

Abstract
Sporulation in the filamentous bacteria Streptomyces coelicolor is a tightly regulated process involving aerial hyphae growth, chromosome segregation, septation and spore maturation. Genetic studies have identified numerous genes that regulate sporulation, including WhiA and the sigma factor WhiG. WhiA, which has been postulated to be a transcriptional regulator, contains two regions typically associated with DNA binding: an N-terminal domain similar to LAGLIDADG homing endonucleases, and a C-terminal helix-turn-helix domain. We characterized several in vitro activities displayed by WhiA. It binds at least two sporulation-specific promoters: its own and that of parABp2. DNA binding is primarily driven by its HTH domain, but requires full-length protein for maximum affinity. WhiA transcription is stimulated by WhiG, while the WhiA protein binds directly to WhiG (leading to inhibition of WhiG-dependent transcription). These separate activities, which resemble a possible feedback loop, may help coordinate the closely timed cessation of aerial growth and subsequent spore formation.
S coelicolor is a filamentous soil-dwelling bacteria that initiates a sporulation program in response to certain environmental cues1. This process of differentiation begins with the formation of aerial hyphae that sprout from the vegetative mycelium, which then undergo multiple rounds of DNA replication in the absence of cell division to generate multigenomic syncitia (recently reviewed in2). Subsequently a coordinated process that includes the cessation of hyphal growth, chromosome segregation and septum formation results in the formation of pre-spore compartments containing individual bacterial chromosomes. A key point in this pathway appears to be the tight coupling of growth cessation with subsequent segregation and compartmentalization of individual bacterial chromosomes into early spore precursors. Later maturation events, including thickening of the cell wall, rounding of the spore's shape, and the accumulation of the WhiE polyketide pigment, culminates in long chains of robust, desiccation-resistant spores.
A variety of genetic screens have identified genes that are required for this tightly choreographed differentiation process (Figure 1). Those required for the initial formation of aerial hyphae are termed ‘Bld' (corresponding to bld mutants that display a ‘bald' phenotype under spore-inducing environmental conditions)3,4. Genes required for the subsequent generation of spores are termed ‘Whi' (corresponding to whi mutants that remain white, rather than accumulating the normal grey color associated with production of polyketide pigment)5,6. whi mutants can be further classified into early (WhiA, -B, -G, -H and –I) and late (WhiD, -E) genes, depending on whether mutations result in the complete inability to septate or only an inability to form mature spores1,7. Several additional genes that function during vegetative growth also play important roles in spore formation, including ParA and ParB (an ATPase and a DNA binding protein required for proper chromosome segregation)8,9, FtsZ (a bacterial tubulin homologue required for septation)10,11,12 and several spore-specific surface proteins13.
Figure 1. Sporulation regulatory network in Streptomyces coelicolor.
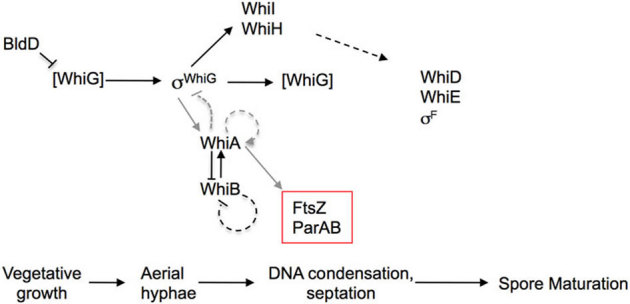
Previous studies described in the introduction have indicated that the sigma factor whiG (σwhiG), which orchestrates the transition from aerial hyphal growth to a sporulation-specific program, is expressed in all stages of growth but becomes active only during a short window early in the sporulation process. “σwhiG” indicates active WhiG; inactive WhiG is in brackets. The transcriptional regulator BldD is required to limit WhiG activity prior to sporulation, although the exact mechanism is not understood. Once active, WhiG directs the transcription of at least two sporulation factors, WhiI and WhiH, which themselves do not become active until later in sporulation. WhiA and the Fe-S cluster protein WhiB mutually regulate each other's expression, and WhiA is also required in vivo to activate its own sporulation-specific transcription. Two other factors—the ParAB and FtsZ genes—are required for hyphal chromosome segregation and septation, respectively. WhiA has been shown in vivo to be required for the sporulation-specific expression of both ParAB and FtsZ. Other sporulation factors required for the subsequent spore maturation include WhiD, WhiE and σF. As described in this paper and indicated with grey lines in the figure, WhiA physically binds to its own promoter and to the Parp2 promoter (i.e. sporulation-specific), consistent with a role in gene activation, and also binds to WhiG. WhiG activates expression of WhiA. A model consistent with those findings is that when WhiA accumulates, it binds to and inhibits WhiG activity, thereby forming a feedback loop that contributes to the inactivation of WhiG and itself. Other factors may also be required to fully limit WhiG activity.
WhiG is a bacterial sigma factor (also termed σWhiG) that directs the transition to a sporulation-specific mode in aerial hyphae7,14. It is expressed throughout all stages of vegetative growth and differentiation15, but undergoes a burst of transcription during the onset of aerial hyphal formation16. WhiG activates transcription of least two genes—the autoregulatory ‘early' Whi-genes WhiI and WhiH16,17. However, there is a significant lag between the burst of WhiG transcription and subsequent expression of WhiH16, suggesting that WhiG is regulated post-transcriptionally. Several groups have speculated that an anti-sigma factor may be involved in inhibiting WhiG activity until the proper window during sporulation7,15,16,18. Consistent with this possibility, WhiG is a member of the family of sigma factors that regulate flagellar formation in Salmonella, including FliA (which shares 41% sequence identity to WhiG); FliA is negatively regulated by an anti-sigma factor encoded by the FlgM gene19. Constraining WhiG activity to the proper time period during sporulation is clearly critical, because overexpression of WhiG results in hypersporulation20.
The WhiA gene contains both a low-level constitutive upstream promoter and a second promoter that is strongly transcribed during growth of the aerial mycelium21. Other sporulation factors, including WhiB, ParAB and FtsZ also contain multiple promoters that include a sporulation-specific one8,9,22,23. WhiA and WhiB influence each others expression by an unknown mechanism8,23, and WhiA is required for its own sporulation-specific transcription21. whiA and whiB mutants share nearly identical phenotypes—tightly coiled, abnormally long aerial hyphae lacking sporulation septae or detectable FtsZ7,14,21,24. Thus, the activity of both proteins appears to be required specifically for cessation of aerial hyphae growth and perhaps subsequent chromosome segregation and/or septation. Ainsa et al. utilized a heterologous in vitro transcription assay to test whether purified WhiG protein could directly promote WhiA transcription21. Those results indicated that WhiA expression was not dependent on WhiG.
