

Non-technical summary
The islet of Langerhans secretes the hormone insulin in response to elevated glucose. Interactions between cells within the islet is important for the regulation of insulin secretion, to both suppress basal insulin secretion and enhance the glucose-stimulated response. We show that multiple mechanisms of cell–cell communication are required for the suppression of basal insulin release. First, gap junctions suppress spontaneous calcium signals which suppresses triggering of insulin release. Second, other juxtacrine mechanisms, regulated by cAMP and glucose, suppress more distal steps in the regulation of insulin granule exocytosis. Each mechanism is sufficiently robust to compensate for a loss of the other and still fully suppress basal insulin release. This new insight into the function of islet of Langerhans is important for understanding the development and treatment of diabetes.
Abstract
Abstract
Cell–cell communication in the islet of Langerhans is important for the regulation of insulin secretion. Gap-junctions coordinate oscillations in intracellular free-calcium ([Ca2+]i) and insulin secretion in the islet following elevated glucose. Gap-junctions can also ensure that oscillatory [Ca2+]i ceases when glucose is at a basal levels. We determine the roles of gap-junctions and other cell–cell communication pathways in the suppression of insulin secretion under basal conditions. Metabolic, electrical and insulin secretion levels were measured from islets lacking gap-junction coupling following deletion of connexion36 (Cx36−/−), and these results were compared to those obtained using fully isolated β-cells. KATP loss-of-function islets provide a further experimental model to specifically study gap-junction mediated suppression of electrical activity. In isolated β-cells or Cx36−/− islets, elevations in [Ca2+]i persisted in a subset of cells even at basal glucose. Isolated β-cells showed elevated insulin secretion at basal glucose; however, insulin secretion from Cx36−/− islets was minimally altered. [Ca2+]i was further elevated under basal conditions, but insulin release still suppressed in KATP loss-of-function islets. Forced elevation of cAMP led to PKA-mediated increases in insulin secretion from islets lacking gap-junctions, but not from islets expressing Cx36 gap junctions. We conclude there is a redundancy in how cell–cell communication in the islet suppresses insulin release. Gap junctions suppress cellular heterogeneity and spontaneous [Ca2+]i signals, while other juxtacrine mechanisms, regulated by PKA and glucose, suppress more distal steps in exocytosis. Each mechanism is sufficiently robust to compensate for a loss of the other and still suppress basal insulin secretion.
Introduction
The pancreatic islets of Langerhans play a central role in the regulation of blood glucose homeostasis through the regulated secretion of the hormones insulin and glucagon. Glucose-stimulated insulin secretion (GSIS) from β-cells is regulated by a series of molecular events including an elevated ATP/ADP ratio following glucose metabolism, subsequent ATP-sensitive K+ (KATP) channel closure, membrane depolarization, Ca2+ influx to increase intracellular free-calcium activity ([Ca2+]i), and the triggering of insulin granule exocytosis. Other important steps independent of this ‘KATP-dependent’ or ‘triggering pathway’ include cAMP elevations which elevate insulin granule trafficking to the plasma membrane and amplify exocytosis (Henquin, 2000). The ability of endocrine cells within the islet to communicate with one another is an important factor for the regulation of insulin secretion (Halban et al. 1982). Thus, in intact islets, the dynamic range of GSIS from β-cells is enhanced many-fold compared to the responses observed from isolated β-cells (Lernmark, 1974; Halban et al. 1982): in intact islets, β-cells exhibit both reduced insulin secretion at low glucose and enhanced insulin secretion at elevated glucose. Isolated β-cells also exhibit heterogeneous and irregular responses to glucose for many variables (Pipeleers, 1992), including NAD(P)H elevations (Bennett et al. 1996), oscillations in [Ca2+]i (Zhang et al. 2003), and the levels of insulin release (Vanschravendijk et al. 1992). Therefore understanding how cells communicate within the intact islet is important to understand precisely how insulin secretion is regulated.
Gap junction channels formed by connexin36 (Cx36) couple β-cells in the islet (Theis et al. 2004; Moreno et al. 2005), which can mediate ionic currents and the diffusion of small molecules (Quesada et al. 2003; Charpantier et al. 2007). As a result, Cx36 gap junctions are important for coordinating the oscillatory dynamics of membrane depolarization and [Ca2+]i (Calabrese et al. 2003; Ravier et al. 2005; Benninger et al. 2008) and for generating pulsatile insulin secretion (Ravier et al. 2005). Additionally, Cx36 gap junctions are important to ensure that there is uniform suppression of spontaneous membrane depolarizations and [Ca2+]i bursts that can occur in a subpopulation of β-cells, as a result of cellular differences in KATP channel activity and other heterogeneous processes of the β-cell. This is especially the case following expression of a KATP channel loss-of-function mutation (Rocheleau et al. 2006), or following KATP channel inhibition with ATP (Speier et al. 2007). This has led to a model whereby the less excitable β-cells in the islet can suppress, via gap junctions, spontaneous electrical activity induced in neighbouring, excited β-cells, (Rocheleau et al. 2006).
The subsequent impact of this regulatory mechanism on physiological insulin secretion is less clear. Thus, Rupnik and colleagues showed that in the absence of Cx36, when switching from high to low glucose the switch-off of insulin secretion is less rapid (Speier et al. 2007), suggesting that Cx36 can at least transiently suppress insulin release. However, Meda and colleagues showed that only minimal changes in insulin secretion occur at low glucose levels (their Fig. 7C), despite a more significant increase in [Ca2+]i (their Fig. 7A) seen following the knockout of Cx36 (Ravier et al. 2005). Therefore, additional mechanistic understanding of the role of islet gap junction coupling in the regulation of basal insulin secretion is needed.
Figure 1. Glucose-dependent [Ca2+]i in Cx36−/− islets.
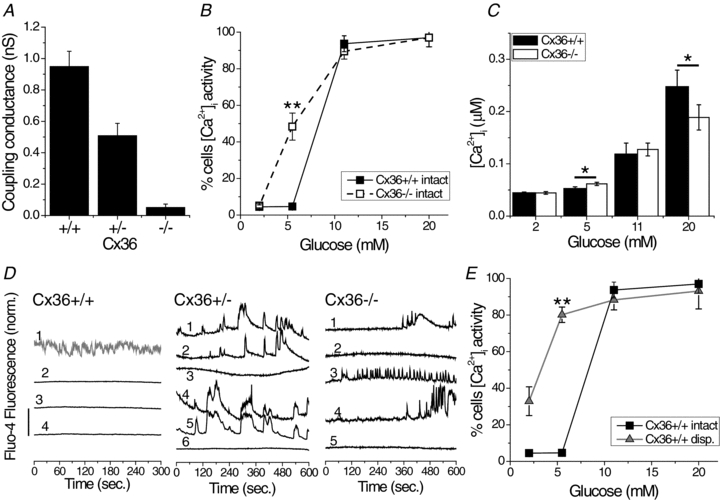
A, mean coupling conductance of peripheral islet β-cells during single-cell current and voltage clamp recording. Measurements were made in islets isolated from Cx36+/+, Cx36+/− and Cx36−/− mice, from 3 mice for each group. B, mean percentage of cells displaying dynamic changes in [Ca2+]i as a function of glucose stimulation for intact Cx36+/+ islets (filled squares) and Cx36−/− islets (open squares). Data averaged over n = 5 mice. **Significant difference of P < 0.01 (Student's unpaired t test) at each glucose concentration comparing intact Cx36+/+ and Cx36−/− islet data. C, mean [Ca2+]i as a function of glucose stimulation for intact Cx36+/+ islets (filled bars) and Cx36−/− islets (open bars). Data averaged over n = 7 mice. *Significant difference of P < 0.05 (Student's paired t test) comparing each experimental group as indicated. D, representative time courses of [Ca2+]i at 5.5 mm glucose, as measured from Fluo4 fluorescence, for a number of non-adjacent cells (numbered) from a single Cx36+/+, Cx36+/− and Cx36−/− islet as indicated. Time courses are offset for clarity and vertical scale bar indicates 2-fold change in Fluo4 fluorescence. Time course 1 in Cx36+/+ (in grey) represents a cell showing no activity at elevated glucose and therefore classified as an α-cell. E, mean percentage of cells displaying dynamic changes in [Ca2+]i as a function of glucose stimulation for intact wild-type islets (black squares) and dissociated wild-type cells (grey triangles). Data averaged over n = 3 mice. **Significant difference of P < 0.01 (Student's unpaired t test) at each glucose concentration comparing intact islet and dissociated cell data. The difference in means ± 95% confidence interval for C is shown in Fig. S2A.
