

Non-technical summary
Intermittent hypoxia is known to increase oxidative stress and decrease nitric oxide metabolism. These two responses, which are involved in hypoxia-induced hypertension, may be mediated by angiotensin II. Using a novel human experimental model, we show that blockade of the type 1 angiotensin II receptors by a medication called losartan prevented the increase in oxidative stress and the decrease in nitric oxide metabolism induced by 6 h of intermittent hypoxia. These results show that the upregulation of angiotensin II contributes to the overproduction of free radicals associated with intermittent hypoxia and help us better understand why blood pressure increases in medical disorders associated with intermittent hypoxia, such as obstructive sleep apnoea.
Abstract
Abstract
The aim of this study was to assess the role of the type 1 angiotensin II (AT1) receptor in the increase of oxidative stress and NO metabolism during a single 6 h exposure to intermittent hypoxia (IH). Nine healthy young men were exposed, while awake, to sham IH, IH with placebo medication, and IH with the AT1 receptor antagonist, losartan, using a double-blind, placebo-controlled, randomized, crossover study design. In addition to blood pressure, oxidative stress, peroxynitrite activity, uric acid, global antioxidant status and the end-products of NO (NOx) metabolism were measured in plasma before and after 6 h of IH. Oxidative stress and peroxynitrite activity increased and NOx decreased during IH with placebo. In contrast, neither sham IH nor IH with losartan affected these parameters. With respect to each condition, blood pressure had the same profile as oxidative stress. These results demonstrate that blockade of AT1 receptors prevented the increase in oxidative stress and peroxynitrite activity and the decrease in NO metabolism induced by IH. Finally, this study suggests that the renin–angiotensin system may participate in the overproduction of reactive oxygen species associated with IH by upregulation of the actions of angiotensin II.
Introduction
Chronic exposure to intermittent hypoxia (IH) in patients suffering from obstructive sleep apnea (OSA) may be responsible for an increased risk of cardiovascular disease (Dyugovskaya et al. 2002; Peng et al. 2006). In order to exclude confounding factors, such as hypertension, obesity or diabetes, human models of intermittent hypoxia have been designed using healthy subjects exposed to IH in either hypoxic chambers (Foster et al. 2009) or hypoxic tents (Tamisier et al. 2009). We recently demonstrated that 4 days of diurnal IH significantly increased blood pressure in healthy subjects (Foster et al. 2009), and Tamisier et al. (2009) reported a slightly higher increase after 2 weeks of nocturnal IH.
It has been proposed that oxidative stress induced by angiotensin II (Ang II) (Taniyama & Griendling, 2003; Fliser et al. 2004) may be one mechanism for the elevation of blood pressure. An elevated concentration of Ang II in the plasma has been reported in patients with OSA (Kraiczi et al. 2000a), and we recently described an increase in oxidative stress after IH in healthy subjects (Pialoux et al. 2009a). Angiotensin II modulates vasoconstriction by acting through the angiotensin II type 1 (AT1) receptors located in vascular smooth muscle (Mehta & Griendling, 2007), and growing evidence suggests that such detrimental effects of AT1 receptor activation are mediated, at least in part, by oxidative stress through the activation of NADPH oxidase (Mehta & Griendling, 2007). Moreover, an increase in reactive oxygen species (ROS) through the activation of Ang II is known to decrease nitric oxide bioavailability in the vasculature and further leads to endothelial dysfunction (Thomas et al. 2008). More specifically, ROS inactivate NO synthesized by endothelial nitric oxide synthase (eNOS) and can induce an uncoupling of eNOS (Thomas et al. 2008).
Although it has been demonstrated that AT1 receptor blockers inhibit oxidative stress and reverse endothelial dysfunction in patients with hypertension (Dohi et al. 2003), there have been no studies on Ang II-induced oxidative stress in OSA patients or in subjects exposed to IH. Nevertheless, 6 weeks of treatment with an AT1 receptor blocker significantly reduced nocturnal systolic (SBP) and diastolic blood pressure (DBP) in OSA patients (Kraiczi et al. 2000b); however, daytime blood pressure was not significantly altered. Co-morbid disease known to increase blood pressure may explain this lack of change and mask the potential effects of AT1 receptor blockade on blood pressure induced by IH.
In this context, we recently demonstrated that the administration of an AT1 receptor blocker in healthy humans prevented the increase in blood pressure induced by a single 6 h exposure to IH (Foster et al. 2010). Therefore, the present study was part of a larger project specifically designed to test the effect of AT1 receptor blockade on blood pressure change induced by acute exposure to IH during wakefulness, the first part of which has been published previously (Foster et al. 2010). The second part is presented in this article, where we hypothesized that blockade of the AT1 receptor blunted both the increase in oxidative stress and the subsequent decrease in NO activity induced by the 6 h exposure to IH. The model of IH was designed to replicate the cyclical pattern of hypoxaemia and reoxygenation that occurs in patients with OSA and to exclude potential confounding factors which may affect blood pressure, oxidative stress or NO metabolism. This hypothesis was tested using a double-blind, placebo-controlled, randomized, crossover study design in healthy humans.
