is interesting to consider whether other transcriptionally induced yet poorly translated mRNAs identified in our analysis might also be subject to localization-dependent translational control.
The most striking example of gene function partitioning according to post-transcriptional regulatory behavior was the separation of cytoplasmic ribosomal protein genes (RPGs) from ribosome biogenesis factors (RBGs) (Fig. 2A, 3, S4). Genes from both functional groups were moderately to strongly reduced in both the total and polysomal mRNA pools after glucose withdrawal. This down-regulation is likely due to the greatly reduced demands for new ribosome synthesis as the cells transition from rapid growth and division, requiring the assembly of ~200,000 new ribosomes every 90 minutes (Warner, 1999), to cellular differentiation and slower growth in the invasive filamentous form (Cullen and Sprague, 2000). The two groups diverged in their post-transcriptional responses to glucose starvation. The P/T ratios of RBG mRNAs increased after glucose withdrawal and remained relatively high throughout the two-hour experiment. In contrast, the mRNAs encoding RPGs showed very low P/T ratios between 10 and 30 minutes after glucose withdrawal, and these ratios increased between 30 and 120 minutes (Fig. 2A). Low P/T ratios indicate that a population of non-translating mRNA exists in the cell. To test this interpretation directly, we performed qRT-PCR on polysome gradient fractions after 10 minutes of glucose starvation. RPG mRNAs (low P/T genes) accumulated in ribosome-free fractions at the top of the gradient, whereas mRNAs from genes with high P/T ratios did not (Fig. S4). In principle, the apparent increase in ribosome occupancy for RPGs after 60–120 minutes could result from improved translation (increased P) or from degradation of the non-polysomal pool of mRNA. Given that P/P and T/T ratios for RPGs were divergent at early times minus glucose and converged after 60 minutes of starvation (Fig. 3), the second interpretation is more plausible, suggesting that this sub-population of mRNA is only transiently stable as a non-translating pool. Notably, the messages that were most dramatically down-regulated upon glucose withdrawal and had low P/T values in acutely starved cells are the most abundant (Fig. 4). Transient preservation of these mRNAs in a non-translating pool (Fig. S4) may reflect a bet-hedging strategy (see Discussion).
Figure 3. RPGs and RBGs differ in their post-transcriptional responses to glucose withdrawal.

Kinetics of total mRNA abundance changes compared to polysomal mRNA abundance changes following glucose withdrawal. RPG mRNAs (red triangles) were preferentially depleted from the polysomal RNA pool compared to the total mRNA pool at early times. RBGs (gold squares), like most genes (blue diamonds), disappeared from the totals in concert with their loss from polysomes. After 120 minutes –glucose, the RPGs T/T versus P/P ratios more closely resembled the population as a whole, indicating a loss of a non-polysomal pool of mRNA. The results of linear regression analysis for each group of genes are shown by color-coded lines. See also Figure S4.
Figure 4. Highly expressed mRNAs are preferentially retained in the non-translating pool.
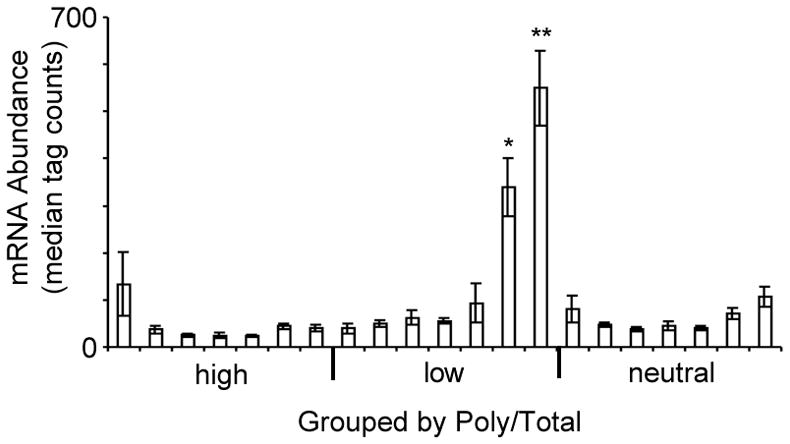
Mean mRNA abundance by group, as determined for each ORF by tag counts from next-generation sequencing of mRNA from cells grown in rich media (Nagalakshmi et al., 2008). Groups are as in Fig. 2A. The two T/T groups that were strongly decreased following glucose withdrawal and also showed low P/T ratios are comprised of significantly more abundant mRNAs (p < 0.05) than all other groups. Significance was assessed by Student’s t-test with Bonferroni correction. Within each P/T classification, groups are arranged from left to right from highest T/T ratio to lowest. Error bars indicate standard error of the mean (SEM).