Figure 7. How multiple cell–cell communication mechanisms regulate basal insulin release.
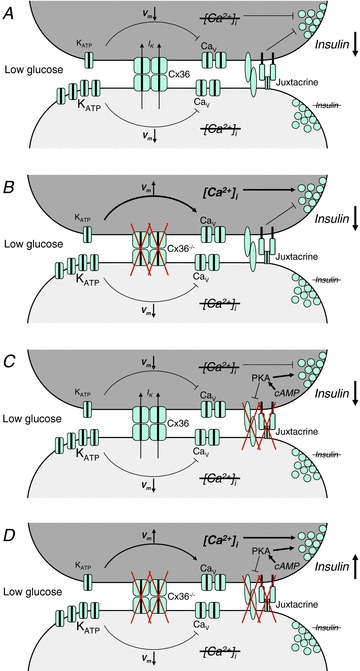
A, two representative cells in an islet have different threshold for glucose activation of Ca2+-signalling, either intrinsic to the cell as illustrated here, or induced by mosaic Kir6.2[AAA] expression. At basal glucose levels, one cell (upper, dark grey) has reduced KATP channel activity making it more excitable. The other cell (lower, light grey) has increased KATP channel activity making it less excitable. Cx36 gap junction coupling mediates a hyperpolarizing current (IK) to the more excitable cell, preventing transient depolarization and voltage-gated calcium channel activation. This suppresses any [Ca2+]i elevations and Ca2+ triggering of insulin secretion in the more excitable cell. More distal, other juxtacrine mechanisms (‘Juxtacrine’) putatively including EphA forward signalling and NCAM signalling also suppress insulin granule trafficking and/or exocytosis to additionally suppress insulin secretion. B, in the absence of gap junctions, the more excitable cell depolarizes and elevates [Ca2+]i, but an elevation in insulin secretion is still blocked downstream of Ca2+ signalling, dependent on other juxtacrine mechanisms. C, cAMP acting via PKA overcomes the effect of other suppressive juxtacrine mechanisms, but in the presence of gap junction coupling a suppression of any [Ca2+]i elevation prevents an elevation in insulin secretion. D, only when gap junction coupling is inhibited and other juxtacrine mechanisms are overcome by cAMP via PKA do more excitable cell show elevated Ca2+-triggering and elevated basal insulin secretion as in isolated cells.
In addition to gap junctions, other juxtacrine mechanisms dependent on cell–cell contact may also help to regulate insulin release. EphA–ephrinA bi-directional signalling between β-cells (Konstantinova et al. 2007) has been suggested to regulate remodelling of the cortical F-actin network, which affects distal points in the GSIS pathway to regulate insulin release, including insulin granule trafficking and docking to the plasma membrane. Similarly, neuronal cell-adhesion molecule (NCAM) between β-cells has also been suggested to regulate the cortical F-actin network and insulin secretion (Olofsson et al. 2009). Therefore understanding the role of juxtacrine mechanisms and how they do or do not interact with gap junctions still needs to be determined.
In this study, we sought to discover the role of gap junction coupling in regulating the suppression of insulin secretion at low glucose in intact islets. We also asked how gap junction coupling quantitatively compares with other mechanisms dependent on cell–cell contact in regulating this suppression, by comparing intact islets lacking gap junctions with dissociated isolated β-cells. We test whether the disruption of gap junction coupling alone recapitulates the behaviour of dissociated β-cells, or whether other mechanisms dependent on cell–cell contact may persist despite elimination of the gap junction mediated coupling. To test this, we quantify islet metabolic, electrical as well as insulin secretory responses to glucose. We also utilize islets in which electrical activity has been perturbed by the mosaic expression of a loss-of-function mutation in KATP channels (Koster et al. 2000; Rocheleau et al. 2006). In this mouse model, elevations of basal [Ca2+]i and insulin secretion due to the KATP loss-of-function are suppressed by mechanisms dependent on cell–cell contact in the intact islet. These islets therefore allow us to specifically study how cellular differences in electrical activity in the intact islet are suppressed by gap junction coupling and other mechanism dependent on cell–cell contacts, as well as to understand how these mechanisms suppress basal insulin secretion.
Methods
Ethical approval
All experiments were performed in compliance with relevant laws and institutional guidelines, and were approved by Vanderbilt University Institutional Animal Care and Use Committee. The authors have read, and the experiments comply with the policies and regulations of The Journal of Physiology given by Drummond (2009).
Animal care
Mice were housed in a temperature controlled facility with 12 h light–dark cycle and access to food and water ad libitum. Generation of Cx36-LacZ knockout mice (Cx36−/−) is described by Degen et al. (2004). Generation of Kir6.2[AAA] transgenic mice is described by Koster et al. (2000). Mice were genotyped by PCR and gel electrophoresis using primers described in Koster et al. (2000) and Degen et al. (2004).
Islet isolation
Islets were isolated by established protocols (Scharp et al. 1973; Koster et al. 2002) from pancreata of 12- to 24-week-old mice. Mice were anaesthetized under i.p. injection of ketamine and xylazine (4.16 mg and 0.84 mg respectively) and killed by cervical dislocation prior to pancreas dissection. Following isolation, islets were maintained in RPMI-1640 medium containing 10% fetal bovine serum, 11 mm glucose, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, at 37°C under humidified 5% CO2 for 24–48 h before imaging. To obtain dissociated β-cells, islets were washed in Ca2+-free Hanks’ balanced salt solution, then incubated with Trypsin–EDTA (both 0.01%) at 37°C for ∼5 min. Trypsin treated islets were dispersed by gently resuspending in islet medium, then plated on glass coverslips in 8-well plates (from 15 islets per well) and maintained for 24–48 h at 37°C, 5% CO2. Single isolated cells constituted 92 ± 4% of dissociated islets, with a mean nearest-neighbour separation of ∼60 μm. Of dissociated islet cells 97.4 ± 1.5% were viable, assessed by propidium iodide exclusion and AM-dye activation.
Electrophysiology
Measurements of β-cell junctional conductance were performed as described in Mears et al. (1995) and Sherman et al. (1995). Isolated islets were perfused with an extracellular solution containing 115 mm NaCl, 5 mm KCl, 3 mm CaCl2, 2 mm MgCl2, 10 mm Hepes, 2 mm glucose, pH 7.2. Membrane potential and current were monitored using the perforated-patch method (Zhang et al. 2003). Pipettes were filled with an intracellular solution containing 11.8 mm NaCl, 28.4 mm K2SO4, 63.7 mm KCl, 1 mm MgCl2, 20.8 mm Hepes, 0.5 mm EGTA, pH 7.2, plus 0.1 mg ml−1 amphotericin-B. After obtaining a gigaohm seal on a patched peripheral β-cell, glucose levels were stepped from 2 mm to 11 mm. Membrane potential was recorded in current clamp mode, then coupling current was measured in voltage clamp mode at –65 mV. The coupling conductance (gcoup) was determined by the change in coupling current (ΔIcoup) divided by the change in membrane potential (ΔV).
Hormone secretion
Following overnight incubation, islets (10 per column) or dispersed cells (from 15 islets per well) were pre-incubated in Krebs–Ringer buffer (128.8 mm NaCl, 5 mm NaHCO3, 5.8 mm KCl, 1.2 mm KH2PO4, 2.5CaCl2, 1.2 mm MgSO4, 10 mm Hepes, 0.1% BSA, pH 7.4) plus 2 mm glucose for 60 min at 37°C, then incubated for 60 min at 37°C in Krebs–Ringer buffer plus different glucose concentrations and/or reagents as indicated. After the incubation period, the medium was removed and assayed for insulin release. To estimate insulin content, islets were lysed in 1% Triton X-100 and frozen at –20°C overnight. Insulin concentrations were measured by rat-insulin radioimmunoassay.