Bioinformatics and structural studies have demonstrated that the WhiA protein is comprised of two separate structural regions (an N-terminal domain containing two LAGLIDADG motifs and a C--turn-helix domain) that are each traditionally associated with highly specific DNA recognition and binding25,26. The former domain is associated predominantly in eukaryotic and archael homing endonucleases (invasive genes that are typically embedded within self-splicing, mobile introns or inteins)27. While the WhiA proteins do not possess active sites or DNA cleavage activities that are associated with homing endonucleases, their clear evolutionary relationship with these mobile genes, and their broad distribution throughout gram positive bacteria provide intriguing possibilities for the evolutionary origins of the core of the Whi gene family.
Given (1) the central role of WhiA in sporulation, (2) its ubiquitous distribution across a host range that greatly surpasses the spore-forming Streptomycetes, and (3) the recent determination of its three-dimensional structure, a biochemical examination of the molecular activities of this protein seemed appropriate. Here we describe several in vitro DNA- and protein-binding and transcriptional activities of purified WhiA from Streptomyces coelicolor that are consistent with WhiA coordinating cessation of aerial hyphal growth with later sporulation events, both by confining WhiG activity to a short window at the onset of sporulation and presumably acting as a general transcription factor in regulating genes required for subsequent steps.
Results
Promoter Binding
Because WhiA is a putative transcriptional regulator and contains two protein domains that are widely utilized for DNA recognition (an N-terminal domain present in the LAGLIDADG class of homing endonucleases and a C-terminal HTH domain), we tested the ability of purified WhiA protein to bind several candidate promoter sequences in vitro using an electrophoretic mobility shift assay (EMSA, see materials and methods). We expressed full-length WhiA as a soluble protein in E. coli containing an N-terminal, 6XHis affinity tag that was removed during purification (Supplementary Figure S1 and Materials and Methods). On the final step of purification--size exclusion chromatography--the protein eluted as a single monodisperse peak with an elution time consistent with a monomer (data not shown). Further analyses using dynamic light scattering and small angle X-ray scattering (SAXS) also demonstrated that full-length WhiA is a monomer in solution (data not shown). For the gel-shift assays we selected candidate promoters from genes whose sporulation-specific expression had been shown to be significantly decreased in ΔWhiA strains, including WhiA itself21 and the Parp2 promoter, which directs the sporulation-specific expression of the ParAB genes8,9. We also tested two promoter sequences in which there was no prior published evidence of WhiA dependence: the WhiI promoter (characterized in17); and the Parp1 promoter, which lies downstream of the Parp2 promoter but is not up-regulated in a sporulation-specific manner8. The Parp2 promoter is identical to the Parp1 promoter but contains an additional 185 basepairs (see Supplemental Figure S2 for a list and summary of promoter regions).
WhiA bound its own promoter and the ParP2, but not the ParP1 or WhiI promoters (Figure 2a; Supplementary Figure S3). Based on the averaging of numerous independent experiments, we estimate the Kd of WhiA for its own promoter to be approximately 300 nM (Supplementary Figure S3). The affinity for the WhiA promoter was at least 2 to 3 fold tighter than for ParP2. In binding experiments with the WhiA promoter, several discrete bands of lower mobility appeared with increasing concentrations of WhiA protein, perhaps indicative cooperative binding (Figure 2A, lanes 1–5). A single shifted band was consistently observed in binding reactions with the ParP2 promoter, but not the multiple bands as seen with the WhiA promoter. The observed binding of the WhiA protein to the Parp2 but not the Parp1 promoter region is consistent with in vivo expression analyses, where the sporulation-specific expression of the ParAB genes is driven by the Parp2 promoter in a WhiA-dependent manner8. These in vitro results of WhiA binding provide a biochemical correlation to previous in vivo expression studies showing that WhiA was required for sporulation-specific expression. We note that the affinity of WhiA for these promoters is low, perhaps indicating that WhiA collaborates with other factor(s) in regulating transcription.
Figure 2. WhiA binds to its own promoter and the Parp2 sporulation-specific promoter.
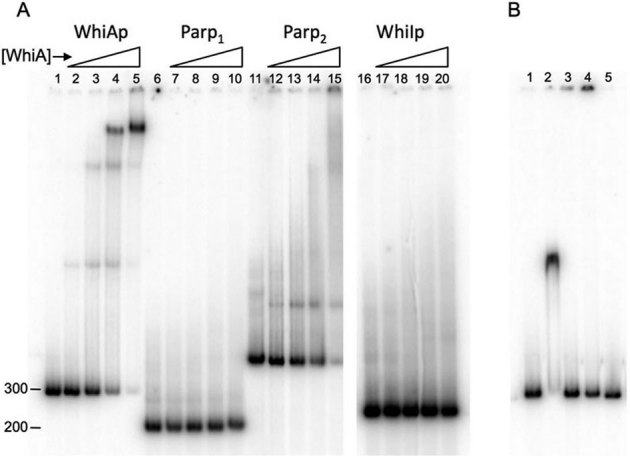
(A) EMSA assays with full-length WhiA protein.32P-labeled DNA sequences corresponding to the WhiA promoter (basepairs −154 to + 86 relative to the transcriptional start as defined by21, lanes 1–5), the Parp1 promoter (lanes 6–10), the Parp2 promoter (lanes 11–15) and the WhiI promoter (lanes 16–20) were incubated with 0, 44, 133, 400 and 1200 nM WhiA protein for 15 minutes at room temperature, separated by non-denaturing polyacrylamide gel electrophoresis and visualized by a phosphorimager. All reactions were run on the same gel; the WhiI promoter lanes (16–20) were cropped and placed adjacent to the other reactions. (B) Gel-shift assay in which the WhiA promoter was incubated with 500 nM or 2 µM h6-WhiA221–328 (HTH domain, lanes 1 and 2) or h6-WhiA1–216 (LAGLIDADG domain, lanes 3 and 4), respectively. Lane 5 is the promoter by itself.
We next tested if either isolated structural domain of WhiA was able to bind the WhiA promoter by itself. For this purpose we purified N-terminal His-tagged versions of the LAGLIDADG domain of WhiA (amino acids 1–216) and the HTH domain (amino acids 221–328; Supplementary Figure S1). Both islated domains were easily overexpressed and purified, and both were well-behaved in solution at high concentrations. The HTH domain, but not the LAGLIDADG domain, bound the WhiA promoter (Figure 2b). The affinity of the HTH domain was significantly lower than full-length WhiA—a gel-shift was observed at 2 µM but not 500 nM WhiA protein--suggesting that while the HTH domain may play a dominant role in DNA binding, the full-length protein is required for maximum affinity (although we cannot formally exclude the possibility that the isolated HTH domain binds more weakly simply due to poor folding behavior). As well, in the binding assay with the HTH domain only a single discrete shifted band is observed, as opposed to the multiple shifted bands can clearly be visualized with full-length WhiA.