Methods
Ethical approval
The research study was approved by the Conjoint Health Research Ethics Board (protocol ID E-21922) at the University of Calgary, by the Office of Clinical Trials – Therapeutic Products Directorate – Health Canada (Clinical Trial Application for LOSARTAN, control no. 123826), and conformed to the Declaration of Helsinki. Written informed consent was obtained from each subject prior to participation in the study.
Subjects
Based on a priori sample size calculations conducted for the primary outcome variable, i.e. changes in mean arterial pressure (MAP) in response to IH, from previously published data (Foster et al. 2009), we calculated that eight or more subjects were required to provide sufficiently high power (β = 0.10) for two-sided tests (α = 0.05). Consequently, 10 healthy male subjects, all residents of Calgary, Alberta, Canada (elevation 1103 m) for at least 1 year, participated in the study. All were non-smokers, did not take medication, and had no history of cardiovascular, cerebrovascular, respiratory or kidney disease. Women were not studied because fluctuations in oestrogen throughout the menstrual cycle may affect the oxidative stress response to intermittent hypoxia (Castelao & Gago-Dominguez, 2008).
Experimental protocols
All subjects were submitted to three experimental protocols (IH+losartan, IH+placebo and sham IH) separated by 1 week minimum, and occurring in random order at the same time of day (i.e. between 9:00 and 15:00 hours). The IH+losartan and IH+placebo protocols were designed in a double-blind fashion. In order to avoid differences in the consumption of exogenous antioxidants, subjects were asked to follow the same diet during the 3 days prior to each experimental protocol. Dietary compliance was monitored by food diaries and urinary sodium excretion.
All protocols involved a 6 h exposure to either IH or sham IH. For the IH+losartan protocol, subjects took 25 mg of losartan on day 1, 50 mg on day 2, and 100 mg on days 3 and 4. Preceding the IH+placebo protocol, all subjects took placebo tablets for four consecutive days in the morning before IH exposure. Venous blood samples were collected from the antecubital vein and placed in EDTA tubes at the beginning and immediately at the end of each experimental protocol (less than 10 min after the cessation of IH) and analysed for measures of oxidative stress and NO activity. The plasma was obtained by centrifugation of the samples at 1000g for 10 min at 4°C. Plasma was separated into aliquots and frozen at –80°C until assays could be performed.
An expanded description of the experimental protocol can be found in the article by Foster et al. (2010).
Experimental techniques
Before and after each IH and sham IH session, arterial oxyhaemoglobin saturation (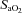 ), MAP, SBP and DBP were measured as the subject rested quietly and comfortably for 10 min while breathing room air. All blood pressure measurements were determined in the right arm as the mean of three values using an automated blood pressure monitoring device (Dinamap; Johnson and Johnson Medical, Inc., New Brunswick, NJ, USA). The IH session consisted of 6 h of continuous cycles of 1 min of isocapnic hypoxia [end-tidal
), MAP, SBP and DBP were measured as the subject rested quietly and comfortably for 10 min while breathing room air. All blood pressure measurements were determined in the right arm as the mean of three values using an automated blood pressure monitoring device (Dinamap; Johnson and Johnson Medical, Inc., New Brunswick, NJ, USA). The IH session consisted of 6 h of continuous cycles of 1 min of isocapnic hypoxia [end-tidal 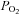 (
(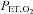 ) nadir = 45.0 mmHg] and 1 min of normoxia (peak
) nadir = 45.0 mmHg] and 1 min of normoxia (peak 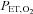 = 88.0 mmHg). The
= 88.0 mmHg). The 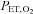 , end-tidal
, end-tidal 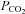 (
(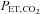 ) and
) and 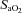 were recorded continuously during all experimental protocols. Intermittent hypoxia was delivered by using a purpose-built normobaric chamber, where the gas composition was maintained to ensure a
were recorded continuously during all experimental protocols. Intermittent hypoxia was delivered by using a purpose-built normobaric chamber, where the gas composition was maintained to ensure a 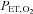 of 45.0 mmHg and a constant
of 45.0 mmHg and a constant 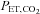 (+1.5 mmHg above rest). Periods of normoxia were constituted by delivering 100% oxygen to the subject's inspirate through a facemask connected to a two-way non-rebreathing valve and a 25-cm-long section of wide-bore tubing. During periods of normoxia, oxygen flowed through the inspired circuit at a rate that provided a
(+1.5 mmHg above rest). Periods of normoxia were constituted by delivering 100% oxygen to the subject's inspirate through a facemask connected to a two-way non-rebreathing valve and a 25-cm-long section of wide-bore tubing. During periods of normoxia, oxygen flowed through the inspired circuit at a rate that provided a 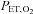 of 88.0 mmHg. The
of 88.0 mmHg. The 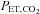 was also controlled during IH by adding 100% CO2 to the subject's inspirate at a flow rate that maintained
was also controlled during IH by adding 100% CO2 to the subject's inspirate at a flow rate that maintained 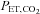 constant. During exposure to IH, respired gas was sampled from a nasal cannula and analysed by a dual oxygen and carbon dioxide analyser (NormocapOxy; Datex-Ohmeda, Louisville, CO, USA) for
constant. During exposure to IH, respired gas was sampled from a nasal cannula and analysed by a dual oxygen and carbon dioxide analyser (NormocapOxy; Datex-Ohmeda, Louisville, CO, USA) for 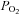 and
and 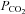 . Sleep was not permitted during any of the experimental protocols.