Fluorescence microscopy and image analysis
All imaging experiments and data analysis were performed using published procedures (see e.g. Rocheleau et al. 2006; Benninger et al. 2011).
To measure [Ca2+]i dynamics, isolated islets or dispersed β-cells were loaded for 1.5 h with 4 μm Fluo4-AM (Invitrogen) at room temperature, in imaging medium (125 mm NaCl, 5.7 mm KCl, 2.5CaCl2, 1.2 mm MgCl2, 10 mm Hepes, 2 mm glucose 0.1% BSA, pH 7.4), then equilibrated at 37°C for 10 min. Intact islets were imaged in a polydimethylsiloxane (PDMS) microfluidic flow device (Rocheleau et al. 2006). Dispersed β-cells were imaged in labtex dishes (Nunc). Islets or β-cells were imaged on an LSM5Live or LSM510-Meta microscope (Zeiss) with a 20× 0.8NA Fluar objective, using a humidified temperature controlled chamber, maintained at 37°C. A series of images were acquired 10 min or 60 min after application of each glucose level studied, for 10 min duration. Fluo4 fluorescence was detected using a 488 nm diode laser for excitation and a 495–555 nm band-pass filter to detect fluorescence emission.
To measure the absolute mean [Ca2+]i concentration, islets were loaded for 30 min with 2 μm Fura2-AM (Invitrogen), equilibrated at 37°C for 10 min, then imaged in a microfluidic device on a TE-300 widefield microscope (Nikon) with a 20× 0.45NA Fluar objective, at 37°C. Images were acquired 10 min after application of each glucose level studied. Islets were sequentially excited at 340 nm and 380 nm (±10 nm band-pass filter), and fluorescence detected with a 470–550 nm band-pass filter. Background fluorescence was measured from unstained islets. The 340 nm and 380 nm fluorescence images were background subtracted and averaged across each islet studied. The 340 nm/380 nm fluorescence ratio was then calibrated according to the Fura2 Calcium Calibration Kit (Invitrogen) instructions.
To measure NAD(P)H, islets were incubated for 1.5 h, then imaged in a microfluidic device on an LSM510-Meta microscope with a 40× 1.3NA Fluar oil-immersion objective, at 37°C. NAD(P)H autofluorescence was detected using a 710 nm mode-locked Ti:sapphire laser (Chameleon, Coherent, Santa Clara, CA, USA) for two-photon excitation, and custom 380–550 nm band-pass filter (Chroma, Brattleboro, VT, USA) and non-descanned detector for emission. Z-stacks of 6 images were acquired at 2 μm spacing. All microscope and laser settings were kept constant between experiments. The NAD(P)H fluorescence intensity was averaged across each islet and each z-position. The mean NAD(P)H fluorescence intensity was normalized to that measured in Cx36+/+ islets at 2 mm glucose.
All image analysis was performed in Matlab (The Mathworks, Natick, MA, USA) and Image Examiner (Zeiss). To calculate the proportion of the islet showing [Ca2+]i bursts, the Fluo4 time course of 5 min duration was examined on a cell-by-cell basis. An electrically active area was defined as showing variations in the fluorescence intensity >3 standard deviations above that calculated in silent cells at 2 mm glucose. The proportion of the islet showing [Ca2+]i bursts was defined as the total electrically active area as a proportion of the total islet area. A small number of cells (5–10%) which were active at low glucose and were no longer active at elevated glucose are likely α-cells (Quesada et al. 2006), and were discounted from subsequent analysis (e.g. Fig. 1D).
Data are presented as means ± SEM. Student's t test for paired or unpaired data was used as appropriate to assess significance between data in each experimental group, as indicated in figure captions. The difference in means ± 95% confidence level was calculated where indicated, and are displayed in Supplemental Fig. S2.
Results
Dependence of islet [Ca2+]i on gap junctions and mechanisms dependent on cell–cell contact
In isolated islets of homozygous connexin36-LacZ knockout mice (Cx36−/−) (Degen et al. 2004) LacZ was uniformly expressed, and islet architecture was unaltered, with α-cells remaining on the islet mantle, and β-cells forming the core (not shown). To confirm the absence of functional gap junction channels, electrical coupling between β-cells was measured using patch clamping (Fig. 1A). β-Cells in wild-type Cx36+/+ islets had a mean junctional conductance of 950 ± 96 pS, which is reduced to 53.6 ± 8.2% in Cx36+/− islets and to 5.5 ± 2.2% in Cx36−/− islets (Benninger et al. 2008).
Cx36−/− islets lacked synchronous and well coordinated oscillations in intracellular free-calcium activity ([Ca2+]i), as previously established (Ravier et al. 2005; Benninger et al. 2008). We quantified [Ca2+]i in Cx36+/+ and Cx36−/− islets at a range of glucose concentrations by measuring the proportion of islet cells that exhibit [Ca2+]i bursts (Fig. 1B). At 2 mm glucose most β-cells were silent in both Cx36+/+ and Cx36−/− islets. At 5.5 mm glucose we observed similar results to 2 mm glucose in Cx36+/+ islets, but in Cx36−/− islets ∼50% of the β-cells exhibited spontaneous [Ca2+]i bursts under what is usually a subthreshold condition. Similar [Ca2+]i bursts were observed either 10 or 60 min after switching to 5.5 mm glucose (not shown). Similarly, in 5.5 mm glucose, there was a significant elevation in the absolute [Ca2+]i concentration (Fig. 1C). Figure 1D shows representative measurements of [Ca2+]i bursts in Cx36+/+, Cx36+/− and Cx36−/− islets bathed in 5.5 mm glucose. The mostly quiescent [Ca2+]i activity observed in Cx36+/+ islets contrasts strongly with our observation of [Ca2+]i bursts in Cx36+/− and Cx36−/− islets, and marked variations in the pattern of oscillations were observed between cells. At 11 mm or 20 mm glucose, a similar proportion of islet cells exhibited [Ca2+]i bursts in Cx36+/+ and Cx36−/− islets (Fig. 1B); however the [Ca2+]i concentration was reduced in the Cx36−/− islets at 20 mm glucose. Dissociated isolated β-cells showed similar response to intact Cx36−/− islets, namely an elevated [Ca2+]i between 2 mm and 5.5 mm glucose in a proportion of cells (Fig. 1E).
Glucose metabolism in Cx36−/− islets was assessed using two-photon excitation imaging of NAD(P)H. There was no significant difference in the glucose-stimulated NAD(P)H response (Fig. 2A) and NAD(P)H levels (Fig. 2B) between Cx36+/+ and Cx36−/− islets. Therefore a loss of islet gap junction mediated electrical coupling leads to elevated [Ca2+]i at basal glucose levels, which is similar to what is observed in dissociated β-cells that lack any cell–cell contacts. This effect is independent of more proximal changes in glucose metabolism.
Figure 2. NAD(P)H response in Cx36−/− islets.
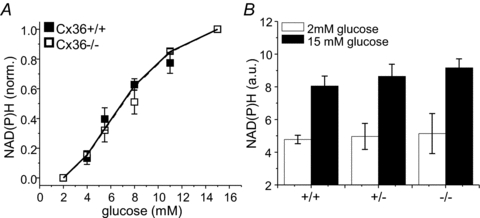
A, normalized (norm.) increase in NAD(P)H autofluorescence as a function of glucose stimulation, for islets isolated from Cx36+/+ (filled squares) and Cx36−/− mice (open squares). Data normalized to the level of NAD(P)H at 2 and 15 mm glucose. A Hill-curve fit is included for each group of data (continuous and dashed line, respectively). B, level of NAD(P)H autofluorescence in arbitrary units (a.u.) at 2 mm glucose and 15 mm glucose in Cx36+/+, Cx36+/− and Cx36−/− islets. Data averaged over n = 4 mice for each group.