We then employed several independent experimental strategies to more precisely define the binding region of WhiA to its own promoter. First, we performed gel-shift competition assays in which the labeled WhiA promoter used in Figure 2 was incubated with a 10-fold excess of a series of 40 basepair, double-stranded unlabeled oligonucleotides corresponding to individual regions of the WhiA promoter (Figure 3a). The competing oligonucleotides were designed to overlap their neighbor by 10 basepairs so that the entire promoter region could be tested for competition. We reasoned that 40 basepairs would span a sufficient length to encompass the WhiA binding site if both domains of WhiA were involved in binding to DNA. Using this approach, an oligonucleotide corresponding to −4 to −44 basepairs relative to the WhiA transcription start site significantly competed for binding with the full-length probe. The neighboring 5′ oligonucleotide (basepairs −34 to +6) also competed, albeit to a lesser extent.
Figure 3. WhiA binding specificity across the WhiA promoter.
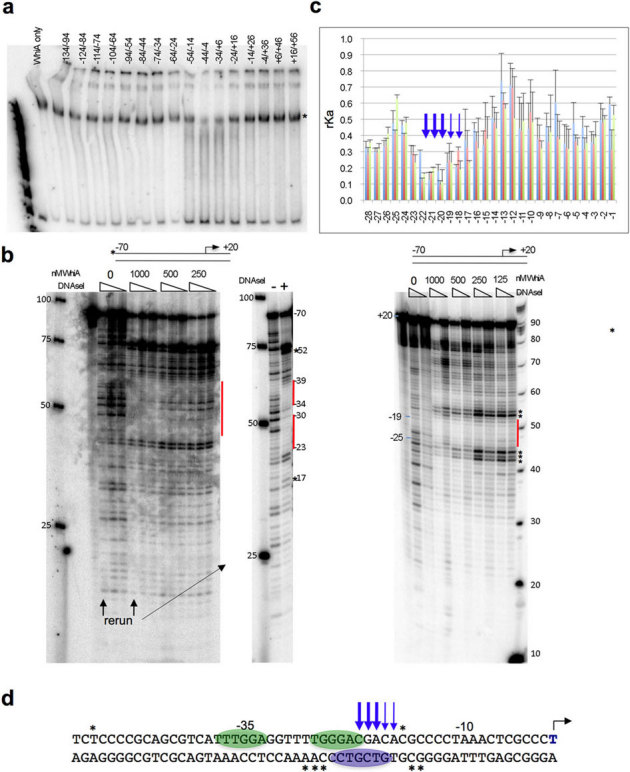
(a) WhiA(FL) binds to its own promoter in a region between −4 and −44 relative to the transcriptional start. 40mer double-stranded oligonucleotides corresponding to the WhiA promoter (overlapping each neighbor by 10 basepairs) were used in competition gel-shift assays at a 10-fold higher concentration than the 32P-labeled WhiA promoter in Figure 2. The “*” indicates the shifted band seen with no competitor (left lane). Numbers above lanes indicate the region of the competing oligonucleotide relative to the transcription start. (b) DNA footprinting assay with the WhiA promoter. A duplex oligo corresponding to the WhiA promoter (−70/+20 relative to the transcriptional start) was labeled on the plus strand (left) or the minus strand (right) and incubated with increasing concentrations of WhiA. For the plus-stranded reaction three dilutions of DNAse I were used (1∶100, 1∶300 and 1∶1000); for the minus-stranded reaction two concentrations were used (1∶300 and 1∶1000). For the plus-strand, reactions with no WhiA or 1000 nM WhiA were rerun for clarity with the highest concentration of DNAse I. For the plus-strand reactions, a 25 basepair standard ladder is indicated on the left; on the minus-stranded reactions a 10 basepair ladder is on the right. Bars indicate regions of protection; hypersensitive sites are indicated by “*”. (c) The specificity of WhiA DNA recognition was tested across basepair positions −44 to −1 using a fluorescent competition binding assay. WhiA displays sequence specificity at positions −22, −21 and −20, and to a lesser extent −19 and −18 (arrows). The relative affinity (rKa) for each competitor was calculated as described in Methods; a reduced value indicates a lower binding affinity and preference for the wild-type basepair. Each position was run in triplicate (error bars are standard deviation). Individual colors correspond to the base at each position: A = blue, T = red, G = green, C = purple. (d) Summary of WhiA specificity for the WhiA promoter. Ovals indicate regions of protection identified by DNAse I footprinting; asterisks indicate hypersensitive regions in the DNaseI assay; arrows indicate basepair positions identified by fluorescence competition as being essential for recognition by WhiA.
Next, we examined the specificity of WhiA binding to its promoter by DNAse I footprint analyses. We used a double-stranded oligonucleotide probe corresponding to basepairs −70 to +20 relative to the transcriptional start site (defined by21; Figure 3b, materials and methods). With increasing concentration of WhiA several regions were protected from DNAse I digestion, including −33 to −39 and −22 to −27 on the plus strand; and −19 to −25 on the minus strand (Figure 3B, 3D). Additionally, several sites were hypersensitive to DNAse I digestion, including −52 and −17 on the plus strand; and −15, −16, −26, −27 and −28 on the minus strand (Figure 3B and 3D).
Finally, we assayed the specificity of the DNA binding interaction exhibited by WhiA against its own promoter. Specificity was tested at each individual basepair position from basepair −44 to −1, using a high-throughput, parallel assay previously developed in our laboratory to describe the binding specificity of homing endonucleases (see Materials and Methods and references28,29). Using this assay, we determined that the identity of five basepairs in particular (positions −22, −21, and −20, and to a lesser extent −19 and −18) are most important for WhiA binding affinity (Figure 3c and 3d). The deleterious effect of any basepair substitution on binding affinity is relatively uniform at four out of five of these positions (−18 through −21). At the fifth position (−22) the protein tolerates either the wild-type G:C basepair or a substitution to A:T. The length of the DNA sequence region that is specifically recognized by WhiA appears to correspond to the typical profile of sequence-specific recognition displayed by a helix-turn-helix DNA binding domain. The sequence of the most strongly recognized DNA region bound by WhiA was non-palindromic, consistent with the apparent monomeric structure of the WhiA protein.
A comparison of the sequence in the WhiA promoter region that is bound by WhiA with the corresponding region of Parp2 that was also sufficient for WhiA binding does not clearly indicate an obvious consensus binding site motif. This is not entirely unexpected, given that correct identification of consensus binding sites is often difficult, even when a large number of DNA binding sites have been identified that are recognized by a single transcription factor, and particularly if a DNA binding protein displays relaxed or nonuniform fidelity at individual positions in its DNA target site recognition profile30,31. It is also possible that DNA recognition by WhiA is strongly dependent on DNA bending and corresponding indirect readout of DNA structure, which might also result in a less obvious DNA target site consensus sequence.
Transcriptional regulation
Having established that WhiA binds at least two sporulation-specific promoter regions in a manner consistent with in vivo results, we next tested the function of WhiA using a heterologous in vitro transcription assay. For this assay we used purified E. coli core RNA polymerase and his-tagged WhiG (H6-WhiG) that we purified as a soluble protein from E. coli (Materials and methods; Supplementary Figure 1). We first tested the validity of the assay using the WhiI promoter, which was previously shown to be transcribed in a WhiG-dependent manner17. When H6-WhiG was incubated with E. coli RNA polymerase and the WhiI promoter, we observed a run-off transcript of the predicted size (Figure 4a), verifying that that our H6-WhiG was active as a sigma factor in this assay.