. Sleep was not permitted during any of the experimental protocols.
Assays
Plasma levels of oxidative stress [i.e. 8-hydroxy-2′-deoxyguanosine (8-OHdG) and uric acid], protein nitrosylation (nitrotyrosine), total antioxidant power (ferric reducing antioxidant power; FRAP), and the end-product of nitric oxide metabolism (NOx) levels were measured at the beginning and end of each experimental day.
Concentrations of plasma 8-OHdG were determined using an ELISA kit from Cell BioLabs (Cell Biolabs, Inc., San Diego, CA, USA). The limits of detection for this assay are 1–200 μg l−1. As DNA is known to be very sensitive to ROS (Loft et al. 2008b), 8-OHdG, an end-product of DNA oxidation, is one of the most reliable markers for oxidative stress in disease processes (Loft et al. 2008a) and was selected as the primary marker of oxidative stress in this study. Data drawn from resting healthy subjects indicated that normal plasma 8-OHdG values ranged between 55.5 and 61.5 ng l−1 (Pialoux et al. 2009b). In addition, Lin et al. (2004) reported a 5.2% intraday coefficient of variation calculated in 12 subjects over 5 days.
Concentrations of plasma uric acid were measured in the plasma using a commercially available kit (Bio-Quant Inc., San Diego, CA, USA). The limits of detection for this assay are 1–100 μmol l−1. Uric acid is produced by purine metabolism during reoxygenation and reflects ROS production through activation of the xanthine oxidase pathway.
Concentrations of plasma nitrotyrosine, an end-product of protein nitrosylation, were measured using an ELISA kit from Cell BioLabs. The limits of detection for this assay are 1–8000 nmol l−1. Since, tyrosine is sensitive to nitrosilation by peroxynitite (ONOO−), the amount of nitrotyrosine reflects the activity of ONOO−. Peroxynitite is formed by the reaction of the superoxide anion (O2·−) with NO.
Plasma FRAP was assessed according to the manual method of Benzie & Strain (1996) and was measured by spectrophotometry at 37°C. The FRAP concentration was calculated using an aqueous solution of known Fe2+ concentration (FeSO4.7H2O) as standard at a wavelength of 593 nm. Ferric reducing antioxidant power is a global indicator of antioxidant power. It was reported that FRAP was negatively correlated with oxidative stress after hypoxic exposure (Pialoux et al. 2009c), highlighting the potential sensitivity of FRAP to oxidative stress.
The end-products of endothelial nitric oxide (NOx), nitrites and nitrates, were measured in the plasma using a commercially available kit (Cayman Chemical Company, Ann Arbor, MI, USA).
Statistical analysis
All data are expressed as means ± SD. Physiological variables (MAP, SBP and DBP, 8-OHdG, uric acid, nitrotyrosine, FRAP and NOx) were analysed using a one-way (treatment effect: IH+losartan, IH+placebo and sham IH) repeated-measures (time: AM and PM; AM and PM refer to the morning and afternoon individual values for each subject) ANOVA, followed by Sidak post hoc test, which is corrected for multiple comparisons. Statistical analyses were performed with analytical software (SPSS v. 15.0; SPSS Inc., Chicago, IL, USA). Differences were considered significant at a value of P < 0.05.
Results
Subjects
Nine of the 10 subjects completed the three experimental protocols and complied with the medication instructions. Consequently, our results are based on data from nine volunteers. Subjects were 29.9 ± 0.6 years old and were free from cardiopulmonary and renal disease, were not obese (body mass index 24.7 ± 0.7 kg m−2) and did not suffer from sleep-disordered breathing (respiratory disturbance index 2.3 ± 0.6 events h−1; mean 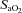 94.3 ± 0.4%), hypertension (MAP 88.9 ± 2.5 mmHg) or diabetes (fasting blood glucose level 5.29 ± 0.18 mmol l−1).
94.3 ± 0.4%), hypertension (MAP 88.9 ± 2.5 mmHg) or diabetes (fasting blood glucose level 5.29 ± 0.18 mmol l−1).
Resting cardiovascular data
Detailed results of the resting cardiovascular data can be found in the article by Foster et al. (2010). Briefly, MAP significantly increased during IH with placebo (+8.0%; P < 0.05) but not with losartan (–0.6%; P > 0.05) or sham IH (+2.2%; P > 0.05).
Oxidative stress
Results of oxidative stress are reported in Table 1.
Table 1.