Insulin secretion dependence on gap junctions and mechanisms dependent on cell–cell contact
To study the impact of the altered [Ca2+]i on the function of Cx36−/− islets, we measured insulin secretion over a range of glucose concentrations. Similar levels of insulin secretion were found in Cx36+/+, Cx36+/− and Cx36−/− islets for all levels of glucose applied (Fig. 3A). Insulin content per islet was not significantly different (Fig. 3B). Transient changes in insulin secretion when switching to low glucose can occur following a disruption of Cx36 gap junctions (Speier et al. 2007). We therefore compared the role of gap junctions on steady-state insulin secretion levels with cell–cell contact-dependent mechanisms, by measuring insulin secretion in both intact and dissociated Cx36+/+ and Cx36−/− islets. The fold response in insulin secretion between 2 mm glucose and 20 mm glucose was reduced in dissociated β-cells compared to intact islets (Fig. 3C), with no difference comparing either intact Cx36+/+ and Cx36−/− islets or dissociated Cx36+/+ and Cx36−/−β-cells. In both Cx36+/+ and Cx36−/− islets, dissociated isolated β-cells showed elevated insulin secretion compared to intact islets at 2 mm and 5.5 mm glucose (Fig. 3D and E, respectively). No difference was observed at 11 mm or 20 mm glucose. Therefore a loss of islet gap junction mediated electrical coupling alone has a negligible impact on insulin secretion compared to the elevated basal insulin secretion observed from dissociated β-cells that lack any cell–cell contact. This is despite the elevated [Ca2+]i that is observed in each case.
Figure 3. Insulin secretion in Cx36−/− islets and dissociated β-cells.
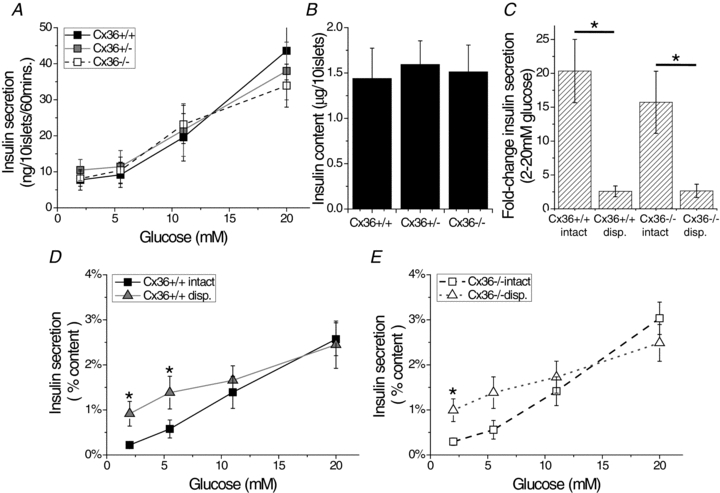
A, glucose-stimulated insulin secretion from isolated islets of Cx36+/+ (black squares), Cx36+/− (grey squares), Cx36−/− (open squares) mice. B, insulin content of Cx36+/+, Cx36+/− and Cx36−/− islets. Data in A and B averaged over n = 7 mice for each group. C, mean fold-increase in insulin secretion between 2 mm and 20 mm glucose stimulation for intact Cx36+/+ and Cx36−/− islets and dissociated (disp.) Cx36+/+ and Cx36−/−β-cells. *Significant difference of P < 0.05 (Student's paired t test) comparing each experimental group as indicated. D, glucose-stimulated insulin secretion from intact Cx36+/+ islets (black squares) and dissociated Cx36+/+β-cells (grey triangles), displayed as fractional insulin secretion per hour normalized by insulin content. E, glucose-stimulated insulin secretion from intact Cx36−/− islets (open squares) and dissociated Cx36−/−β-cells (open triangles). Measurements for all groups in C, D and E made in parallel experiments, averaged over n = 11 littermate Cx36+/+ and Cx36−/− mice for each group. *Significant difference of P < 0.05 (Student's paired t test) at each glucose concentration comparing intact islet and dissociated cell data. The difference in means ± 95% confidence interval for D and E is shown in Fig. S2B.
Elevation in [Ca2+]i, but not insulin secretion, in KATP loss-of-function islets lacking gap junctions
So far we have found that in intact Cx36−/− islets, the elevated [Ca2+]i at low glucose is similar to dissociated β-cells. However, insulin secretion is unchanged, in contrast to the elevated insulin secretion seen at low glucose and the reduced fold-response in isolated β-cells. To further test these divergent [Ca2+]i and insulin response, we studied the role of Cx36 gap junctions in islets of mice that have a loss-of-function to the KATP channel, via expression of a dominant-negative mutant-Kir6.2 subunit (Kir6.2[AAA]; Koster et al. 2002). This mutation renders the pore-forming subregion of the channel non-functional, essentially eliminating KATP conductance independent of glucose. Therefore increased variation in cellular electrical activity occurs, defined by the penetrance of the Kir6.2[AAA] transgene (visualized by a GFP tag). Isolated β-cells of Kir6.2[AAA] islets exhibit elevated and largely glucose-independent [Ca2+]i and insulin secretion responses, as expected given the substantial reduction in KATP conductance. However, within intact Kir6.2[AAA] islets, β-cells exhibit essentially normal [Ca2+]i and insulin secretion responses to glucose (Rocheleau et al. 2006). Therefore the maintained cell–cell contacts in these islets robustly suppresses the expected elevations in [Ca2+]i and insulin release at low glucose due to the KATP mutation. These islets therefore allow us to quantify and further distinguish how gap junctions and other means of cell–cell communication can specifically regulate basal electrical activity and insulin secretion in the intact islet. They also allow us to determine how insulin secretion may be suppressed by cell–cell contacts in a more mechanistic manner.
We therefore crossed Kir6.2[AAA] transgenic mice with Cx36−/− mice. At glucose concentrations ranging from 2 to 6 mm, there was a substantial increase in [Ca2+]i bursts observed in Kir6.2[AAA]:Cx36−/− islets compared to more typically quiescent [Ca2+]i changes observed in Kir6.2[AAA]:Cx36+/+ and WT control islets (Fig. 4A). Kir6.2[AAA]:Cx36+/− islets exhibited an intermediate number of [Ca2+]i bursts (Fig. 4B), indicating a progressive increase in [Ca2+]i activity as the level of electrical coupling is gradually reduced (Supplemental Fig. S1). Similarly, in Kir6.2[AAA]:Cx36−/− islets, the [Ca2+]i concentration observed at 2 mm glucose was significantly increased, while the [Ca2+]i concentration at 20 mm glucose was significantly reduced (Fig. 4C). Isolated β-cells dissociated from Kir6.2[AAA]:Cx36+/+ or Kir6.2[AAA]:Cx36−/− islets exhibited similar glucose-stimulated [Ca2+]i responses (Fig. 4D and E), with elevated [Ca2+]i at 2 mm glucose restricted to GFP+ cells expressing the mutant Kir6.2[AAA] transgene.
Figure 4. Gap junction-dependent [Ca2+]i in Kir6.2[AAA] islets.