Figure 4. σWhiG activates transcription from the WhiA promoter and is inhibited by the WhiA protein.
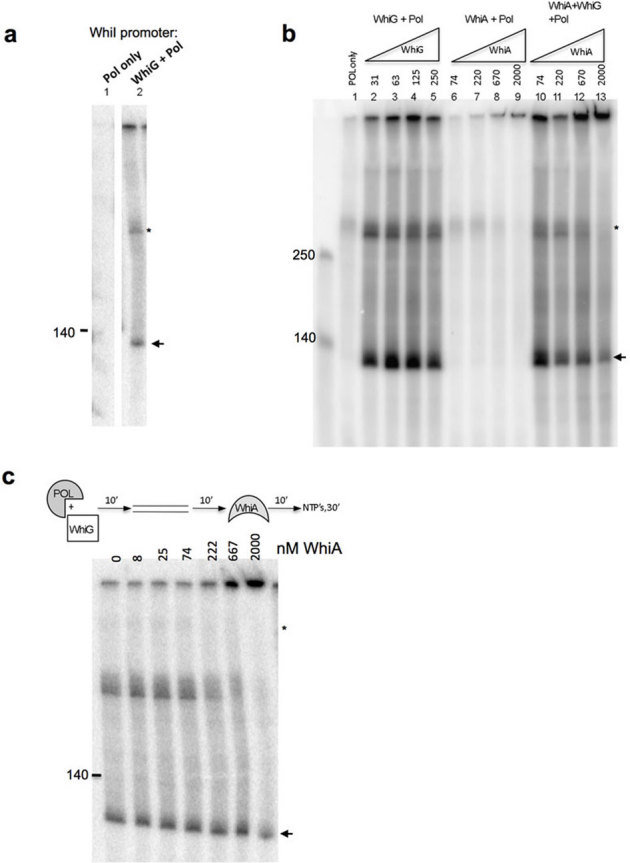
(a) WhiG directs transcription from the WhiI promoter. 1 unit of E. coli RNA polymerase was pre-incubated by itself (lane 1) or with H6-WhiG (lane 2) at room temperature for 10 minutes, followed by a 10 minute incubation with 0.3 pmol of PCR-amplified WhiI promoter (expected transcript size is 112 basepairs). Transcription was initiated by adding a ribonucleotide mix containing 32P-α-CTP and incubated for 30 minutes at 37°C. An arrow indicates the transcript of the expected size. The upper band (indicated by a smaller “*”) runs at the same size as the input oligo and is likely end-labeled by RNA polymerase. (b) Lanes 1–5: WhiG directs transcription from the WhiA promoter. Reactions were carried out as described in A with 0.3 pmol of PCR-amplified WhiA promoter (−154 to + 86, expected transcript size is 122 basepairs) and the indicated concentrations of H6-WhiG (in nM). Lanes 6–9: WhiA does not activate transcription from its own promoter. WhiA was incubated at the indicated concentrations with it's own promoter as described for lanes 1–5. Note that H6-WhiG was not present in these reactions. Lanes 10–13: WhiA inhibits WhiG-directed transcription in a dose-dependent manner. H6-WhiG (63 nM, same concentration as in lane 3), E. coli RNA Pol (1 U) and WhiA (indicated concentrations) were preincubated 10′ at room temperature, followed by a 10′ incubation with the WhiA promoter prior to adding ribonucleotides to start the reaction as described in lanes 2–5. (c) Order of addition influences the effect of WhiA on WhiG-directed transcription. H6-WhiG (50 nM) and RNA Polymerase (1 U) were preincubated for 10′, followed by the addition of the WhiA promoter template for 10′, followed by the addition of the indicated concentration of WhiA for 10′, followed by ribonucleotides to start the reaction.
We next tested whether WhiG could promote transcription from the WhiA promoter. Although the WhiA promoter closely resembles other WhiG-dependent promoters, including WhiI and WhiH, WhiG was previously reported not to direct transcription from the WhiA promoter in a run-off transcription assay21. However, we found that WhiG did indeed direct transcription from the WhiA promoter (Figure 4b, lanes 2–5). A possible explanation for this discrepancy is that we purified WhiG as a soluble protein from E. coli under native buffer conditions, whereas in the prior studies WhiG was expressed in E. coli from insoluble inclusion bodies and was subsequently denatured and then refolded. Proteins purified in such a manner often fail to regain full activity. That same paper also reported that WhiA transcription was detected in a whiG deletion mutant using an S1 nuclease assay21. However, it is possible that those transcripts could have arisen from transcription from WhiA's constitutive, whiG-independent promoter, rather than its sporulation-specific promoter. Regardless, the in vitro biochemical results in this current study (Figure 4b) clearly demonstrate WhiG-dependent transcription from WhiA's sporulation-specific promoter.
Given that WhiA binds to its own promoter, and is required in vivo for transcription from its promoter21, we next tested whether WhiA could itself function as a sigma factor and direct its own transcription in vitro. This possibility has previously been postulated32. However, WhiA clearly did not direct transcription from its own promoter (Figure 4b, lanes 6–9). We also tested WhiA for sigma factor activity using the Parp2 promoter and again did not observe transcription activity (data not shown). It should be noted that although the E. coli RNA polymerase worked well for H6-WhiG-directed transcription in our assays, and has been used successfully with many other cross-species sigma factors in the literature, these results do not rule out the possibility that WhiA indeed functions as a sigma factor but has a strict requirement for the S. coelicolor RNA polymerase.
We next tested whether WhiA affected WhiG-directed transcription from the WhiA promoter. WhiA, H6-WhiG and RNA polymerase were pre-incubated with each other, followed by an incubation with the PCR-amplified promoter template prior to addition of ribonucleotides to initiate the reaction. Under this reaction scheme, WhiA inhibited transcription in a dose-dependent manner (Figure 4B, lanes 10–13). The inhibition of transcription by WhiA was not complete in this assay--in three different experiments where H6-WhiG was used at a concentration of 63 nM and WhiA at 200 nM, the average inhibition was 45% (with a standard deviation of 14%).
We then tested whether order of addition of WhiG and WhiA in the experiment above influenced inhibition of transcription by first preincubating H6-WhiG and RNA polymerase, followed by addition of the DNA template, then WhiA, then ribonucleotides to initiate the reaction (Figure 4c). Under this reaction scheme we observed no inhibitory effect by WhiA. Therefore, WhiA appears to inhibit transcription by blocking the association between WhiG and RNA Polymerase.