Oxidative stress and nitric oxide metabolism data
| Sham IH | IH+placebo | IH+losartan | ||||
|---|---|---|---|---|---|---|
| AM | PM | AM | PM | AM | PM | |
| 8-OHdG (μg l−1) | 62.8 ± 8.7 | 56.1 ± 5.4 | 57.2 ± 4.2 | 63.0 ± 4.5*†‡ | 60.4 ± 6.8 | 54.5 ± 4.0* |
| Nitrotyrosine (nmol l−1) | 37.2 ± 11.8 | 30.8 ± 9.7 | 31.7 ± 10.0 | 42.0 ± 13.5*‡ | 37.6 ± 11.9 | 38.6 ± 12.2 |
| Uric acid (mg l−1) | 6.4 ± 0.6 | 6.1 ± 0.4 | 5.4 ± 0.5 | 6.6 ± 0.6* | 5.7 ± 0.4 | 7.1 ± 0.5* |
| NOx (mmol l−1) | 40.7 ± 5.7 | 42.9 ± 5.1 | 45.6 ± 7.3 | 34.9 ± 4.6*‡ | 45.5 ± 4.2 | 39.3 ± 3.7 |
| FRAP (mmol l−1) | 743 ± 108 | 768 ± 133 | 855 ± 163 | 716 ± 155*§ | 844 ± 188 | 782 ± 143* |
Abbreviations: AM and PM refer to the morning and afternoon individual values for each subject; FRAP, ferric reducing antioxidant power; IH, intermittent hypoxia; NOx, end-products of NO metabolism; and 8-OHdG, 8-hydroxy-2′-deoxyguanosine.
P < 0.05 compared with corresponding AM value
P < 0.05 compared with corresponding IH+losartan value
P < 0.05 compared with corresponding sham IH value
P = 0.06 compared with corresponding sham IH value.
Nitrotyrosine and 8-OHdG followed the same profile as MAP with respect to each experimental condition. Nitrotyrosine and 8-OHdG did not display a significant time-of-day effect. However there was a time × treatment effect (P = 0.02 for nitrotyrosine and P = 0.04 for 8-OHdG). There were no significant differences in AM between the three treatments for 8-OHdG and nitrotyrosine.
After IH exposure (i.e. in PM), 8-OHdG was higher in IH+placebo compared with IH+losartan. Moreover, 8-OHdG increased between AM and PM in IH+placebo (+10%; P = 0.04) but not in IH+losartan and sham IH, which tended to decrease (P = 0.06 and P = 0.07, respectively). Likewise, in IH+placebo, nitrotyrosine was lower in PM than in sham IH (P = 0.04). Additionally, nitrotyrosine increased between AM and PM in IH+placebo (+32%; P = 0.02) but not in IH+losartan or sham IH (Fig. 1).
Figure 1. Nitrotyrosine responses to sham intermittent hypoxia (IH), IH+placebo and IH+losartan.
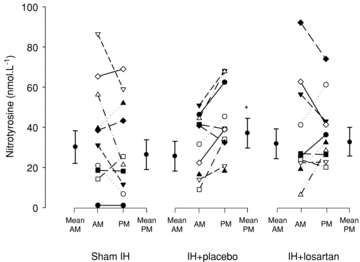
Abbreviations: AM, morning or pre-exposure measurements; and PM, afternoon or postexposure measurements. Mean AM and PM refer to the group means ± SD for the morning and afternoon measurements. AM and PM refer to the morning and afternoon individual values for each subject. *P < 0.05 compared with AM.
Anaysis of variance revealed significant time (AM lower than PM; P = 0.001) and treatment × time (P = 0.003) effects for uric acid.
There was no significant difference in AM between the three treatments for uric acid and FRAP.
Uric acid increased between AM and PM in IH+placebo (+22%; P = 0.004) and IH+losartan (+24%; P = 0.002) but not in sham IH. Finally, time (P = 0.01) and treatment × time (P = 0.01) were significant for FRAP. Ferric reducing antioxidant power decreased between AM and PM in IH+placebo (–16%; P = 0.004) and IH+losartan (–7%; P = 0.03) but not in sham IH. In addition, FRAP had a tendency to be higher in PM in IH+losartan compared with IH+placebo (P = 0.06).
Nitric oxide metabolism
Results for the end-product of nitric oxide metabolism are reported in Table 1. There were significant time (AM greater than PM; P = 0.03) and treatment × time effects (P = 0.047). There were no significant differences in AM between the three treatments for uric acid and FRAP.
In PM, NOx was lower (P = 0.04) in IH+placebo compared with sham IH (Fig. 2). Moreover, NOx decreased between AM and PM in IH+placebo (–23%; P = 0.02) but not in IH+losartan or sham IH. This pattern of response is opposite to the change in nitrotyrosine and 8-OHdG during these protocols.
Figure 2. End-products of nitric oxide metabolism responses to sham IH, IH+placebo and IH+losartan.
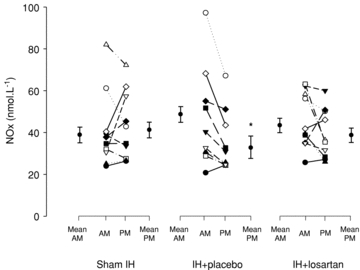
Abbreviations: AM, morning or pre-exposure measurements; NOx, end-product of nitric oxide metabolism; and PM, afternoon or postexposure measurements. Mean AM and PM refer to the group means ± SD for the morning and afternoon measurements. AM and PM refer to the morning and afternoon individual values for each subject. *P < 0.05 compared with AM.