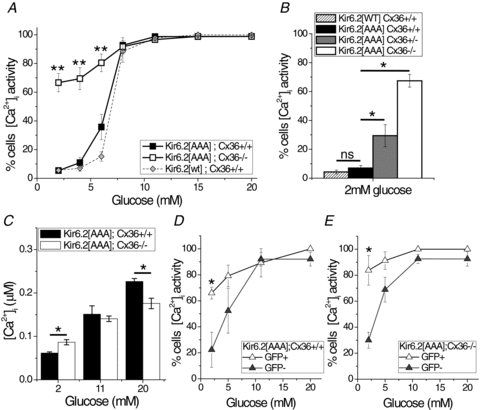
A, mean percentage of cells displaying dynamic changes in [Ca2+]i as a function of glucose stimulation, for islets isolated from Kir6.2[AAA]:Cx36+/+ (black squares), Kir6.2[AAA]:Cx36−/− (open squares), Kir6.2[WT]:Cx36+/+ (wild-type, grey diamonds) mice. Data averaged over n = 5 mice. **Significant difference of P < 0.01 (Student's unpaired t test) at each glucose concentration comparing Kir6.2[AAA]:Cx36+/+ and Kir6.2[AAA]:Cx36−/− data. B, mean percentage of islet cells displaying dynamic changes in [Ca2+]i at 2 mm glucose, for intact Kir6.2[WT]:Cx36+/+ (hatched), Kir6.2[AAA]:Cx36+/+ (black), Kir6.2[AAA]:Cx36+/− (grey), and Kir6.2[AAA]:Cx36−/− (white) islets. Data averaged over n = 6 mice. *Significant difference of P < 0.05 and ‘ns’, non-significant difference (P > 0.05), comparing each experimental group as indicated. C, mean [Ca2+]i concentration as a function of glucose stimulation for intact Kir6.2[AAA]:Cx36+/+ islets (black bars) and Kir6.2[AAA]:Cx36−/− islets (white bars). Data averaged over n = 3 mice. *Significant difference of P < 0.05 (Student's paired t test) comparing each experimental group as indicated. D, mean percentage of cells, dissociated from Kir6.2[AAA]:Cx36+/+ islets, displaying dynamic changes in [Ca2+]i as a function of glucose stimulation for GFP positive cells (open triangles) and GFP negative cells (grey triangles). E, as in D for cells dissociated from Kir6.2[AAA]:Cx36−/− islets. Data in D and E averaged over n = 4 experiments, from 2 mice. *Significant difference of P < 0.05 (Student's unpaired t test) at each glucose concentration comparing GFP positive and GFP negative cells.
We next determined if the altered [Ca2+]i observed in the absence of Cx36 impacts insulin secretion in Kir6.2[AAA] islets. GSIS observed in Kir6.2[AAA]:Cx36−/− islets is slightly left shifted to glucose compared to control transgenic islets, but there was no significant difference measured at 2 mm or 5.5 mm glucose compared to Kir6.2[AAA]:Cx36+/+ and WT controls (Fig. 5A and B). Significant elevation of insulin secretion was measured at 8 mm and 11 mm glucose, but not at 20 mm glucose (Fig. 5A), and insulin content was not significantly different (Fig. 5C). GSIS in dissociated β-cells from Kir6.2[AAA]:Cx36+/+ or Kir6.2[AAA]:Cx36−/− islets was also not significantly different (Fig. 5D), consistent with the pattern previously reported for Kir6.2[AAA] cells (Rocheleau et al. 2006). Thus in intact Kir6.2[AAA] islets, the substantially elevated [Ca2+]i at low (2–6 mm) glucose caused by an elimination of Cx36 gap junction coupling does not lead to a significant elevation in insulin secretion. Elevated insulin secretion does occur, however, in dissociated β-cells at low glucose, as well as in intact islets at moderately higher glucose levels.
Figure 5. Insulin secretion in Kir6.2[AAA]:Cx36−/− islets.
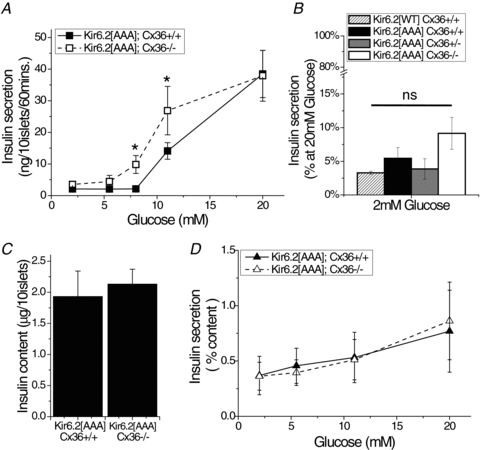
A, glucose-stimulated insulin secretion from isolated islets of Kir6.2[AAA]:Cx36+/+ mice (black squares) and Kir6.2[AAA]:Cx36−/− mice (open squares). Data averaged over n = 7 mice. *Significant difference of P < 0.05 (Student's paired t test) at each glucose concentration comparing Kir6.2[AAA]:Cx36+/+ and Kir6.2[AAA]:Cx36−/− data. B, insulin secretion at 2 mm glucose, for intact Kir6.2[WT]:Cx36+/+ (hatched), Kir6.2[AAA]:Cx36+/+ (black), Kir6.2[AAA]:Cx36+/− (grey), and Kir6.2[AAA]:Cx36−/− (white) islets. Data are expressed as a percentage of insulin secretion in Kir6.2[AAA]:Cx36+/+ islets at 20 mm glucose to facilitate comparison with Fig. 4B. ‘ns’ indicates non-significant difference (P > 0.05) comparing each experimental group to Kir6.2[AAA]. C, insulin content of Kir6.2[AAA]:Cx36+/+ and Kir6.2[AAA]:Cx36−/− islets. Data averaged over n = 7 mice. D, glucose-stimulated insulin secretion from dissociated Kir6.2[AAA]:Cx36+/+ (grey triangles) and Kir6.2[AAA]:Cx36−/− (open triangles) β-cells. Displayed is the fractional insulin secretion per hour normalized by insulin content. Data averaged over n = 6 mice. The difference in means ± 95% confidence interval for A and C is shown in Fig. S2C.
Gap junction-independent suppression of insulin by cell–cell contact is cAMP and PKA dependent
We then asked what molecular events underlie the gap-junction independent suppression of insulin secretion in intact Kir6.2[AAA]:Cx36−/− islets at 2 mm glucose? The suppression still occurs despite a large elevation in [Ca2+]i, suggesting that a step distal to [Ca2+]i elevation in GSIS is involved. A number of Ca2+-independent pathways underlying insulin secretion regulate the trafficking and exocytosis of insulin granules at the plasma membrane (Henquin, 2000), including elevated cAMP (Wang & Thurmond, 2009). To test whether a step distal to [Ca2+]i is involved, we elevated cAMP in Kir6.2[AAA] islets by applying 3-isobutyl-1-methylxanthine (IBMX; 100 μm) and forskolin (50 μm). At 2 mm glucose, insulin secretion was suppressed in Kir6.2[AAA]:Cx36+/+ islets. There was a significant elevation in insulin secretion from Kir6.2[AAA]:Cx36−/− islets (Fig. 6A), approaching that seen at 20 mm glucose and correlating with the measured [Ca2+]i changes in these islets. Application of the GLP1-receptor (GLP1R) agonist exendin4 (100 nm, Tocris Bioscience, Ellisville, MO, USA), which raises cAMP levels by Gs activation of adenyl cyclase, also led to an elevation of insulin secretion at 2 mm glucose, but only in Kir6.2[AAA]:Cx36−/− islets (Fig. 6B). To test for potential alterations in the sensitivity of Ca2+-triggered insulin secretion which could also explain the Ca2+-independent suppression, we maximally elevated [Ca2+]i using high KCl (+30 mm). This led to a similar elevation in insulin secretion in both sets of islets (Fig. 6C).
Figure 6. Gap junction-independent regulation of insulin secretion in Cx36−/− islets.