WhiA may inhibit WhiG-directed transcription by binding directly to either WhiG or to RNA Polymerase. To test this we utilized a pulldown assay in which untagged WhiA was incubated with His-tagged WhiG, pulled down by Ni-NTA resin, resolved by SDS PAGE and visualized by silver staining (materials and methods). Under these conditions WhiA and WhiG clearly interacted physically (Figure 5). WhiA and with E. coli RNA polymerase did not react in a parallel assay in which His-tagged-WhiA was pulled down with Ni-NTA beads (data not shown). As discussed above, it remains a possibility that WhiA interacts with the S. coelicolor RNA Polymerase but not the E. coli RNA polymerase used in these assays; or that WhiA and the E. coli polymerase interact with a low affinity not detectable in this assay. In summary, the binding results are consistent with WhiA-mediated inhibition of transcription directed by σWhiG.
Figure 5. WhiA binds directly to h6-WhiG.
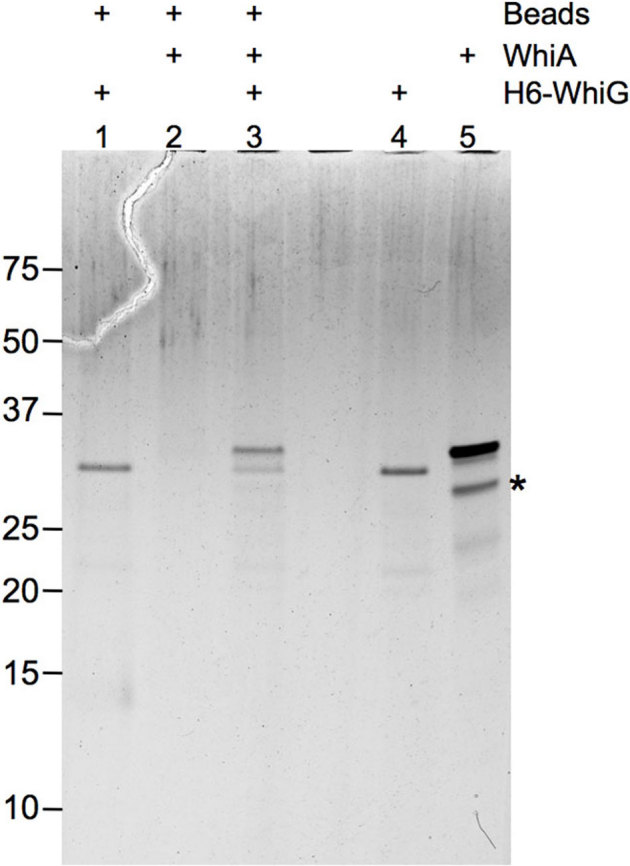
2 µg of untagged WhiA was incubated with 2 µcro;g h6-WhiG, purified with Ni-NTA resin, washed 5 times and separated on SDS PAGE followed by silver staining. Lanes 4 and 5 contain 1/20 the amount of protein used in the pull-down reactions. An “*” indicates a breakdown product of WhiA that corresponds to the LAGLIDADG domain.
Discussion
Sporulation in S. coelicolor is tightly regulated, both temporally and spatially, and clearly involves one or more discrete points in the organism's lifecycle where the progression through the differentiation program is carefully coordinated. One particularly important transition is the cessation of growth and of DNA replication in the aerial hyphae, which is closely followed by segregation of individual bacterial chromosomes and septation. Our in vitro characterization of WhiA, coupled with published in vivo results, suggests that it plays an important role in coordinating these events.
A notable feature of the early Whi genes (-A, -B-, -H, -I, -G) is that they encode regulatory factors, and it is critical that they are active only during the window in which they are required for the sporulation program. All of these factors except WhiG are expressed in a sporulation-specific manner, but other regulatory mechanisms are likely to be in place to also ensure their proper timing of activation. This appears to be the case for WhiH and WhiI—with both genes there is a delay between when they are transcribed and when they become active16,17. WhiG transcript levels are relatively constant throughout differentiation, with the exception of a short burst of WhiG transcription around the time of aerial hyphal formation16, and WhiG transcripts are not restricted to the aerial hyphae, as is found for many other sporulation factors. These features of WhiG have led others to postulate that WhiG activity is likely to be regulated post-transcriptionally, perhaps by an anti-sigma factor7,15,16. Two features of WhiA and WhiG that we have demonstrated in vitro—the ability of WhiG to direct transcription of WhiA, and the ability of the WhiA protein to inhibit WhiG-directed transcription—are sufficient to create a feedback loop to confine WhiG activation to a short window during the transition from hyphal growth to a sporulation program (Figure 1). It is also possible that WhiA plays a role in constraining WhiG activity during vegetative growth since is constitutively expressed (albeit at a much lower level than during sporulation). While we consistently observed WhiA-mediated inhibition of WhiG in the in vitro transcription assays, the inhibition was not complete. Given the central role that WhiG plays in directing the sporulation program, regulation of its activity is likely to be complex and require multiple regulatory factors. One such factor is the transcriptional regulator BldD, which binds to the WhiG promoter and appears to repress its expression prior to sporulation onset33.
While WhiA accumulation might suppress the whiG-dependent “early” steps in sporulation, an additional activity that WhiA displays—binding to several sporulation specific promoters--could be quite relevant to subsequent events in sporulation. We found that in addition to binding its own promoter, WhiA bound the ParABp2 promoter, which directs the sporulation-specific (and WhiA-dependent) expression of ParA and ParB genes8,9. ParA and B are required for proper chromosome segregation during sporulation and are among the earliest markers of sporulation. In vivo WhiA was also required for the sporulation-specific expression of FtsZ22, which is essential for proper septation. Therefore, WhiA might be involved in regulating the sporulation-specific expression of (at least) three critical sporulation factors: ParA, ParB and FtsZ. An apparent paradox from our results—i.e. that WhiA is required both of activation of its own expression, as shown by characterization of a whiA mutant21, but also appears to turn it off by binding and inhibiting WhiG—might indicate a regulatory mechanism that would confine WhiA's activity to a window during sporulation. In this model, as WhiA protein is produced it could carry out its function as a putative transcriptional activtator, but once it accumulated to a certain level would then inhibit WhiG and turn off its own expression.
Although WhiA does not share homology with previously characterized anti-sigma factors, its ability to bind WhiG and inhibits its transcription are consistent with structural and mechanistic features of other bacterial anti-sigma factors (reviewed in34). It is formally possible that WhiA might inhibit the initiation of transcription by other sigma factors, but that issue has not yet been examined experimentally. WhiA is thought to act specificially in S. coelicolor to regulate sporulation, so at least in that organism it appears more likely to act primarily in concert with WhiG.
Anti-sigma factors are notable for often containing modular structures with multiple folded domains, and for their ability to form simultaneous interactions to several distinct surfaces of the relevant sigma factor, in order to prevent each of those regions from interacting with the core RNA polymerase. To the best of our knowledge, there is no direct precedent for a single genetic regulator acting simultaneously as an anti-sigma factor and as a general transcription factor in bacteria, although such an ability presents an attractive scenario wherein such a single protein could coordinate a complex developmental switch at a decision point within a differentiation pathway. One recent set of studies has demonstrated the ability of a single bacterial regulator (the HipB persistence factor) to directly inhibit a cell growth regulator (HipA) while simultaneously acting as a transcription factor via binding to the HipBA operator35.