Only a Subset of mRNAs Can Return to Polysomes Following Starvation and Re-Feeding
Previous reports showed that the global reduction in protein synthesis caused by ten minutes of glucose withdrawal can be reversed within five minutes of glucose re-addition (Ashe et al., 2000; Brengues et al., 2005). It was proposed that this rapid recovery of translation is due to mobilization of stored, translationally silenced mRNPs from P-bodies (cytoplasmic RNP granules) to polysomes. Only two reporter genes were tested for the capacity to return to polysomes in the absence of new transcription (Brengues et al., 2005). Our data suggest that most mRNAs are depleted from the total mRNA pool coincident with their loss from polysomes (Fig. 1C, S4D), and that the capacity for translational resurrection of pre-existing messages is thus narrowly restricted to a select sub-population that includes the RPG mRNAs. To test this hypothesis directly, we examined global as well as gene-specific recovery of translation following glucose re-addition to starved cells. New transcription was inhibited with the drug thiolutin (Grigull et al., 2004), to assess the capacity of pre-existing mRNAs to return to polysomes. Treatment with thiolutin slightly blunted translational recovery upon glucose re-addition, resulting in fewer heavy polysomes, more disomes, and persistence of a larger 80S monosome peak (Fig. 5A). These results indicate that initiation of translation on newly synthesized mRNAs accounts for some of the ‘recovery’ of translation after glucose re-addition, even after only 10 minutes of glucose starvation.
Figure 5. Translational resurrection is restricted to a subset of genes for a limited time.

(A) Polysome profiles of cells subjected to 10 minutes of glucose starvation followed by 5 minutes of glucose repletion in the presence (+T) or absence of thiolutin to inhibit new transcription. Thiolutin treatment slightly reduced polysome recovery. Note the relative heights of the disome (open arrowhead) and polysome (filled arrowhead) peaks and the widths of the monosome peaks ± thio. Analytical polysome assays (A, E, F) were repeated 2–4 times. Representative traces are shown for each. (B) Ratio values shown by color scale from microarray analysis of non-polysomal (NP) and polysomal (P) mRNA from cells starved for 10 minutes (YP 10′ +thio) or starved and re-fed for 5 minutes (+D 5′ +thio). Polysomal mRNA from YPD cultures served as the reference sample for each array. Genes are organized according to T/T and P/T groups from Figure 2. Group 5 is shown at 50% vertical scale. Genes that showed low P/T ratios in starved cells were less depleted from the non-polysomal fraction than genes with high or neutral P/T ratios and showed greater mobilization into polysomes upon re-feeding. “Δpoly” values were derived from the ratio of ratios (YPD5_P versus YP10_P). (C) Graphs show quantification of the Δpoly values from B. Error bars in C and D indicate SEM. Asterisks (*) indicate Student’s t-test p-value < 0.0001. (D) RPGs as a class preferentially recovered in polysomes upon glucose re-addition. “All” = all genes from T/T 5–7. (E) Prolonged glucose starvation in the absence of new transcription leads to reduced polysome recovery upon re-feeding. Cells were starved for 45 (top row) or 60 (bottom row) minutes in the presence of thiolutin before re-feeding for 5 minutes. Polysome profiles of recovery (+D 5′) after only 10 minutes –glucose +thio are overlaid for comparison (red lines). (F) New transcription contributes substantially to polysome recovery after prolonged starvation. Polysome profiles of recovery (+D 5′) after 45′ or 60′ of starvation are shown in black. Polysome recovery after starvation for 45 or 60 minutes +thiolutin is shown (blue lines) for comparison. See also Figure S5.
Nevertheless, substantial polysome recovery occurred even when new transcription was inhibited (Fig. 5A). To determine which genes participate in this recovery, the polysomal and non-polysomal mRNA pools were examined using microarrays following 10 minutes of glucose starvation and again after 5 minutes of glucose repletion, with thiolutin present throughout. A gene’s P/T ratio during glucose starvation predicted its ability to return to polysomes upon glucose re-addition. Genes that showed high or neutral P/T ratios following glucose removal (Fig. 2A) were depleted from the non-polysomal as well as the polysomal mRNA pools after 10 minutes of glucose starvation, and showed very little mobilization into polysomes 5 minutes after glucose re-addition (Fig. 5B, C). This argues that genes like the RBGs, which have high P/T ratios under conditions of decreasing P, do so because of rapid depletion of the non-polysomal mRNA. Consistent with this interpretation, we found by qRT-PCR that 80–95% of the total mRNA from these high P/T genes is gone from the total mRNA pool by 10 minutes after glucose starvation. In contrast, the genes that showed low P/T after glucose withdrawal were preferentially depleted from the polysomal compared to the non-polysomal pool (Fig. S4D). This group of genes also showed significantly greater mobilization into polysomes upon glucose re-addition by microarray (p < 0.0001), and by qRT-PCR analysis of select genes’ mRNA abundance in polysome gradient fractions (Fig. 6). Similar results were observed for more moderately down-regulated genes, with the low P/T sub-population showing significantly greater recovery than either the high or neutral P/T genes, although the extent of recovery was somewhat less. The RPGs as a class showed ~6-fold more recovery than all other starvation-repressed genes (T/T clusters 5, 6 and 7 from Fig. 2A) combined (p < 6.0 × 10−54), whereas RBGs did not recover (Fig. 5D).
Figure 6. Quantitative RT-PCR validation of select genes’ mRNA abundance in polysome fractions following glucose starvation and re-feeding.