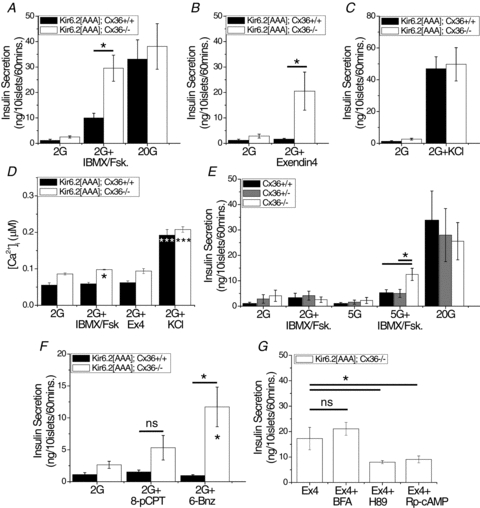
A, insulin secretion from isolated islets of Kir6.2[AAA]:Cx36+/+ (black bars) and Kir6.2[AAA]:Cx36−/− (white bars) mice. Islets were incubated at 2 mm glucose (2G), plus the cAMP raising agents IBMX and forskolin (+IBMX/Fsk, 100 μm and 50 μm, respectively), or with 20 mm glucose (20G). Data averaged over n = 5 littermate mouse pairs. B, insulin secretion from isolated islets, as in A, plus the GLP1R agonist exendin4 (100 nm). Data averaged over n = 7 littermate mouse pairs. C, insulin secretion from isolated islets as in A, plus high KCl (+KCl, +30 mm). Data averaged over n = 5 littermate mouse pairs. D, mean [Ca2+]i concentration in isolated islets, treated with 2 mm glucose alone, or plus IBMX and forskolin, exendin4 (+Ex4), or high KCl, as in A–C. Data averaged over n = 3 mice. E, insulin secretion from isolated islets of Cx36+/+ (black bars), Cx36+/− (grey bars), Cx36−/− (white bars) mice, incubated at 2 mm or 5.5 mm glucose alone (2G, 5G), plus IBMX and forskolin (+IBMX/Fsk, 100 μm and 50 μm, respectively), or at 20 mm glucose alone (20G). Data averaged over n = 4 littermate mice. F, insulin secretion from islets, as in A, plus either the Epac-specific cAMP analogue 8-pCPT-2-O-Me-cAMP (8-pCPT, 300 μm) or the PKA-specific cAMP analogue 6-Bnz-cAMP (6-Bnz, 300 μm). Data averaged over n = 7 littermate mice pairs. G, insulin secretion from isolated islets of Kir6.2[AAA]:Cx36−/− mice incubated at 2 mm glucose plus exendin4 alone (Ex4, 100 nm) or exendin4 plus either the Epac inhibitor brefeldin A (BFA, 100 μm), the PKA antagonist H89 (10 μm) or the specific PKA antagonist Rp-cAMP (100 μm). Data averaged over n = 8 mice. *Significant difference of P < 0.05, ***significant difference of P < 0.001, ‘ns’, non-significant difference (P > 0.05) (Student's paired t test), comparing each experimental group as indicated, or compared to 2 mm glucose alone (in D and F indicated within the bar).
The high KCl treatment was associated with a substantial elevation in [Ca2+]i in both Kir6.2[AAA]:Cx36+/+ and Kir6.2[AAA]:Cx36−/− islets (Fig. 6D). However, cAMP elevations induced by IBMX and forskolin led to a very small elevation in [Ca2+]i in the Kir6.2[AAA]:Cx36−/– islets, and exendin4 treatment had no significant effect on [Ca2+]i (Fig. 6D). We also measured insulin secretion in Cx36+/+, Cx36+/−, or Cx36−/− islets (Kir6.2 wild-type) treated with IBMX and forskolin. At 5.5 mm, but not 2 mm glucose, there was increased insulin secretion in the Cx36−/− islets compared to Cx36+/+ islets (Fig. 6E), again correlating with the measured [Ca2+]i changes in these islets (Fig. 1).
We then used two different approaches to determine the target of cAMP that enables the β-cells to overcome the suppression of Ca2+-triggered insulin secretion at low glucose. First, we applied cAMP analogues specific to either protein kinase A (PKA) or exchange protein activated by cAMP 2 (Epac2), the major targets of cAMP in the β-cell. Application of the PKA-specific agonist 6-Bnz-cAMP (300 μm, Sigma) produced a significant elevation of insulin secretion from Kir6.2[AAA]:Cx36−/− islets (Fig. 6F), but no elevation was observed upon application of the Epac-specific agonist 8-Me-2-O-pCPT-cAMP (300 μm, Biolog, Bremen, Germany). In each case, insulin secretion from Kir6.2[AAA]:Cx36+/+ islets was unchanged. Second, the PKA antagonist H89 (10 μm) and the specific PKA antagonist Rp-cAMP (100 μm, Biolog) both reduced the exendin4-dependent elevation in insulin secretion in Kir6.2[AAA]:Cx36−/− islets (Fig. 6G). Application of brefeldin A (100 μm), previously used as an Epac inhibitor (Zhong & Zucker, 2005), has no effect on the exendin4-dependent elevation of insulin secretion at low glucose. Thus at low glucose, elevated cAMP acting via PKA, leads to elevated insulin secretion only in the absence of gap junction electrical coupling, and only when this absence of electrical coupling leads to elevated [Ca2+]i.
Discussion
The ability of cells to communicate with one another in the intact islet of Langerhans is important for the enhancement and coordination of glucose-stimulated insulin secretion (GSIS), as well as for the uniform suppression of insulin secretion at low glucose. Gap junction coupling coordinates the dynamics underlying GSIS, and can mediate the suppression of electrical activity induced in a population of islet β-cells. We asked whether gap junction coupling alone mediates a suppression of electrical activity in intact islets, and whether this impacts insulin secretion at low glucose. To account for other mechanisms that could be suppressing basal insulin secretion, we compared islets that lack functional gap junction coupling (Cx36−/−) with dissociated β-cells that lack any cellular proximity. We also utilized islets with altered KATP channel activity, in which elevations in [Ca2+]i and insulin release are suppressed in the intact islet but not isolated β-cells. Together, these experiments allow us to specifically quantify the relative role of gap junctions in regulating and suppressing basal electrical activity and insulin secretion.
Gap junctions alone suppress basal excitability and enhance glucose-stimulated excitability
The near total elimination of junctional conductance by a Cx36 deletion indicates a lack of functional gap junction coupling between β-cells. The spontaneous [Ca2+]i bursts which emerge in a subpopulation of β-cells at low glucose indicates a variation between cells (heterogeneity) in the threshold at which glucose stimulates [Ca2+]i. Similar heterogeneity in [Ca2+]i oscillations occurs in both dissociated β-cells (Zhang et al. 2003) and islets lacking gap junctions (Ravier et al. 2005; Benninger et al. 2008). Therefore β-cells within the islet are intrinsically heterogeneous in terms of the electrical response to glucose, and gap junction acts to uniformly suppress any subthreshold response. A distribution for the glucose threshold for [Ca2+]i bursting can be interpolated from the response (Fig. 1B), a median of ∼5 mm, with lower/upper quartiles of ∼3.5 mm/∼9 mm, respectively. Although significant data exist on the heterogeneities of isolated β-cells (Pipeleers, 1992; Vanschravendijk et al. 1992; Bennett et al. 1996; Zhang et al. 2003; Wojtusciszyn et al. 2008), it remains to be determined whether this heterogeneity arises from a variation between β-cells in ATP-synthesis, KATP channel density, or other factors.
The similar [Ca2+]i response to glucose in Cx36−/− islets and isolated β-cells suggests gap junctions are the predominant mechanism present to suppress elevations in basal [Ca2+]i that rise due to cellular heterogeneity. We quantitatively confirm this in Cx36−/− islets that express a loss-of-function KATP channel mutation: the elevated [Ca2+]i at low glucose is similar to the penetrance of the Kir6.2[AAA] transgene and similar to the elevated [Ca2+]i in isolated β-cells from these islets (Fig. 4). We therefore extend the previous model (Rocheleau et al. 2006) to normal islets whereby gap junctions alone overcome the intrinsic β-cell heterogeneity such that the less-excitable cells act to suppress the more-excitable cells and uniformly control the overall electrical response across the islet.
In the absence of Cx36, the heterogeneity in [Ca2+]i oscillations means that some β-cells exhibit transient repolarizing events even at high glucose; in contrast to the sustained elevation in [Ca2+]i in the presence of Cx36. This can explain the decrease in mean [Ca2+]i concentration at high glucose stimulation in the absence of Cx36, and points to the role of gap junctions to also maximize the [Ca2+]i response.