Methods
Constructs for protein expression and in vitro transcription assays
The sequence of S. coelicolor WhiA (WhiASc) was codon-optimized for expression in E. coli (Blue Heron) and subcloned into the pET15 vector that was modified to contain a thrombin-cleavable, N-terminal 6Xhis tag (pET15(HE)-WhiA)). WhiA truncation constructs (amino acids 221–328 and 1–216) were subcloned by PCR into a pET22b+vector-modified to also contain a cleavable N-terminal 6Xhis tag. WhiG was amplified from genomic S. violaceoruber DNA (ATCC) (having the same sequence as annotated for S. coelicolor A3(2)) and cloned into pET22b+ with an N-terminal H6-tag.
Promoter sequences used for EMSA and in vitro transcription assays were PCR amplified from genomic DNA (Supplemental Figure S2) and cloned into the BamHI site of pGEM3T.
Protein Production
Codon-optimized WhiA constructs were expressed in BL21(DE3) cells, and WhiG was expressed in BL21 (DE3) RIL cells. For induction, a 1 mL starter culture was grown at 37°C overnight in LB media supplemented with 100 µg/mL ampicillin, 0.5% glucose and 1 mM MgCl2. The next morning this culture was diluted into ∼200 mL of prewarmed media with ampicillin, grown 5–6 hours, then 25 mL of this culture was diluted into six flasks containing 1.5 L of prewarmed media with ampicillin. When the OD600 reached 0.6 the cultures were chilled on ice for 20 minutes, followed by induction with 0.5 mM IPTG (final concentration). The cultures were grown overnight at 16°C, pelleted by centrifugation and stored at −20°C. Pellets were thawed and lysed by sonication on ice in 300 mM NaCl, 50 mM Tris pH 7.5, 20 mM imidazole, 0.1% Triton X-100 and 1 mM PMSF. The lysate was centrifuged 45 min in an SS34 rotor at 19,000 rpm and the cleared lysate was loaded onto column containing Talon resin (Qiagen; for a 9 L prep ∼2 mL of resin was used) using a peristaltic pump (Biorad) in the cold room. The column was then hooked up to an FPLC (Akta, Amersham) and washed with at least 30 column volumes of 50 mM Tris (pH 7.5), 300 mM NaCl, 20 mM imidazole. When the A280 trace flat-lined the protein was eluted in 50 mM Tris, pH 7.5, 300 mM NaCl, 200 mM imidazole. The peak fractions were immediately diluted 1∶1 with 50 mM Tris pH 7.5, loaded onto a 1 mL Heparin HiTrap column (GE Healthcare) in 200 mM NaCl/50 mM Tris (pH 7.5), and eluted using 200 mM to 1 M NaCl gradient (in 50 mM Tris, 7.5) over 20 column volumes. h6-WhiA elutes at ∼500 mM NaCl. The peak fraction was then concentrated to ∼4 mL in a centrifugal filter with 10 K MWCO (Millipore). The N-terminal 6xHis tag was cleaved by adding biotinylated thrombin (Novagen, 1 U per ∼0.5 mg of h6-WhiA) and incubating at 15°C overnight. The next morning 15 µl of packed streptavidin agarose (Novagen) was added per U of thrombin and incubated at room temperature for 15 minutes, then spin filtered through a 0.22 µm centrifugal filter (Millipore) and loaded onto a prep grade HiLoad 16/60 Superdex200 gel filtration column (GE Healthcare) equilibrated in 150 mM (NH4)2SO4/50 mM Tris, 7.5. The peak fractions were pooled, concentrated to ∼2–3 mg/mL, supplemented with 10% glycerol, and stored at −80°C. A typical final yield of untagged WhiA under these conditions was ∼0.5 mg per L of culture.
The purification of h6-tagged WhiA variants (1–216 and 221–328) was as described above, except the thrombin-cleavage and Heparin HiTrap column steps were omitted. H6-WhiG was expressed as a soluble protein under the same conditions, except that BL21 (DE3) RIL bacteria were used. After the metal affinity column, h6-WhiG was concentrated in a 0.5 mL 10 k MWCO Amicon Ultra Centrifugal filter (Millipore) and buffer-exchanged by three rounds of ∼4 fold dilutions with 150 mM NaCl, 50 mM Tris pH7.5, 20% glycerol. Single use aliquots were stored at −80°C. A typical final yield of h6-WhiG under these conditions was ∼0.1 mg per L of culture.
Electrophoretic mobility shift assays
For EMSA assays DNA probes corresponding to the various promoters were PCR-amplified, digested with BamHI (to increase the efficiency of end-labeling), gel-purified, and then labeled with T4 polynucleotide kinase (New England Biolabs) with 0.2 µl of γ32P-ATP according to the manufacturers instructions. Labeled oligos were then spin-filtered through G50 Sephadex resin (Roche) to remove unincorporated label. WhiA protein (or WhiA truncations) were incubated with 30 nM (∼50 ng) of cold oligo and ∼1 ng of labeled oligo in a 10 µl reaction containing 10 mM HEPES (pH 7.9), 50 mM NaCl, 2.5% sucrose, 2.5 mM MgCl2, 1 mM DTT and 25 ng/µl λ/HindIII fragments (to limit non-specific binding) for 15′ at room temperature. Samples were then loaded on a 6% polyacrylamide gel (made with 37.5∶1 acrylamide:bisacrylamide) in 0.5X TBE that had been prerun for at least 30 minutes at 7 W (constant wattage). The samples were electrophoresed for 2 hrs at 8 W (constant wattage), with temperature monitoring not to exceed 27°C. The gels were dried and exposed to a phosphorimager plate.
DNAse I footprinting assay
Double-stranded oligonucleotides corresponding to −70/+20 relative to WhiA's sporulation-specific transcription start site were generated by annealing single-stranded oligonucleotides of the plus and minus strands (synthesized by IDT technologies). The ss-oligonucleotides were labeled with 32P-γATP using T4 polynucleotide kinase (NEB) prior to annealing to generate double-stranded oligos specifically labeled on one strand. The WhiA protein (at final concentrations indicated in Figure 3) was incubated with 2.5 ng of labeled oligonucleotide, 25 ng of cold ds-oligonucleotide and 500 ng of λ/HindIII fragments in EMSA binding buffer (20 µl final volume) for 10′ at room temperature. 1 µl of DNAseI was then added at various dilutions (optimized in prior reactions), incubated for 5 minutes, and the reaction was stopped by adding 100 µl of 20 mM EDTA followed by 100 µl of phenol/chloroform. Samples were centrifuged 5 minutes at top speed in an Eppendorf centrifuge, and the aqueous layer was extracted with 100 µl of chloroform followed by ethanol precipitation and two washes with 70% ethanol. The dried pellet was resuspended in 5 µL of water followed by four volumes of 95% formamide/10 mM EDTA containing bromphenol blue/xylene cyanol. The reactions were then separated on an 8% acrylamide sequencing gel (SequaGel) at 20W (constant wattage, ∼30 minutes), dried and exposed to film.