(A) Polysome gradient fractions from plus glucose (left), starved (10 minutes minus glucose, with thiolutin, center), and re-fed (10 minutes minus glucose and 5 minutes plus glucose, with thiolutin, right). (B, C) (left) mRNA abundance per fraction, relative to 1/12th input and normalized to Fluc dope-in control RNA. (center, right) Adjusted mRNA abundance per fraction determined by qRT-PCR comparison with plus glucose fractions, shown on the same scale as plus glucose samples. Error bars indicate standard deviation of the mean of three replicates.
We verified that P-bodies formed under these conditions based on the localization of previously characterized protein and RNA reporters (Fig. S5). In addition, we examined the localization of RPS26A-U1A, a low P/T gene capable of returning to polysomes upon re-feeding, and of RRP4-U1A and LSG1-U1A, high P/T genes that are largely depleted within 10 minutes of glucose withdrawal and are not capable of returning to polysomes in the absence of new transcription. We detected P-body localization for RPS26A-U1A and RRP4-U1A. We did not detect LSG1-U1A, which was very lowly expressed by Northern blot (data not shown). These reporter experiments don’t distinguish whether the P-body localized portion of RPS26A mRNA is the fraction of intact mRNA that returns to polysomes upon re-feeding (~50%, Fig. 6B), or alternatively, if the P-body localized portion is comprised of decay intermediates of the fraction (~50%, Fig. S4D) that disappeared from the total mRNA pool, based on qRT-PCR using primers within the ORF. We found that stabilizing 3′UTR decay intermediates led to prominent P-body localization (Fig. S5), which is consistent with previous reports (Sheth and Parker, 2003). Thus, P-bodies likely contain endogenous mRNAs undergoing decay. Whether or not they also contain stored translationally repressed RPG mRNAs remains an open question. Wherever translationally repressed mRNAs reside in the cell, only select mRNAs are able to persist in a non-translating pool and resume active translation following a short period of glucose starvation.
The glucose starvation microarray time course data suggest that the non-translating sub-population of mRNAs that accumulates at early times is depleted after more prolonged glucose starvation (Fig. 2A, 3). This interpretation of the data predicts a turning point after which any recovery of polysomes upon glucose re-addition would require new transcription. To test this prediction, we subjected cells to varying periods of glucose starvation, with and without inhibition of new transcription by the drug thiolutin, and examined global translation activity after five minutes of glucose re-addition. After 45 or 60 minutes of starvation in the absence of new transcription, polysome recovery was greatly reduced compared to cells starved for only 10 minutes (Fig. 5E). If new transcription was allowed to occur during extended starvation, translational recovery upon glucose re-addition increased (Fig. 5F). These observations, together with the indication that specific mRNAs (RPGs) are lost between the 10- and 60-minute time points (Fig. 3), suggests that translational activation of these silenced mRNAs contributes substantially to polysome recovery upon re-feeding of briefly starved cells. These data argue that cells’ inability to rapidly restore translation without new transcription after longer periods of starvation is due to the disappearance of a population of non-translating mRNAs. Between 30 and 60 minutes appears to be a switch point for P/T ratios in our data, regardless of whether the ratios are increasing or decreasing. This suggests that the timing of the ‘turning point’ for cells to recover translation of stored RPGs mRNAs may relate to widespread changes in the activity of factors that influence P/T through effects on mRNA stability.
Molecular Insights Into Selective Preservation of Non-Translating RPG Transcripts
How are certain mRNAs able to persist in the cell, even transiently, following glucose starvation, when the majority of mRNAs are depleted from the total RNA pool_coincident, within the time resolution of our experiments, with their loss from polysomes? Comparison with genome-wide mRNA half-life measurements indicates that stability in glucose-replete conditions is probably not the determining factor for an mRNA’s capacity to persist in a stable non-translating pool after glucose withdrawal (Fig. S6). In particular, the RPGs as a class have short half-lives in glucose-replete conditions compared to most genes (Grigull et al., 2004; Wang et al., 2002).
Alternatively, the ‘post-transcriptional operon’ hypothesis posits a role for RNA-binding proteins (RBPs) in coordinating the post-transcriptional fate of functionally related genes (Keene, 2007; Keene and Tenenbaum, 2002). To investigate the possibility that specific RBPs affect the post-transcriptional behavior of mRNAs following glucose withdrawal, we examined data from a recent genome-wide association study that identified mRNA targets of 40 yeast RBPs (Hogan et al., 2008). Comparing the ‘transcriptional’ (changes in total mRNA abundance) and post-transcriptional (P/T) regulatory groups identified in our study with RBP-mRNA target groups revealed concordance between RBP association patterns and P/T but not T/T (Fig. 7A). For simplicity, only the seven RBPs that showed significant enrichment or de-enrichment (p < 0.01, Bonferroni corrected) of target mRNAs within at least one group are displayed. Similar to GO terms, which rarely spanned opposing P/T groups, RBP-association profiles were similar for “high” and “neutral” P/T groups for many RBPs, and for “low” and “neutral” for Puf3, whereas the “high” and “low” groups had almost no enrichments in common and frequently appeared to be mirror images of each other. In contrast, organizing RBP-association patterns by T/T group resulted in a ‘checkerboard’ appearance, despite the fact that T/T groups contain functionally and cytotopically related genes. This suggests that the coherence of the association between P/T behavior and particular RBPs might be the result of direct effects of these RBPs on mRNA translatability and/or mRNA stability outside of polysomes.