Discussion
This is the first human study examining the effect of AT1 receptor blockade on the acute IH-induced increase in oxidative stress and decrease in nitric oxide metabolism. Our results are consistent with data from previous human (Pialoux et al. 2009a) and animal studies (Peng et al. 2006) showing that IH increases oxidative and nitrosative stresses and decreases nitric oxide metabolism. Furthermore, these data demonstrate that losartan blunted the increase in oxidative stress and peroxynitrite activity and the decrease in nitric oxide metabolism caused by IH. Thus, these results suggest that the renin–angiotensin system could be one of the mechanisms responsible for overproduction of ROS during IH by upregulating the action(s) of Ang II. In addition, the lack of effect of losartan on IH-mediated uric acid production supports the notion that the generation of ROS through the xanthine oxidase pathway is not affected by AT1 receptor blockade, despite previous studies suggesting the contrary (Diez, 2006). Finally, these findings from an experimental human model, which mimics the nocturnal hypoxaemia experienced by patients with OSA, have not been previously reported in humans. Although these data are drawn from a single bout of 6 h of exposure to IH, they do, however, provide further insight into the mechanisms leading to hypertension in OSA.
It has been proposed that the renin–angiotensin system contributes to the pathogenesis of hypertension in OSA because renal nerve denervation, Ang II receptor blockade and suppression of the renin–angiotensin system by a high-salt diet have all prevented the anticipated increase in blood pressure in animal models of chronic IH (Morgan, 2007). More specifically, animal studies support the notion that IH-induced hypertension results, at least partly, from stimulation of the renin–angiotensin system (Fletcher et al. 2002) leading to an increase in Ang II levels (Fletcher et al. 1999). Nevertheless, until now, the signalling pathway by which AT1 receptor blockers reduce the hypertensive response to IH remained uncertain. The increases in 8-OHdG and nitrotyrosine seen with IH+placebo were abolished with IH+losartan, demonstrating that AT1 receptor blockade prevents overproduction of ROS in IH. These results also suggest that upregulation of the renin–angiotensin system, leading to increased Ang II activity, may be one of the mechanisms through which ROS production is increased during intermittent hypoxia.
It has been previously reported that similar doses of losartan to the ones that we used in this study reduced oxidative stress and increased serum levels of NO in patients with essential hypertension (Sosa-Canache et al. 2007). Likewise, generation of superoxide anion (O2·−) was lowered and endothelial function was improved in salt-sensitive hypertensive rats after blockade of AT1 receptors, despite lack of an effect on blood pressure (Wu et al. 2001).
The signalling pathway by which Ang II increases blood pressure still remains speculative (Lacolley et al. 2009). However, in endothelial and smooth muscle cells, Ang II binding to the AT1 receptor subtype activates membrane-bound NADPH oxidase, which is thought to be the most significant source of O2·− in vascular tissue (Griendling et al. 1994; Rajagopalan et al. 1996). Superoxide release has been mechanistically implicated in the pathogenesis of hypertension via the impairment of endothelial function (Touyz & Berry, 2002; Touyz & Schiffrin, 2004). Therefore, in this model, Ang II upregulation secondary to activation of the renin–angiotensin system would induce an increase in O2·− within vascular tissue, resulting in endothelial dysfunction and hypertension.
Previous studies have shown that ROS are generated during periods of IH, mimicking the arterial deoxygenation–reoxygenation profile of OSA patients, and that ROS production increases systemic oxidative stress (Pialoux et al. 2009a) and decreases NO bioavailability (Foster et al. 2009). The changes we found in 8-OHdG, which is an end-product of DNA oxidation, and NOx, an index of NO metabolism, in the IH+placebo protocol are consistent with these previous reports. Moreover, the increase in nitrotyrosine that we observed in the IH+placebo protocol strongly suggests that the oxidative stress caused by IH can blunt NO bioavailability and further contribute to the rise of MAP following IH exposure. This finding is supported by mechanistic evidence whereby superoxide (O2·−) reacts with NO synthesized by eNOS to form the oxidant peroxynitrite, thereby inducing NO inactivation and eNOS uncoupling (Thomas et al. 2008). Finally, upregulation of antioxidant enzymes during IH could lead to an underestimation of ROS overproduction during IH. Indeed, this hypothetical increase of antioxidant enzyme efficiency could explain the small magnitude of change in our oxidative stress marker. However, in a similar protocol (Pialoux et al. 2009a; 6 h of 2 min poikilocapnic hypoxia alternating with 2 min normxia vs. 6 h of 1 min isocapnic hypoxia alternating with 1 min normoxia), we did not find significant changes in glutathione peroxidase and catalase activities after 6 h of IH compared with sham exposure. This strongly suggests that antioxidant enzymes were not altered during IH in the present study.