Fluorescence competition DNA binding specificity assay
Determination of the DNA binding specificity by WhiA across the −44 to −4 region of its promoter was carried out as previously described29. Details of the exact methodology and calculations used for the assay can be found in that reference and its supplementary material. In this assay, a known concentration of H6- tagged WhiA was immobilized in individual wells of a 96-well, Talon-coated plate and incubated with a fluorescently labeled oligonucleotide containing the wild-type −44→−4 region. A competing, unlabeled oligonucleotide corresponding to the same DNA sequence, with the exception of a single basepair substitution at one specific position in the promoter binding site, was then added to each well, followed by a series of wash steps. Any DNA sequence that effectively competes for binding with the wild-type promoter (i.e. that contains a basepair substitution that does not decrease binding affinity) causes a reduction in fluorescence intensity in the corresponding well. In contrast, any DNA sequence that displays a reduced affinity as compared to the wild-type promoter (i.e., that contains a basepair substitution that does decrease affinity) leads to an increase in the fluorescence intensity.
The matrix of DNA duplexes, each containing the −44 to −4 region of the WhiA promoter but containing a single base substitution relative to the wild type promoter region is shown in Supplementary Figure S4. The fluorescently labeled wild-type top strand oligonucleotide, modified with 5′ Cy3TM, was synthesized separately (IDT) and annealed with a complementary unlabeled bottom strand. In addition, a completely unrelated DNA oligonucleotide sequence (5′-ATCGATCATCGTCGCATGATCAT-3′) and its complement were also synthesized and annealed as non-specific binding control.
Each individual competitive binding assay consists of one of three unlabeled oligonucleotides versus the same labeled wild-type target: (1) the unlabeled wild type promoter competing against the labeled version of the same sequence; (2) a completely randomized unlabeled sequence used as a negative control to account for competition by a nonspecific DNA sequence; and (3) each of the target sequences in the substrate matrix, containing a single basepair mismatch, competing against the labeled wild-type promoter. All competition experiments were performed in triplicate.
H6-tagged WhiA was immobilized onto nickel-coated 96 well plates (Ni-NTA HisSorbTMplates) by incubating 100 μl of 300 nM WhiA in TBS/BSA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.2%BSA) in wells for 2 hours at room temperature. The plates were washed four times with TBS/Tween-20 (50 mM Tris pH 7.5, 150 mM NaCl, 0.05%Tween-20) to remove unbound protein prior to addition of DNA. The immobilized WhiA in each well was incubated for two hours against a mixture of 450 nM labeled wild type DNA duplex and 13.5 μM (a 30-fold excess) of unlabeled competitor duplex in 200 μl of binding buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.02 mg/mL poly dI-dC, 10 mM CaCl2).
The plates were washed four times with TBS (50 mM Tris pH 7.5, 150 mM NaCl). The fluorescent signal retained from each test well was counted using a SpectraMax® M5/M5e micro-plate reader (Molecular Devices; excitation: 510 nm, emission: 565 nm, cutoff: 550 nm). All measurements were performed in triplicate. Additional negative control experiments performed in the absence of protein indicated that no significant detectable fluorescent signal was retained after the protocol described above was completed.
The measurements of the retained fluorescent signal for each mismatch sequence variant (F(i,j)) were then converted to relative binding affinities as compared to the wild-type target site as previously described.
In vitro transcription reactions
Promoters for in vitro run-off transcription reactions were generated by PCR from pGEM3T vectors (see Supplemental Figure S2 for expected sizes). 10 µl in vitro transcription reactions were carried out in 40 mM Tris-HCl (pH 7.5), 150 mM KCl, 10 mM MgCl2, 0.01% TX-100, 2 mM DTT; 300 µM ATP, GTP and CTP, 50 µM UTP, and 0.1 µl of α32P-UTP, 1 unit of E. coli core RNA polymerase (Epicentre Biotechnologies; we estimate based on silver-stained gel visualization that 1 unit of polymerase in this reaction volume is roughly equivalent to 100 nM polymerase +/− 50 nM), and 60 nM h6-WhiG. In a typical reaction, h6-WhiG was preincubated with RNA Polymerase for 10′ at room temperature. The DNA template was then added (0.3 pmol) and further incubated for 10′. A cocktail containing the transcription buffer and NTP's (including radioactively-labeled UTP) was then added, and the reaction was incubated for 30′ at 37°C. The reaction was stopped by the addition of an equal volume of 8 M Urea, 50 mM EDTA, 90 mm Tris-borate buffer, pH 8.3, 0.02% bromphenol blue and 0.02% xylene cyanol. The samples were heated to 90°C for 3 min, and 10 µl was then electrophoresed on an 8% polyacrylamide (using 29∶1 acrylamide: bisacrylamide)/8 M urea gel in 1X TBE Buffer. The gel was prerun for at least 20 minutes at 27 W (constant), then ran for 1 hr at 40 W constant with temperature monitoring not to exceed 45°C. The gel was then exposed to a phosphorimager.
Pull-down protein binding assay
2 µg of h6-WhiG was incubated with 2 µg of untagged WhiA protein in 150 mM NaCl/50 mM Tris, 7.5/20 mM Imidazole in a 10 µl volume for 15′ at room temperature. A slurry containing 5 µl of Ni-NTA resin (Qiagen) was then added in a volume of 100 µl of the same buffer, and the sample was nutated for 45′ at 4°C. The resin was then loaded onto a 96-well plate with low protein-binding Durapore Membrane (Millipore) and centrifuged for 1 min, 700 rpm in a swinging bucket table top centrifuge at 4°C. The flow-through was discarded the wells were washed 5 times with 150 µl of 200 mM NaCl/50 mM Tris, 7.5/20 mM imidazole using a multi-channel pipettor. After the last wash the beads were resuspended and transferred to a 0.6 mL eppendorf tube. The beads were briefly centrifuged, the supernatant was carefully aspirated with a 25 gauge needle, and then 15 µl of 2X SDS PAGE sample buffer was added to the beads. The samples were heated at 95°C for 5 minutes, and all of the sample was loaded onto a 12% NuPAGE Bis-Tris Gel (Invitrogen) and electrophoresed in MES buffer (Invitrogen). The gel was stored in 50% ethanol for at least one hour prior to silver staining using standard protocols.
Author Contributions
B.K.K. conducted all the experiments and contributed to writing all sections of the paper; B.L.S. helped design and assisted with the experiments and contributed to writing all sections of the paper.
Supplementary Material
Supplementary Information
Acknowledgments
Funding was provided by the NIH (R01 GM49857) and the FHCRC Division of Basic Sciences. Assistance, reagents, advice and access to space and equipment were provided by Ryo Takeuchi, Lindsey Doyle, Toshi Tsukiyama and the Ferre-D'Amare Lab.