Figure 7. Resurrection-competent mRNAs associate with Pab1 and have longer poly(A) tails.

(A) Comparison of mRNA behavior following glucose withdrawal (7 T/T groups × 3 P/T groups) with RBP association profiles [Hogan et al.]. RBP target lists (rows) were clustered according to p-values for enrichment or de-enrichment within a given mRNA regulatory group. P-values were obtained using Fisher’s exact test with Bonferroni correction for multiple hypothesis testing. Columns are grouped by P/T (left) or T/T (right) similarity, ordered from left to right: high, low, neutral, and from most increased to most decreased T/T. Note that the ‘strong down’ T/T category includes genes that are not strongly reduced until after 60 minutes, although they are strongly reduced in the P/P at earlier times. (B) mRNAs with low P/T ratios have longer poly(A) tails as a group than mRNAs with high P/T. This effect is most pronounced for the group of genes that was most strongly down regulated (T/T) following glucose withdrawal. Poly(A) tail lengths were determined genome-wide and classified as ‘long’, ‘short’, or ‘no call’ (neither long nor short compared to most genes) [Beilharz and Preiss]. + and − indicate the presence of significantly too many or too few long- or short-tailed mRNAs within each of the 21 mRNA groups. Significance was assessed by Fisher’s exact test (Bonferroni corrected p-value < 0.01). (C) mRNAs with long poly(A) tails show greater capacity for translational recovery upon glucose re-addition than mRNAs with short or average length tails. Polysome recovery was assessed as described in Figure 5. Error bars indicate SEM. Asterisks (*) indicate p < 0.05 (Bonferroni corrected, Student’s t-test for difference compared to all other transcripts in the same T/T group). See also Figures S6 and S7.
Pab1-associated mRNAs were notably enriched with groups having low P/T ratios, and correspondingly depleted in groups with high and neutral P/T ratios (Fig. 7A). Comparison of P/T groups with genome-wide measurements of poly(A) tail lengths (Beilharz and Preiss, 2007) revealed a lack of short-tailed and an enrichment of long-tailed genes among mRNAs with low P/T values. Conversely, genes with long poly(A) tails were significantly under-represented among groups with high P/T ratios (Fig. 7B). The association of Pab1 and long poly(A) tails with groups of mRNAs with low P/T ratios was counterintuitive given Pab1’s role in enhancing translation initiation and the positive correlation between poly(A) tail length and ribosome density (Beilharz and Preiss, 2007). However, genome-wide ribosome occupancy (P/T), as measured in this study or by others (Arava et al., 2003), does not correlate with measures of translational efficiency (number of ribosomes/mRNA) determined by polysome fractionation (Arava et al., 2003) or by ribosome footprint profiling (Ingolia et al., 2009) (Fig. S7). Thus, we interpret the low P/T values of RPGs and high P/T values of RBGs after 10–30 minutes without glucose as consequences of differences in the stabilities of non-translating mRNAs, rather than as differences in translational activity following glucose withdrawal.
Consistent with a role for Pab1/poly(A) in stabilizing non-translating mRNAs, poly(A) tail length was positively correlated with the capacity for translational resurrection following glucose re-addition (Fig. 7C). The intersection between the two groups that show increased total mRNA levels, low P/T ratios, and enrichment with Pab1 (as well as Puf3) is dominated by nuclear-encoded mitochondrial protein genes, previously described as having long poly(A) tails (Beilharz and Preiss, 2007) and thought to transit the cytoplasm in a translationally repressed state before being translated on peri-mitochondrial ribosomes (Eliyahu et al., 2010; Marc et al., 2002; Sylvestre et al., 2003). A potentially unifying explanation for the enrichment of Pab1 with various low P/T groups is that Pab1 association may stabilize non-translating mRNAs.
DISCUSSION
Translation is Reduced, Not ‘Inhibited’, Following Glucose Withdrawal
Since the initial discovery of a rapid and dramatic signal-mediated reduction in polysomes following glucose withdrawal (Ashe et al., 2000), subsequent studies have investigated the mechanisms responsible for ‘global inhibition’ of translation in glucose-starved cells (for example, (Brengues and Parker, 2007; Brengues et al., 2005; Hilgers et al., 2006; Holmes et al., 2004; Hoyle et al., 2007)). Thus, we were initially surprised to observe widespread agreement between the relative abundance of mRNAs in the polysomal and total mRNA pools following glucose withdrawal. The data reported here show that mRNAs from more than 1,000 genes rapidly increased their relative association with polysomes in glucose-starved cells. Many of these genes are known to be transcriptional targets of well-characterized glucose-regulated transcription factors, and encode proteins required for survival and growth in the absence of glucose. Furthermore, the kinetics of accumulation of these mRNAs in polysomes was nearly indistinguishable from their appearance/accumulation in the total mRNA pool. The simplest interpretation of these data is that cells respond to glucose starvation by up-regulating expression of genes whose products promote adaptation to low glucose environments. This view agrees with extensive genetic and genome-wide gene expression studies (reviewed in (Zaman et al., 2008)), and is consistent with an earlier study, which reported (as data not shown) that protein synthesis, measured by [35S]methionine incorporation rates, decreased to 20% after 5 minutes without glucose, and returned to 40% by 15 minutes without glucose (Kuhn et al., 2001). We conclude that there is widespread translation at a reduced level in glucose-starved cells.