Interestingly, losartan prevented the concomitant decrease in NOx and the 9 mmHg increase in MAP observed after IH. Collectively, these data support the hypothesis that the Ang II–ROS–NO pathway contributes to the increase in blood pressure during IH. Increases in blood pressure of 4 and 5 mmHg following 4 and 14 days of exposure to IH, respectively, have been previously reported in healthy subjects (Foster et al. 2009; Tamisier et al. 2009). The 9 mmHg increase in blood pressure observed in our study during a single 6 h of IH suggests that this protocol provided a greater stimulus than those previously described (Foster et al. 2009; Tamisier et al. 2009). Indeed, our model prevented the fall in 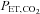 (see Foster et al. 2010, their figure 4, for detailed information) observed with other models of IH, which may explain the difference in the blood pressure response.
(see Foster et al. 2010, their figure 4, for detailed information) observed with other models of IH, which may explain the difference in the blood pressure response.
The absence of an effect of losartan on uric acid release during IH suggests that the renin–angiotensin system does not alter the production of uric acid during IH, contrary to what has been previously suggested (Diez, 2006). Enzymatic production of both superoxide and uric acid from the xanthine oxidase pathway occurs during the cycle of hypoxia–reoxygenation, similar to what occurs during ischaemia–reperfusion (Xu et al. 2004; Yuan et al. 2004) and does not theoretically involve the renin–angiotensin system. These physiological mechanisms are clearly supported by the similar uric acid increases in both IH protocols (i.e. IH+placebo and IH+losartan). If ROS generation can occur independently from pathways other than the renin–angiotensin system, why did both 8-OHdG and nitrotyrosine not increase in our IH+losartan protocol? One possible explanation is that the capacities of the antioxidant systems, here evaluated by FRAP, were sufficient to buffer the ROS overproduced during IH+losartan, whereas they were not sufficient to buffer the greater amount of ROS produced during the IH+placebo protocol. As previously discussed, the blockade of the AT1 receptor eliminates one pathway of ROS production in IH. This hypothesis is supported by a greater decrease in total antioxidant status (measured by FRAP) in the IH+placebo protocol compared with IH+losartan.
Limitations
Our model does have some limitations with respect to the pathogenesis of hypertension in OSA. Indeed, the study was performed by alteration of the environmental gas mixtures in awake young and healthy subjects, in contrast to OSA, which occurs during sleep. The negative intrathoracic pressure, the arousals resulting from apnoea and the associated sleep fragmentation have been shown to increase sympathoexcitation (Kara et al. 2003; Yamauchi & Kimura, 2008), which should cause a further upregulatation of Ang II (Narkiewicz et al. 1998). In addition, 1 day of exposure to IH induces less oxidative stress than longer exposure to IH (Pialoux et al. 2009a). In this context, our model may underestimate the full impact of IH on blood pressure control. However, our approach of using healthy humans exposed to IH allowed us to distinguish the effect of IH per se from other potentially confounding factors (outlined above) associated with OSA.
Finally, it could be also hypothesized that the reduction in blood pressure induced by losartan led to a decrease of ROS generation through a lower shear stress on the vascular wall and subsequent NADPH oxidase activation. Indeed, oscillatory shear stress seems a particularly potent stimulus of superoxide production in arterial hypertension (Harrison et al. 2003; McNally et al. 2003). However, increased shear stress was also shown to upregulate NO metabolism (Noris et al. 1995; Nadaud et al. 1996), which was not found in our study. As subjects in the IH+placebo protocol remained normotensive, it is unlikely that shear stress contributed to production of ROS.
Conclusion
This study demonstrated significantly increased oxidative stress and decreased NO activity following exposure to isocapnic IH in healthy humans. These changes were abolished by blockade of the AT1 receptor with losartan. Our results suggest that the renin–angiotensin system may play a significant role in mediating the increase in oxidative stress and the subsequent decrease in NO that occurs during IH. Furthermore, this improves our understanding of the underlying mechanisms responsible for the development of hypertension in OSA. Further research is required to determine whether these experimental findings apply in patient populations that experience intermittent hypoxia, which may impact on their clinical management and outcomes.
Acknowledgments
The authors would like to thank our dedicated subjects for their participation in this study. Support was provided by the Alberta Heritage Foundation for Medical Research (AHFMR; M.J.P.), the Canadian Institutes of Health Research (CIHR; P.J.H. and M.J.P.) and the Canadian Foundation for Innovation (M.J.P.). V.P. was supported by postdoctoral research awards from the AHFMR, the Heart and Stroke Foundation of Canada (HSFC) and the CIHR Focus-on-Stroke Program. G.E.F. was a CIHR Strategic Training Fellow in TORCH (Tomorrow's Research Cardiovascular Health Professionals) and was supported by research awards from the AHFMR, the HSFC and the Natural Sciences and Research Council of Canada. S.B.A. is an AHFMR Clinical Investigator and CIHR New Investigator. A.E.B. and was supported by a research award from the AHFMR. M.J.P. is an AHFMR Senior Medical Scholar.