References
- Chater K. F. Streptomyces inside-out: a new perspective on the bacteria that provide us with antibiotics. Philos Trans R Soc Lond B Biol Sci 361, 761–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flardh K. & Buttner M. J. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol 7, 36–49 (2009). [DOI] [PubMed] [Google Scholar]
- Hopwood D. Genetic analysis and genome structure in Streptomyces coelicolor. Bacteriol Rev 31, 373–403 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick M. J. A morphological and genetic mapping study of bald colony mutants of Streptomyces coelicolor. J Gen Microbiol 96, 299–315 (1976). [DOI] [PubMed] [Google Scholar]
- Chater K. F. A morphological and genetic mapping study of white colony mutatnts of Streptomyces coelcolor. J. Gen. Microbiol. 72, 9–28 (1972). [DOI] [PubMed] [Google Scholar]
- Hopwood D. A., Wildermuth H. & Palmer H. M. Mutants of Streptomyces coelicolor defective in sporulation. J Gen Microbiol 61, 397–408 (1970). [DOI] [PubMed] [Google Scholar]
- Flardh K., Findlay K. C. & Chater K. F. Association of early sporulation genes with suggested developmental decision points in Streptomyces coelicolor A3(2). Microbiology 145 (Pt 9)</vol>, 2229–43 (1999). [DOI] [PubMed] [Google Scholar]
- Jakimowicz D., Mouz S., Zakrzewska-Czerwinska J. & Chater K. F. Developmental control of a parAB promoter leads to formation of sporulation-associated ParB complexes in Streptomyces coelicolor. J Bacteriol 188, 1710–20 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J., Calcutt M. J., Schmidt F. J. & Chater K. F. Partitioning of the linear chromosome during sporulation of Streptomyces coelicolor A3(2) involves an oriC-linked parAB locus. J Bacteriol 182, 1313–20 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall B. & Lutkenhaus J. FtsZ in Bacillus subtilis is required for vegetative septation and for asymmetric septation during sporulation. Genes Dev 5, 447–55 (1991). [DOI] [PubMed] [Google Scholar]
- Grantcharova N., Lustig U. & Flardh K. Dynamics of FtsZ assembly during sporulation in Streptomyces coelicolor A3(2). J Bacteriol 187, 3227–37 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin W. Bacterial sporulation: FtsZ rings do the twist. Curr Biol 12, R391–2 (2002). [DOI] [PubMed] [Google Scholar]
- Claessen D., de Jong W., Dijkhuizen L. & Wosten H. A. Regulation of Streptomyces development: reach for the sky! .Trends Microbiol 14, 313–9 (2006). [DOI] [PubMed] [Google Scholar]
- Chater K. F. Construction and phenotypes of double sporulation deficient mutants in Streptomyces coelicolor A3(2). J Gen Microbiol 87, 312–25 (1975). [DOI] [PubMed] [Google Scholar]
- Kelemen G. H. et al.. The positions of the sigma-factor genes, whiG and sigF, in the hierarchy controlling the development of spore chains in the aerial hyphae of Streptomyces coelicolor A3(2). Mol Microbiol 21, 593–603 (1996). [DOI] [PubMed] [Google Scholar]
- Ryding N. J. et al.. A developmentally regulated gene encoding a repressor-like protein is essential for sporulation in Streptomyces coelicolor A3(2). Mol Microbiol 29, 343–57 (1998). [DOI] [PubMed] [Google Scholar]
- Ainsa J. A., Parry H. D. & Chater K. F. A response regulator-like protein that functions at an intermediate stage of sporulation in Streptomyces coelicolor A3(2). Mol Microbiol 34, 607–19 (1999). [DOI] [PubMed] [Google Scholar]
- Losick R. & Shapiro L. Changing views on the nature of the bacterial cell: from biochemistry to cytology. J Bacteriol 181, 4143–5 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes K. T. & Mathee K. The anti-sigma factors. Annu Rev Microbiol 52, 231–86 (1998). [DOI] [PubMed] [Google Scholar]
- Chater K. F. et al.. The developmental fate of S. coelicolor hyphae depends upon a gene product homologous with the motility sigma factor of B. subtilis. Cell 59, 133–43 (1989). [DOI] [PubMed] [Google Scholar]
- Ainsa J. A. et al.. WhiA, a protein of unknown function conserved among gram-positive bacteria, is essential for sporulation in Streptomyces coelicolor A3(2). J Bacteriol 182, 5470–8 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flardh K., Leibovitz E., Buttner M. J. & Chater K. F. Generation of a non-sporulating strain of Streptomyces coelicolor A3(2) by the manipulation of a developmentally controlled ftsZ promoter. Mol Microbiol 38, 737–49 (2000). [DOI] [PubMed] [Google Scholar]
- Soliveri J., Brown K. L., Buttner M. J. & Chater K. F. Two promoters for the whiB sporulation gene of Streptomyces coelicolor A3(2) and their activities in relation to development. J Bacteriol 174, 6215–20 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwedock J., McCormick J. R., Angert E. R., Nodwell J. R. & Losick R. Assembly of the cell division protein FtsZ into ladder-like structures in the aerial hyphae of Streptomyces coelicolor. Mol Microbiol 25, 847–58 (1997). [DOI] [PubMed] [Google Scholar]
- Knizewski L. & Ginalski K. Bacterial DUF199/COG1481 proteins including sporulation regulator WhiA are distant homologs of LAGLIDADG homing endonucleases that retained only DNA binding. Cell Cycle 6, 1666–70 (2007). [DOI] [PubMed] [Google Scholar]
- Kaiser B. K., Clifton M. C., Shen B. W. & Stoddard B. L. The structure of a bacterial DUF199/WhiA protein: domestication of an invasive endonuclease. Structure 17, 1368–76 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard B. L. Homing endonuclease structure and function. Q Rev Biophys 38, 49–95 (2005). [DOI] [PubMed] [Google Scholar]
- Thyme S. B. et al.. Exploitation of binding energy for catalysis and design. Nature 461, 1300–4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Pellenz S. & Stoddard B. L. Activity and specificity of the bacterial PD-(D/E)XK homing endonuclease I-Ssp6803I. J Mol Biol 385, 1498–510 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo G. D. DNA binding sites: representation and discovery. Bioinformatics 16, 16–23 (2000). [DOI] [PubMed] [Google Scholar]
- Tompa M. et al.. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 23, 137–44 (2005). [DOI] [PubMed] [Google Scholar]
- Chater K. Evaluation of Kaiser BK . et al.. in Faculty of 1000 (Microbiology) (F1000.com, 20 Jan 2010). [Google Scholar]
- Elliot M. A., Bibb M. J., Buttner M. J. & Leskiw B. K. BldD is a direct regulator of key developmental genes in Streptomyces coelicolor A3(2). Mol Microbiol 40, 257–69 (2001). [DOI] [PubMed] [Google Scholar]
- Campbell E. A., Westblade L. F. & Darst S. A. Regulation of bacterial RNA polymerase sigma factor activity: a structural perspective. Curr Opin Microbiol 11, 121–7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M. A. et al.. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323, 396–401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information