Most mRNAs are Depleted Coincident with Translational Repression
Previous studies suggested that widespread degradation of mRNAs is not part of the response to glucose withdrawal based on Northern blots of specific genes (Ashe et al., 2000). Examining the behavior of those genes in our genome-wide experiments reveals that their behavior is not typical. Three of the genes examined, PAB1, ACT1 and PGK1, were barely reduced in polysomes after 10 minutes without glucose (12%, 13% and 5% reductions, respectively), and the fourth gene, RPL28/CYH2, is unusual in its capacity to survive as a non-translating mRNA. Among more than 2,000 genes showing consistently reduced mRNA levels in polysomes following glucose withdrawal, only a small minority (< 20%), dominated by the RPGs, persisted in a stable non-translating pool that could return to polysomes if glucose was restored. Most mRNAs were depleted from the total mRNA population as quickly as they were depleted from polysomes. This observation is consistent with reports showing increased decapping and decay of specific mRNAs in response to decreased translation initiation (LaGrandeur and Parker, 1999; Schwartz and Parker, 1999).
Mechanisms of Post-transcriptional Regulation in Glucose-starved Cells
As our data are consistent with widespread decay of translationally repressed mRNAs, we reconsidered the evidence for and against a model of widespread mRNA decapping as the cause of translational repression following glucose starvation. Deletion of either component of the decapping enzyme complex (dcp1Δ or dcp2Δ) prevents polysome reduction following glucose withdrawal (Holmes et al., 2004). Moreover, the ability to inhibit protein synthesis following glucose removal correlated with the extent of residual decapping activity retained by various dcp1 mutants. Our genome-wide results are equally consistent with the model that activation of decapping causes widespread translational repression and mRNA decay, or alternatively, that translational repression occurs first and mRNA decay follows soon after. We favor the first model because inhibition of decapping prevents translational repression, whereas no known translational repressive mechanism is required for polysome collapse, despite extensive efforts to identify such a mechanism (Holmes et al., 2004). Nevertheless, decapping and 5′ to 3′ mRNA decay can occur on polysome-associated mRNAs (Hu et al., 2009). Thus, for the majority of genes whose mRNAs are depleted from the total pool coincident, within the time resolution of our experiments, with their disappearance from polysomes, we cannot conclude whether they are first evicted from polysomes and then degraded, or whether the initiation of decay prevents subsequent loading of ribosomes (and permits run-off of previously loaded ribosomes).
If widespread decapping is in fact responsible for most of the translational repression in glucose-starved cells, three major questions remain. First, what is the trigger that activates decapping within one minute following glucose withdrawal? The rapidity of the response suggests that it involves post-translational modification of some factor. Mutations that disrupt either the glucose derepression signaling pathway or the Ras/PKA pathway interfere with translational repression (Ashe et al., 2000), but no direct molecular connections to regulation of the decapping machinery have yet been characterized. Second, how do some messages escape decapping? There are two types of escapees: the ‘privileged’ mRNAs, including the RPG messages, that are present at the moment of glucose withdrawal and shift out of polysomes, and all the mRNAs that are produced afterwards. Third, what mechanism accounts for the translational repression of the RPGs?
Our results implicate the unusually long poly(A) tails of some messages and their association with the poly(A) binding protein as likely determinants for the preservation of non-translating mRNA. This is an attractive model given the evidence that deadenylation is the rate-limiting step in most mRNA decay (Parker and Song, 2004). For the second class of escapees – mRNAs whose abundance increases in glucose-starved cells – it is conceivable that they are endowed with longer poly(A) tails in the nucleus and/or that they are assembled into RNPs with features that render them inherently resistant to decapping. We do not favor such a model because these mRNAs are depleted from polysomes within 5 minutes following glucose re-addition (see complete array data available in Supplemental Table S2). An alternative explanation is that the ‘special’ feature of newly transcribed mRNAs is simply that they enter the cytoplasm after the decapping machinery is sequestered in P-bodies.
How are the RPG mRNAs translationally repressed? The observation that this group of mRNAs readily returns to active translation upon glucose re-addition strongly suggests that they are not decapped. Furthermore, our model that their long poly(A) tails and stable association with Pab1 are important for escaping decapping argues that they retain all of the features thought to be required for ribosome recruitment under normal conditions. The mTOR pathway is implicated in nutrient regulation of RPG translation in mammalian cells (Hamilton et al., 2006), but no such mechanism was thought to exist in yeast (Warner, 1999). Although the molecular details of the yeast mechanism almost certainly differ, our data suggest that a translational control mechanism targeting RPGs does exist. Genome-wide investigations of yeast translational responses to other stresses including high salinity (Melamed et al., 2008), transfer to a non-fermentable carbon source (Kuhn et al., 2001), and rapamycin treatment (Preiss et al., 2003) have also reported translational repression of RPGs, suggesting that this mechanism acts downstream of a common stress response signaling pathway.