Glossary
Abbreviations
- Ang II
angiotensin II
- AT1 receptor
type 1 angiotensin II receptor
- DBP
diastolic blood pressure
- eNOS
endothelial nitric oxide synthase
- FRAP
ferric reducing antioxidant power
- IH
intermittent hypoxia
- MAP
mean arterial pressure
- NOx
end-products of NO metabolism
- O2·−
superoxide anion
- 8-OHdG
8-hydroxy-2′-deoxyguanosine
- ONOO−
peroxynitrite
- OSA
obstructive sleep apnoea
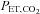
end-tidal
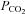
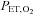
end-tidal
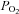
- ROS
reactive oxygen species
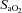
arterial oxyhaemoglobin saturation
- SBP
systolic blood pressure
Author contributions
Study concept: G.E.F., P.J.H. and M.J.P. Study design: V.P., P.J.H., A.E.B., S.B.A., G.E.F. and M.J.P. Acquisition of data: V.P., G.E.F. and A.E.B. Analysis and interpretation of data: V.P., P.J.H., G.E.F., S.B.A., A.E.B. and M.J.P. Drafting of the manuscript: V.P., P.J.H. and M.J.P. Critical revision of the manuscript for important intellectual content: V.P., G.E.F., S.B.A., A.E.B., P.J.H. and M.J.P. Statistical analysis: V.P., G.E.F. and M.J.P. Obtained funding: P.J.H. and M.J.P. Administrative, technical and material support: P.J.H. and M.J.P. Medical cover: P.J.H. Study supervision: P.J.H. and M.J.P. The authors declare no competing financial interests.
Authors' present addresses
V. Pialoux: Centre de Recherche et d'Innovation sur le Sport, Université Claude Bernard Lyon 1, France.
G. E. Foster: School of Human Kinetics, University of British Columbia, Canada.
References
- Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem. 1996;239:70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- Castelao JE, Gago-Dominguez M. Risk factors for cardiovascular disease in women: relationship to lipid peroxidation and oxidative stress. Med Hypotheses. 2008;71:39–44. doi: 10.1016/j.mehy.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Diez J. Review of the molecular pharmacology of Losartan and its possible relevance to stroke prevention in patients with hypertension. Clin Ther. 2006;28:832–848. doi: 10.1016/j.clinthera.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Dohi Y, Ohashi M, Sugiyama M, Takase H, Sato K, Ueda R. Candesartan reduces oxidative stress and inflammation in patients with essential hypertension. Hypertens Res. 2003;26:691–697. doi: 10.1291/hypres.26.691. [DOI] [PubMed] [Google Scholar]
- Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med. 2002;165:934–939. doi: 10.1164/ajrccm.165.7.2104126. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Bao G, Li R. Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension. 1999;34:309–314. doi: 10.1161/01.hyp.34.2.309. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Orolinova N, Bader M. Blood pressure response to chronic episodic hypoxia: the rennin–angiotensin system. J Appl Physiol. 2002;92:627–633. doi: 10.1152/japplphysiol.00152.2001. [DOI] [PubMed] [Google Scholar]
- Fliser D, Buchholz K, Haller H. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation. 2004;110:1103–1107. doi: 10.1161/01.CIR.0000140265.21608.8E. [DOI] [PubMed] [Google Scholar]
- Foster GE, Brugniaux JV, Pialoux V, Duggan CT, Hanly PJ, Ahmed SB, Poulin MJ. Cardiovascular and cerebrovascular responses to acute hypoxia following exposure to intermittent hypoxia in healthy humans. J Physiol. 2009;587:3287–3299. doi: 10.1113/jphysiol.2009.171553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster GE, Hanly PJ, Ahmed SB, Beaudin AE, Pialoux V, Poulin MJ. Intermittent hypoxia increases arterial blood pressure in humans through a rennin–angiotensin system-dependent mechanism. Hypertension. 2010;56:369–377. doi: 10.1161/HYPERTENSIONAHA.110.152108. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- Kara T, Narkiewicz K, Somers VK. Chemoreflexes–physiology and clinical implications. Acta Physiol Scand. 2003;177:377–384. doi: 10.1046/j.1365-201X.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- Kraiczi H, Hedner J, Peker Y, Carlson J. Increased vasoconstrictor sensitivity in obstructive sleep apnea. J Appl Physiol. 2000a;89:493–498. doi: 10.1152/jappl.2000.89.2.493. [DOI] [PubMed] [Google Scholar]
- Kraiczi H, Hedner J, Peker Y, Grote L. Comparison of atenolol, amlodipine, enalapril, hydrochlorothiazide, and losartan for antihypertensive treatment in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2000b;161:1423–1428. doi: 10.1164/ajrccm.161.5.9909024. [DOI] [PubMed] [Google Scholar]
- Lacolley P, Safar ME, Regnault V, Frohlich ED. Angiotensin II, mechanotransduction, and pulsatile arterial hemodynamics in hypertension. Am J Physiol Heart Circ Physiol. 2009;297:H1567–H1575. doi: 10.1152/ajpheart.00622.2009. [DOI] [PubMed] [Google Scholar]