Redundancy in how cell–cell communication suppresses basal insulin secretion
Despite the elevations in [Ca2+]i at low glucose following a deletion of Cx36, there is no elevation in insulin secretion. This is in contrast to the elevated basal insulin secretion (and lower dynamic range) observed in dissociated β-cells. Thus, mechanisms dependent on cell–cell contacts within the islet robustly suppresses insulin secretion at low glucose independent of gap junction coupling and associated [Ca2+]i elevations. The divergence between basal [Ca2+]i and insulin secretion is most striking in the KATP loss-of-function islets lacking gap junctions (see Figs 4B and 5B), which makes these an ideal model to study the physiological gap junction-independent suppression of insulin secretion (see below).
Elevating cAMP levels under low glucose conditions causes a rise in insulin secretion in the absence of Cx36, but negligibly in the presence of Cx36. This result demonstrates that gap junctions can robustly suppress elevations in basal insulin secretion induced by secretagogues that act independent of Ca2+. Importantly, these data suggest that GLP1 or exendin-4 based treatments to elevate islet cAMP (Lovshin & Drucker, 2009) will be less effective for enhancing islet function in diabetic individuals when gap junction coupling is also dysregulated: the cAMP-induced insulin secretion at low glucose will reduce the dynamic range of the glucose-dependent insulin secretion.
The lack of suppression of basal insulin secretion in the absence of Cx36 following elevated cAMP (Fig. 6) suggests that cAMP, mediated by PKA, is overcoming the gap junction-independent coupling mechanisms that normally suppress insulin secretion. Since no further change in [Ca2+]i is measured upon these treatments (Fig. 6D), the suppressive effect at low glucose acts distal to Ca2+ triggering, likely at the level of trafficking and/or exocytosis of insulin granules (Wang & Thurmond, 2009). This is consistent with other juxtacrine mechanisms that can regulate insulin granule trafficking at elevated glucose, such as EphA–ephrinA (Konstantinova et al. 2007) and NCAM (Olofsson et al. 2009). Interestingly, under basal glucose conditions little or no elevations in insulin release were observed in those studies upon a disruption of EphA or NCAM signalling, despite altered F-actin stability, and in contrast to substantial changes that occurred at elevated glucose. We can account for this since the presence of gap junction coupling would still suppress basal [Ca2+]i elevations and thus insulin release, in a similar manner to when cAMP is elevated. Furthermore, EphA signalling can regulate Rac1 activity (Konstantinova et al. 2007), a key component of the exocytotic machinery that can also be regulated by cAMP and PKA (O'Connor & Mercurio, 2001; Li et al. 2004). Therefore elevated PKA would overcome any suppressive ability of EphA5 signalling. We conclude there is a redundancy in the action of cell–cell communication in suppressing insulin secretion at low glucose levels, with other mechanisms likely acting to regulate steps in insulin granule trafficking and exoctyosis.
The elevated insulin secretion at moderate elevations in glucose (8–11 mm) in Kir6.2[AAA]:Cx36−/− islets suggests that other suppressive mechanisms diminish as glucose levels rise. Furthermore, in isolated islets expressing gain-of-function KATP channels, at high glucose modulation of gap junctions alone can elevate [Ca2+]i and insulin secretion (Benninger et al. 2011). These data are also consistent with the suppressive action of EphA–ephrinA (Konstantinova et al. 2007) being glucose dependent.
Summary
We summarize in Fig. 7 how mechanisms dependent on cell–cell contact suppresses insulin secretion at low glucose with a robust dual regulation. Due to intrinsic β-cell heterogeneity, some of the cells will have elevated excitability. In the absence of any cell–cell contact these β-cells exhibit elevated [Ca2+]i and insulin secretion at low glucose. In the intact islet, gap junctions suppress elevations in [Ca2+]i by coupling hyperpolarizing currents from the less-excitable β-cells to the more-excitable β-cells (Fig. 7A). In addition, other juxtacrine mechanisms dependent on cell–cell contact suppress insulin secretion by regulating insulin granule trafficking and/or exocytosis. In the absence of gap junctions, which leads to elevated basal [Ca2+]i, other juxtacrine mechanisms still suppress insulin release at the level of exocytosis (Fig. 7B). Upon elevated cAMP, mediated by PKA, which overcomes other suppressive juxtacrine mechanisms and promotes exocytosis, gap junctions still control the suppression of basal insulin release via suppressing Ca2+ triggering (Fig. 7C). Only in the absence of Cx36 gap junctions and elevated cAMP/PK, which overcomes other suppressive juxtacrine mechanisms, do cells in the intact islet behave like dissociated β-cells (Fig. 7D).
Cell–cell communication can regulate hormone secretion in other neuroendocrine cellular systems (e.g. Bonnefont et al. 2005; Asa & Tannenbaum, 2006; Ojeda et al. 2006; Colomer et al. 2008). Similar multicellular mechanisms could also be present in these systems and play a role in regulating the response of a wide range of hormone secretions.
Acknowledgments
We would like to thank Maria S. Remedi for critical reviewing of this manuscript. This study was primarily supported by NIH grants K99-DK085145 (to R.K.P.B.), R01-DK053434 and P20-GM072048 (to D.W.P.), R01-DK46409 (L.S.S.); and the Department of Defense Medical Free-Electron Laser Program. Experiments on the LSM5Live were performed in part through the use of VUMC Cell Imaging Shared Resource (DK20593, CA68485, DK58404). Radioimmunoassays for insulin concentration were performed within the VUMC hormone assay core (DK20593).
Glossary
Abbreviations
- αGA
18-α-glycyrrhetinic acid
- [Ca2+]i
intracellular free calcium activity
- Cx36
connexin36
- GFP
green fluorescent protein
- GSIS
glucose-stimulated insulin secretion
- KATP channel
ATP-sensitive potassium channel
- PM
plasma membrane
- Vm
membrane potential
- PKA
protein kinase A
- GLP1R
glucagon-like peptide 1 receptor
- Epac
exchange protein activated by cAMP
Author contributions
R.K.P.B. designed the experiments, researched the data and wrote the manuscript. W.S.H. researched the data. M.Z. designed the experiments and researched the data. L.S.S. reviewed and edited the manuscript. D.W.P. reviewed and edited the manuscript and contributed to the discussion. All authors approved the final version of the manuscript.