Implications for the Interpretation of P-bodies
Glucose starvation in yeast has been extensively studied as a model stress to investigate the relationship between translational repression and mRNA localization to cytoplasmic granules or P-bodies. A central tenet of the model is that P-bodies are not just sites of mRNA decay, but can also be sites for storage of translationally silent intact mRNAs capable of return to the actively translating pool in response to altered cellular conditions (Brengues et al., 2005; Teixeira et al., 2005). Because core P-body components and their roles in mRNA metabolism are well conserved, this model has far-reaching implications for the regulation of gene expression in eukaryotic cells (Anderson and Kedersha, 2006; Parker and Sheth, 2007).
Our findings refute the notion that reciprocal movement between a non-translating mRNA pool and polysomes is a general property of eukaryotic mRNAs (Brengues et al., 2005). This model was based on previous observations of two reporter mRNAs following glucose starvation and repletion. We found that less than 20% of genes have the capacity to persist in a translationally silent state in glucose-starved cells. This raises the possibility that the majority of the proteins accumulated in P-bodies under these conditions are engaged with messages undergoing irreversible decay, although we cannot rule out the possibility that a subset of P-body associated endogenous mRNAs can escape and resume translation. If so, the RPG messages are good candidates.
Bet-Hedging
What advantage might transient preservation of RPG messages confer, and why should their behavior differ from the RBGs? Although the ribosome biogenesis factors and ribosomal proteins (RPs) function together in the assembly of new ribosomes, the factors act catalytically whereas the RPs are required stoichiometrically. Accordingly, the mRNAs encoding the RPs are among the most abundant messages in the cell, comprising up to 30% of cellular mRNA during rapid growth in rich media and requiring a significant investment of energy for their synthesis (Holstege et al., 1998; Nagalakshmi et al., 2008; Warner, 1999). If the cell were to degrade these messages immediately following release from polysomes, reversing this decision would be energetically costly. By retaining these mRNAs in a non-translating state for 10–30 minutes after the perception of glucose withdrawal, the cell might preserve the capacity for rapid resumption of growth should glucose return or the starvation signal turn out to have been a false alarm. Given that the production of new ribosomes largely determines the growth rate and generation time of yeast in nutrient replete conditions (Warner, 1999; Zaman et al., 2008), it is tempting to speculate that the selection of RPG mRNAs for transient preservation is a bet-hedging strategy by which the cell retains the capacity for a return to rapid growth and division while simultaneously initiating preparations for prolonged starvation. Previous investigations of global relationships between transcriptional and translational changes following other environmental stresses (high salt shock, transfer to a non-fermentable carbon source, or treatment with rapamycin to mimic nutrient deprivation) also described translational repression of RPGs through measurements that required the existence of a stable non-translating population of mRNA (Kuhn et al., 2001; Melamed et al., 2008; Preiss et al., 2003). The transient preservation of translationally repressed RPGs may be a general feature of yeast cells’ responses to growth-inhibiting stresses. Although translational activity was not determined, exploration of the common response to diverse environmental stresses revealed reduction in both RBG and RPG mRNAs, with the RPGs showing a delay (Gasch et al., 2000).
An interesting and unexplained phenomenon revealed by our data is the existence of a turning point, beyond which the cell appears to give up on hedging its bets and commits fully to the starvation gene expression program. How is the timing of this turning point determined molecularly? Does the timer run at a constant speed, or can it be adjusted according to features of the environment that might predict how likely it is that the cell will experience a reversal of fortunes in the near future? Do all cells hedge a little, transiently storing 30–50% of their RPG mRNAs, or do some cells store 100% and others none? These questions remain for future studies.
In answer to our original question – why would the cell choose to regulate gene expression at the level of translation – we conclude in part: because translational repression is reversible at relatively low cost. Direct testing of this adaptive fitness model awaits the identification of the molecular mechanism(s) responsible for transient translational repression of the RPGs, in order to assess its impact on competitive fitness under fluctuating conditions.
EXPERIMENTAL PROCEDURES
Yeast Strains and Culture
All experiments used Sigma 1278b background yeast (MATa ura3 leu2 trp1 his3), strain F1950 (a gift from Hiten Madhani). Yeast cultures were grown in YPAD (1% Yeast extract, 2% Peptone, 0.01% Adenine hemisulfate, 2% Dextrose) at 30°C in baffled flasks with vigorous shaking. Glucose-starved cultures were prepared from YPAD cultures grown to OD600 = 1.0 – 1.1, harvested by centrifugation (5 min at 12,000 × g), resuspended in pre-warmed YPA medium lacking glucose and returned to shaking at 30°C for various times. Transcription by RNA polymerase II was inhibited where indicated by the addition of thiolutin (Sigma) to the culture medium to a final concentration of 3 μg/mL.
Extract Preparation, Polysome Gradient Fractionation and RNA isolation
Yeast polysomes were prepared as described (Clarkson et al., 2010). Briefly, cycloheximide was added to cell cultures to a final concentration of 0.1 mg/mL for 2 minutes before harvesting cells by centrifugation (5 min at 12,000 × g). Cells were lysed by vortexing with glass beads in cold 1X PLB (20 mM HEPES-KOH, pH 7.4, 2 mM Magnesium Acetate, 100 mM Potassium Acetate, 0.1 mg/mL cycloheximide, 3 mM DTT). The crude extracts were clarified (20 minutes at 15,500 × g) and the resulting supernatants were applied to linear 10–50% sucrose gradients in 1X PLB. Lysates were typically 100–200 OD260 U/mL. For large-scale gradients, 50 OD260 U were applied to 40 mL gradients and spun for 4 hours at 27,000 rpm in a Beckman SW28 rotor. For small-scale gradients, 20 OD260 U were applied to 11 mL gradients and spun for 3 hours at 35,000 rpm in a Beckman SW41 rotor.