- Lin HS, Jenner AM, Ong CN, Huang SH, Whiteman M, Halliwell B. A high-throughput and sensitive methodology for the quantification of urinary 8-hydroxy-2′-deoxyguanosine: measurement with gas chromatography-mass spectrometry after single solid-phase extraction. Biochem J. 2004;380:541–548. doi: 10.1042/BJ20040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loft S, Hogh DP, Mikkelsen L, Risom L, Forchhammer L, Moller P. Biomarkers of oxidative damage to DNA and repair. Biochem Soc Trans. 2008a;36:1071–1076. doi: 10.1042/BST0361071. [DOI] [PubMed] [Google Scholar]
- Loft S, Moller P, Cooke MS, Rozalski R, Olinski R. Antioxidant vitamins and cancer risk: is oxidative damage to DNA a relevant biomarker? Eur J Nutr. 2008b;47(Suppl 2):19–28. doi: 10.1007/s00394-008-2004-0. [DOI] [PubMed] [Google Scholar]
- McNally JS, Davis ME, Giddens DP, Saha A, Hwang J, Dikalov S, Jo H, Harrison DG. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. 2003;285:H2290–H2297. doi: 10.1152/ajpheart.00515.2003. [DOI] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Morgan BJ. Vascular consequences of intermittent hypoxia. Adv Exp Med Biol. 2007;618:69–84. doi: 10.1007/978-0-387-75434-5_6. [DOI] [PubMed] [Google Scholar]
- Nadaud S, Philippe M, Arnal JF, Michel JB, Soubrier F. Sustained increase in aortic endothelial nitric oxide synthase expression in vivo in a model of chronic high blood flow. Circ Res. 1996;79:857–863. doi: 10.1161/01.res.79.4.857. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Cooley RL, Dyken ME, Somers VK. Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation. 1998;98:772–776. doi: 10.1161/01.cir.98.8.772. [DOI] [PubMed] [Google Scholar]
- Noris M, Morigi M, Donadelli R, Aiello S, Foppolo M, Todeschini M, Orisio S, Remuzzi G, Remuzzi A. Nitric oxide synthesis by cultured endothelial cells is modulated by flow conditions. Circ Res. 1995;76:536–543. doi: 10.1161/01.res.76.4.536. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1α deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pialoux V, Hanly PJ, Foster GE, Brugniaux JV, Beaudin AE, Hartmann SE, Pun M, Duggan CT, Poulin MJ. Effects of exposure to intermittent hypoxia on oxidative stress and acute hypoxic ventilatory response in humans. Am J Respir Crit Care Med. 2009a;180:1002–1009. doi: 10.1164/rccm.200905-0671OC. [DOI] [PubMed] [Google Scholar]
- Pialoux V, Mounier R, Brown AD, Steinback CD, Rawling JM, Poulin MJ. Relationship between oxidative stress and HIF-1α mRNA during sustained hypoxia in humans. Free Radic Biol Med. 2009b;46:321–326. doi: 10.1016/j.freeradbiomed.2008.10.047. [DOI] [PubMed] [Google Scholar]
- Pialoux V, Mounier R, Rock E, Mazur A, Schmitt L, Richalet JP, Robach P, Coudert J, Fellmann N. Effects of acute hypoxic exposure on prooxidant/antioxidant balance in elite endurance athletes. Int J Sports Med. 2009c;30:87–93. doi: 10.1055/s-0028-1103284. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa-Canache B, Hernandez-Hernandez R, Armas-Padilla MC, Armas-Hernandez MJ, Cammarata-Segura R, Pacheco B, Guerrero J, Israili ZH, Valasco M. Effect of losartan therapy on endothelial function in hypertensive patients. Am J Ther. 2007;14:166–171. doi: 10.1097/01.pap.0000249919.44604.e1. [DOI] [PubMed] [Google Scholar]
- Tamisier R, Gilmartin GS, Launois SH, Pepin JL, Nespoulet H, Thomas R, Levy P, Weiss JW. A new model of chronic intermittent hypoxia in humans: effect on ventilation, sleep, and blood pressure. J Appl Physiol. 2009;107:17–24. doi: 10.1152/japplphysiol.91165.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. [DOI] [PubMed] [Google Scholar]
- Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2008;10:1713–1765. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Berry C. Recent advances in angiotensin II signaling. Braz J Med Biol Res. 2002;35:1001–1015. doi: 10.1590/s0100-879x2002000900001. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. 2004;122:339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- Wu R, Millette E, Wu L, de Champlain J. Enhanced superoxide anion formation in vascular tissues from spontaneously hypertensive and desoxycorticosterone acetate-salt hypertensive rats. J Hypertens. 2001;19:741–748. doi: 10.1097/00004872-200104000-00011. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D, Liu R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Yamauchi M, Kimura H. Oxidative stress in obstructive sleep apnea: putative pathways to the cardiovascular complications. Antioxid Redox Signal. 2008;10:755–768. doi: 10.1089/ars.2007.1946. [DOI] [PubMed] [Google Scholar]
- Yuan G, Adhikary G, McCormick AA, Holcroft JJ, Kumar GK, Prabhakar NR. Role of oxidative stress in intermittent hypoxia-induced immediate early gene activation in rat PC12 cells. J Physiol. 2004;557:773–783. doi: 10.1113/jphysiol.2003.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]