Supplementary material
Figure S1
Figure S2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Asa SL, Tannenbaum GS. Cell-cell communication in the pituitary: orchestrator of pulsatile growth hormone secretion? Trends Endocrinol Metab. 2006;17:299–300. doi: 10.1016/j.tem.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Jetton TL, Ying GT, Magnuson MA, Piston DW. Quantitative subcellular imaging of glucose metabolism within intact pancreatic islets. J Biol Chem. 1996;271:3647–3651. doi: 10.1074/jbc.271.7.3647. [DOI] [PubMed] [Google Scholar]
- Benninger RKP, Remedi MS, Head WS, Ustione A, Piston DW, Nichols CG. Defects in β-cell Ca2+ signalling, glucose metabolism and insulin secretion in a murine model of KATP channel-induced neonatal diabetes mellitus. Diabetologia. 2011;54:1087–1097. doi: 10.1007/s00125-010-2039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RKP, Zhang M, Head WS, Satin LS, Piston DW. Gap junction coupling and calcium waves in the pancreatic islet. Biophys J. 2008;95:5048–5061. doi: 10.1529/biophysj.108.140863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefont X, Lacampagne A, Sanchez-Hormigo A, Fino E, Creff A, Mathieu MN, Smallwood S, Carmignac D, Fontanaud P, Travo P, Alonso G, Courtois-Coutry N, Pincus SM, Robinson I, Mollard P. Revealing the large-scale network organization of growth hormone-secreting cells. Proc Natl Acad Sci U S A. 2005;102:16880–16885. doi: 10.1073/pnas.0508202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese A, Zhang M, Serre-Beinier W, Caton D, Mas C, Satin LS, Meda P. Connexin 36 controls synchronization of Ca2+ oscillations and insulin secretion in MIN6 cells. Diabetes. 2003;52:417–424. doi: 10.2337/diabetes.52.2.417. [DOI] [PubMed] [Google Scholar]
- Charpantier E, Cancela J, Meda P. β cells preferentially exchange cationic molecules via connexin 36 gap junction channels. Diabetologia. 2007;50:2332–2341. doi: 10.1007/s00125-007-0807-9. [DOI] [PubMed] [Google Scholar]
- Colomer C, Ore LAO, Coutry N, Mathieu MN, Arthaud S, Fontanaud P, Iankova I, Macari F, Thouennon E, Yon L, Anouar Y, Guerineau NC. Functional remodeling of gap junction-mediated electrical communication between adrenal chromaffin cells in stressed rats. J Neurosci. 2008;28:6616–6626. doi: 10.1523/JNEUROSCI.5597-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degen J, Meier C, Van Der Giessen RS, Sohl G, Petrasch-Parwez E, Urschel S, Dermietzel R, Schilling K, De Zeeuw CI, Willecke K. Expression pattern of lacZ reporter gene representing connexin36 in transgenic mice. J Comp Neurol. 2004;473:511–525. doi: 10.1002/cne.20085. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halban PA, Wollheim CB, Blondel B, Meda P, Niesor EN, Mintz DH. The possible importance of contact between pancreatic-islet cells for the control of insulin release. Endocrinology. 1982;111:86–94. doi: 10.1210/endo-111-1-86. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- Konstantinova I, Nikolova G, Ohara-Imaizumi M, Meda P, Kucera T, Zarbalis K, Wurst W, Nagamatsu S, Lammert E. EphA-ephrin-A-mediated β cell communication regulates insulin secretion from pancreatic islets. Cell. 2007;129:359–370. doi: 10.1016/j.cell.2007.02.044. [DOI] [PubMed] [Google Scholar]
- Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of β cell K-ATP channels induces profound neonatal diabetes. Cell. 2000;100:645–654. doi: 10.1016/s0092-8674(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Koster JC, Remedi MS, Flagg TP, Johnson JD, Markova KP, Marshall BA, Nichols CG. Hyperinsulinism induced by targeted suppression of beta cell K-ATP channels. Proc Natl Acad Sci U S A. 2002;99:16992–16997. doi: 10.1073/pnas.012479199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lernmark A. Preparation of, and studies on, free cell-suspensions from mouse pancreatic-islets. Diabetologia. 1974;10:431–438. doi: 10.1007/BF01221634. [DOI] [PubMed] [Google Scholar]
- Li JS, Luo RH, Kowluru A, Li GD. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 β-cells. Am J Physiol Endocrinol Metab. 2004;286:E818–E827. doi: 10.1152/ajpendo.00307.2003. [DOI] [PubMed] [Google Scholar]
- Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:262–269. doi: 10.1038/nrendo.2009.48. [DOI] [PubMed] [Google Scholar]
- Mears D, Sheppard NF, Atwater I, Rojas E. Magnitude and modulation of pancreatic β-cell gap junction electrical conductance in-situ. J Membr Biol. 1995;146:163–176. doi: 10.1007/BF00238006. [DOI] [PubMed] [Google Scholar]
- Moreno AP, Berthoud VM, Perez-Palacios G, Perez-Armendariz EM. Biophysical evidence that connexin-36 forms functional gap junction channels between pancreatic mouse β-cells. Am J Physiol Endocrinol Metab. 2005;288:E948–E956. doi: 10.1152/ajpendo.00216.2004. [DOI] [PubMed] [Google Scholar]
- O'Connor KL, Mercurio AM. Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J Biol Chem. 2001;276:47895–47900. doi: 10.1074/jbc.M107235200. [DOI] [PubMed] [Google Scholar]
- Ojeda SR, Lomniczi A, Mastronardi C, Heger S, Roth C, Parent AS, Matagne V, Mungenast AE. Minireview: The neuroendocrine regulation of puberty: Is the time ripe for a systems biology approach? Endocrinology. 2006;147:1166–1174. doi: 10.1210/en.2005-1136. [DOI] [PubMed] [Google Scholar]
- Olofsson CS, Hakansson J, Salehi A, Bengtsson M, Galvanovskis J, Partridge C, Sorhedewinzell M, Xian XJ, Eliasson L, Lundquist I, Semb H, Rorsman P. Impaired insulin exocytosis in neural cell adhesion molecule−/− mice due to defective reorganization of the submembrane f-actin network. Endocrinology. 2009;150:3067–3075. doi: 10.1210/en.2008-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipeleers DG. Heterogeneity in pancreatic β-cell population. Diabetes. 1992;41:777–781. doi: 10.2337/diab.41.7.777. [DOI] [PubMed] [Google Scholar]
- Quesada I, Fuentes E, Andreau E, Meda P, Nadal A, Soria B. On-line analysis of gap junctions reveals more efficient electrical than dye coupling between islet cells. Am J Physiol Endocrinol Metab. 2003;284:E980–E987. doi: 10.1152/ajpendo.00473.2002. [DOI] [PubMed] [Google Scholar]
- Quesada I, Todorova MG, Alonso-Magdalena P, Beltra M, Carneiro EM, Martin F, Nadal A, Soria B. Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified α- and β-cells within intact human islets of langerhans. Diabetes. 2006;55:2463–2469. doi: 10.2337/db06-0272. [DOI] [PubMed] [Google Scholar]
- Ravier MA, Guldenagel M, Charollais A, Gjinovci A, Caille D, Sohl G, Wollheim CB, Willecke K, Henquin JC, Meda P. Loss of connexin36 channels alters β-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes. 2005;54:1798–1807. doi: 10.2337/diabetes.54.6.1798. [DOI] [PubMed] [Google Scholar]
- Rocheleau JV, Remedi MS, Granada B, Head WS, Koster JC, Nichols CG, Piston DW. Critical role of gap junction coupled K-ATP channel activity for regulated insulin secretion. PLoS Biol. 2006;4:221–227. doi: 10.1371/journal.pbio.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharp DW, Kemp CB, Knight MJ, Ballinge WF, Lacy PE. Use of Ficoll in preparation of viable islets of Langerhans from rat pancreas. Transplantation. 1973;16:686–689. doi: 10.1097/00007890-197312000-00028. [DOI] [PubMed] [Google Scholar]
- Sherman A, Xu L, Stokes CL. Estimating and eliminating junctional current in coupled cell-populations by leak subtraction – a computational study. J Membr Biol. 1995;143:79–87. doi: 10.1007/BF00232525. [DOI] [PubMed] [Google Scholar]
- Speier S, Gjinovci A, Charollais A, Meda P, Rupnik M. Cx36-mediated coupling reduces β-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes. 2007;56:1078–1086. doi: 10.2337/db06-0232. [DOI] [PubMed] [Google Scholar]
- Theis M, Mas C, Doring B, Degen J, Brink C, Caille D, Charollais A, Kruger O, Plum A, Nepote V, Herrera P, Meda P, Willecke K. Replacement by a lacZ reporter gene assigns mouse connexin36, 45 and 43 to distinct cell types in pancreatic islets. Exp Cell Res. 2004;294:18–29. doi: 10.1016/j.yexcr.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Vanschravendijk CFH, Kiekens R, Pipeleers DG. Pancreatic β-cell heterogeneity in glucose-induced insulin-secretion. J Biol Chem. 1992;267:21344–21348. [PubMed] [Google Scholar]
- Wang ZX, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis – roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:893–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtusciszyn A, Armanet M, Morel P, Berney T, Bosco D. Insulin secretion from human β cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia. 2008;51:1843–1852. doi: 10.1007/s00125-008-1103-z. [DOI] [PubMed] [Google Scholar]
- Zhang M, Goforth P, Bertram R, Sherman A, Satin L. The Ca2+ dynamics of isolated mouse β-cells and islets: Implications for mathematical models. Biophys J. 2003;84:2852–2870. doi: 10.1016/S0006-3495(03)70014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Zucker RS. cAMP acts on exchange protein activated by cAMP/cAMP-regulated guanine nucleotide exchange protein to regulate transmitter release at the crayfish neuromuscular junction. J Neurosci. 2005;25:208–214. doi: 10.1523/JNEUROSCI.3703-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.