RNA was purified from polysomal gradient samples by denaturation with guanidine HCl followed by successive isopropanol and ethanol precipitations. Total RNA was isolated from clarified lysates by hot phenol extraction followed by isopropanol precipitation (Clarkson et al., 2010).
cDNA Synthesis and Labeling, Microarray fabrication and Hybridization
Production of Cy3 and Cy5 labeled cDNA from total and polysomal RNA samples, microarray fabrication, hybridization and washing were performed as described (Clarkson et al., 2010). cDNA synthesis was performed with a 1:1 mixture of oligo dT and random nonamer primers to permit detection of mRNAs with short poly(A) tails. Labeled cDNA was hybridized to custom microarrays containing 70mer oligonucleotide probes to all SGD-annotated ORFs. Details about the Operon YBOX and AROS probe sets are available at the website: http://omad.operon.com/download/index.php.
Image Analysis and Microarray Data Processing
A single ratio value (median pixel intensities for the 635 nm and 532 nm images of each spot) was determined for all microarray features by averaging within-array spot replicates from all biological and technical replicate array experiments. Spot ratio data were normalized within each array using global loess regression as described (Pleiss et al., 2007). Arrays with high background or spatial bias were discarded and the experiment repeated. A maximum of 8 ratio measurements were obtained for each feature: 2 spots × 2 biological replicate experiments × 2 [dye-flipped] technical replicate microarrays [duplicate cDNA synthesis, labeling and microarray hybridization]. Prior to feature identification, poor quality spots were excluded from further analysis if they showed visible defects due to dust or printing irregularities.
Clustering Analysis
Hierarchical clustering was performed on timecourse data of averaged feature ratio values using centroid linkage and Euclidean distance as the distance measure. A standard k-means clustering algorithm was used to identify groups of genes displaying similar changes in total mRNA relative abundance (T/T) or ribosome occupancy (P/T). Briefly, k-means clustering proceeds by first randomly assigning genes to one of an arbitrarily chosen number of groups. The mean vector for all genes in each group is computed, and the genes are then reassigned to the group whose center is closest to the gene. In our analysis, each vector was comprised of five T/T or six P/T values. We used Euclidian distance as the distance metric for evaluating closeness. Clustering proceeds by repeating these two steps until the optimal solution is found. The optimal solution is the one with the lowest possible within-group sum of distances. All clustering analysis was performed using Cluster 3.0.
Supplementary Material
Highlights.
Effects of glucose starvation on mRNA levels and translation activity are similar
Ribosomal protein gene mRNAs are selectively kept in a translationally repressed pool
Translational activation by refeeding is restricted to a few genes for a limited time
Reactivation-competent mRNAs are enriched with Pab1 and have longer poly(A) tails
Acknowledgments
We thank Adam Carrol, Paige Nittler, and Manlin Luo for technical assistance with the microarrays; Gregg Whitworth for advice and custom software for data processing; Joel Greenwood for computer support; Hiten Madhani for yeast strains; Roy Parker and Pam Silver for plasmids; and Megan Bergkessel, Chris Burge, Bryan Clarkson and members of the Gilbert lab for helpful discussions and comments on the manuscript. This work was supported by grant R00GM081399 (NIGMS) to W.G.
Footnotes
Accession Numbers
The microarray data have been deposited at the Gene Expression Omnibus (GEO) repository under the accession number GSE31393.
Supplemental Information includes Supplemental Experimental Procedures, seven figures and two tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe MP, De Long SK, Sachs AB. Glucose depletion rapidly inhibits translation initiation in yeast. Mol Biol Cell. 2000;11:833–848. doi: 10.1091/mbc.11.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz TH, Preiss T. Widespread use of poly(A) tail length control to accentuate expression of the yeast transcriptome. RNA. 2007;13:982–997. doi: 10.1261/rna.569407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake WJ, KAErn M, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- Brengues M, Parker R. Accumulation of polyadenylated mRNA, Pab1p, eIF4E, and eIF4G with P-bodies in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:2592–2602. doi: 10.1091/mbc.E06-12-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brengues M, Teixeira D, Parker R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science. 2005;310:486–489. doi: 10.1126/science.1115791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson BK, Gilbert WV, Doudna JA. Functional overlap between eIF4G isoforms in Saccharomyces cerevisiae. PLoS ONE. 2010;5:e9114. doi: 10.1371/journal.pone.0009114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- Coller J, Parker R. General translational repression by activators of mRNA decapping. Cell. 2005;122:875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen PJ, Sprague GF. Glucose depletion causes haploid invasive growth in yeast. Proc Natl Acad Sci USA. 2000;97:13619–13624. doi: 10.1073/pnas.240345197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliyahu E, Pnueli L, Melamed D, Scherrer T, Gerber AP, Pines O, Rapaport D, Arava Y. Tom20 mediates localization of mRNAs to mitochondria in a translation-dependent manner. Mol Cell Biol. 2010;30:284–294. doi: 10.1128/MCB.00651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigull J, Mnaimneh S, Pootoolal J, Robinson MD, Hughes TR. Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol Cell Biol. 2004;24:5534–5547. doi: 10.1128/MCB.24.12.5534-5547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton TL, Stoneley M, Spriggs KA, Bushell M. TOPs and their regulation. Biochem Soc Trans. 2006;34:12–16. doi: 10.1042/BST20060012. [DOI] [PubMed] [Google Scholar]
- Hilgers V, Teixeira D, Parker R. Translation-independent inhibition of mRNA deadenylation during stress in Saccharomyces cerevisiae. RNA. 2006;12:1835–1845. doi: 10.1261/rna.241006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255. doi: 10.1371/journal.pbio.0060255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes LE, Campbell SG, De Long SK, Sachs AB, Ashe MP. Loss of translational control in yeast compromised for the major mRNA decay pathway. Mol Cell Biol. 2004;24:2998–3010. doi: 10.1128/MCB.24.7.2998-3010.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Hoyle NP, Castelli LM, Campbell SG, Holmes LE, Ashe MP. Stress-dependent relocalization of translationally primed mRNPs to cytoplasmic granules that are kinetically and spatially distinct from P-bodies. J Cell Biol. 2007;179:65–74. doi: 10.1083/jcb.200707010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature. 2009;461:225–229. doi: 10.1038/nature08265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone O, Lasko P. Translational regulation and RNA localization in Drosophila oocytes and embryos. Annu Rev Genet. 2001;35:365–406. doi: 10.1146/annurev.genet.35.102401.090756. [DOI] [PubMed] [Google Scholar]
- Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- Keene JD, Tenenbaum SA. Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell. 2002;9:1161–1167. doi: 10.1016/s1097-2765(02)00559-2. [DOI] [PubMed] [Google Scholar]
- Kuhn KM, DeRisi JL, Brown PO, Sarnow P. Global and specific translational regulation in the genomic response of Saccharomyces cerevisiae to a rapid transfer from a fermentable to a nonfermentable carbon source. Mol Cell Biol. 2001;21:916–927. doi: 10.1128/MCB.21.3.916-927.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGrandeur T, Parker R. The cis acting sequences responsible for the differential decay of the unstable MFA2 and stable PGK1 transcripts in yeast include the context of the translational start codon. RNA. 1999;5:420–433. doi: 10.1017/s1355838299981748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc P, Margeot A, Devaux F, Blugeon C, Corral-Debrinski M, Jacq C. Genome-wide analysis of mRNAs targeted to yeast mitochondria. EMBO Rep. 2002;3:159–164. doi: 10.1093/embo-reports/kvf025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed D, Pnueli L, Arava Y. Yeast translational response to high salinity: global analysis reveals regulation at multiple levels. RNA. 2008;14:1337–1351. doi: 10.1261/rna.864908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Regulation of noise in the expression of a single gene. Nat Genet. 2002;31:69–73. doi: 10.1038/ng869. [DOI] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol. 2007;5:e90. doi: 10.1371/journal.pbio.0050090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss T, Baron-Benhamou J, Ansorge W, Hentze MW. Homodirectional changes in transcriptome composition and mRNA translation induced by rapamycin and heat shock. Nat Struct Biol. 2003;10:1039–1047. doi: 10.1038/nsb1015. [DOI] [PubMed] [Google Scholar]
- Raser JM, O’Shea EK. Noise in gene expression: origins, consequences, and control. Science. 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD. Translational control during early development. Bioessays. 1991;13:179–183. doi: 10.1002/bies.950130406. [DOI] [PubMed] [Google Scholar]
- Saint-Georges Y, Garcia M, Delaveau T, Jourdren L, Le Crom S, Lemoine S, Tanty V, Devaux F, Jacq C. Yeast mitochondrial biogenesis: a role for the PUF RNA-binding protein Puf3p in mRNA localization. PLoS ONE. 2008;3:e2293. doi: 10.1371/journal.pone.0002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, Parker R. Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:5247–5256. doi: 10.1128/mcb.19.8.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova JB, Selley JN, Sanchez-Cabo F, Carroll K, Eddy AA, McCarthy JE, Hubbard SJ, Pavitt GD, Grant CM, Ashe MP. Global gene expression profiling reveals widespread yet distinctive translational responses to different eukaryotic translation initiation factor 2B-targeting stress pathways. Mol Cell Biol. 2005;25:9340–9349. doi: 10.1128/MCB.25.21.9340-9349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestre J, Vialette S, Corral Debrinski M, Jacq C. Long mRNAs coding for yeast mitochondrial proteins of prokaryotic origin preferentially localize to the vicinity of mitochondria. Genome Biol. 2003;4:R44. doi: 10.1186/gb-2003-4-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira D, Sheth U, Valencia-Sanchez MA, Brengues M, Parker R. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA. 2005;11:371–382. doi: 10.1261/rna.7258505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces responds to nutrients. Annu Rev Genet. 2008;42:27–81. doi: 10.1146/annurev.genet.41.110306.130206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.