

Abstract
Cdc14 is an essential phosphatase in yeast but its role in the mammalian cell cycle remains obscure. We report here that Cdc14b-knockout cells display unscheduled induction of multiple cell cycle regulators resulting in early entry into DNA replication and mitosis from quiescence. Cdc14b dephosphorylates Ser5 at the C-terminal domain (CTD) of RNA polymerase II, a major substrate of cyclin-dependent kinases. Lack of Cdc14b results in increased CTD-Ser5 phosphorylation, epigenetic modifications that mark active chromatin, and transcriptional induction of cell cycle regulators. These data suggest a function for mammalian Cdc14 phosphatases in the control of transcription during the cell cycle.
Progression throughout the cell cycle is known to be tightly regulated by cyclin-dependent kinases (Cdks), whose enzymatic activity promotes DNA replication and mitosis1. The relevance of the counteracting phosphatases has been less studied. In Saccharomyces cerevisiae, the protein phosphatase Cdc14 is essential for the inactivation of mitotic Cdks and mitotic exit by dephosphorylating many Cdk substrates2,3. The subcellular localization of ScCdc14 is regulated initially by the FEAR network which functions in early anaphase and subsequently by the Mitotic Exit Network (MEN) which is active in late anaphase. In Schizosaccharomyces pombe, the localization of the Cdc14-like phosphatase Clp1/Flp1, which also antagonizes mitotic Cdk activity, is regulated by the septation-initiation network (SIN), a signaling pathway homologous to the MEN.
Two Cdc14 orthologs, Cdc14a and Cdc14b, exist in all mammals. These proteins are able to rescue the phenotype of Flp1-deficient yeast strains and Cdc14b, in particular, is able to fulfill all essential functions of ScCdc14 suggesting some functional conservation4,5. However, the dephosphorylation of Cdk phosphosites seems to rely mostly on PP2A-B55 complexes during mammalian mitotic exit6,7 and the relevance of Cdc14 phosphatases is not clear. Cdc14a specifically regulates centrosome function and prevents premature Cdk1 activation8 whereas Cdc14b has been proposed to play multiple functions during the cell cycle9. Cdc14b is able to induce malignant transformation in vitro although this activity has no clear links to direct cell cycle regulation10. Recent knockout studies in established cell lines have shown not obvious mitotic defects in the absence of these proteins, although mammalian Cdc14 phosphatases have been suggested to be required for efficient DNA repair11,12. Genetic ablation of Cdc14b exon 2 in the mouse resulted in proliferative defects and increased senescence both in vitro and in vivo13. In this manuscript, we report that lack of mouse Cdc14b results in increased transcription of cell cycle-specific genes. This function correlates with the ability of Cdc14b of removing phosphate residues from Ser5 of the C-terminal domain (CTD) of RNA polymerase II, a site known to modulate several epigenetic marks required for gene transcription.
Results
Cell cycle alterations in Cdc14b-deficient cells
We have generated a new Cdc14b null allele in the mouse by excising exons 8-9 which encode most of the phosphatase catalytic domain. As reported recently13, Cdc14b(−/−) mice are viable although develop some pathologies that will be described elsewhere. Cdc14b(−/−) primary mouse embryonic fibroblasts (MEFs) grew well in culture during the first passages and do not show differences in cell proliferation in asynchronous cultures (Fig. 1a–c). The duration of mitosis was normal and these cells do not show any defect during mitotic exit in the absence (Fig. 1a) or presence (Fig. 1d) of mitotic poisons. Similarly, Cdc14b-deficient MEFs displayed normal levels of γH2AX, a marker of replicative stress or DNA damage, in untreated cultures at different passages (Fig. 1e) or upon γ-irradiation (10 Gy; Fig. 1f). Moreover, these mutant cells did not display obvious deficiencies in checkpoint function or DNA repair since γH2AX signal decay occurred with normal kinetics in the absence of Cdc14b at passage 3 (Fig. 1f). Whether these cells may have additional deficiencies in DNA damage checkpoint of DNA repair under different culture conditions (senescence) or in different genetic backgrounds remains to be explored.
Figure 1. Normal cell cycle progression in asynchronous Cdc14b-null cells.
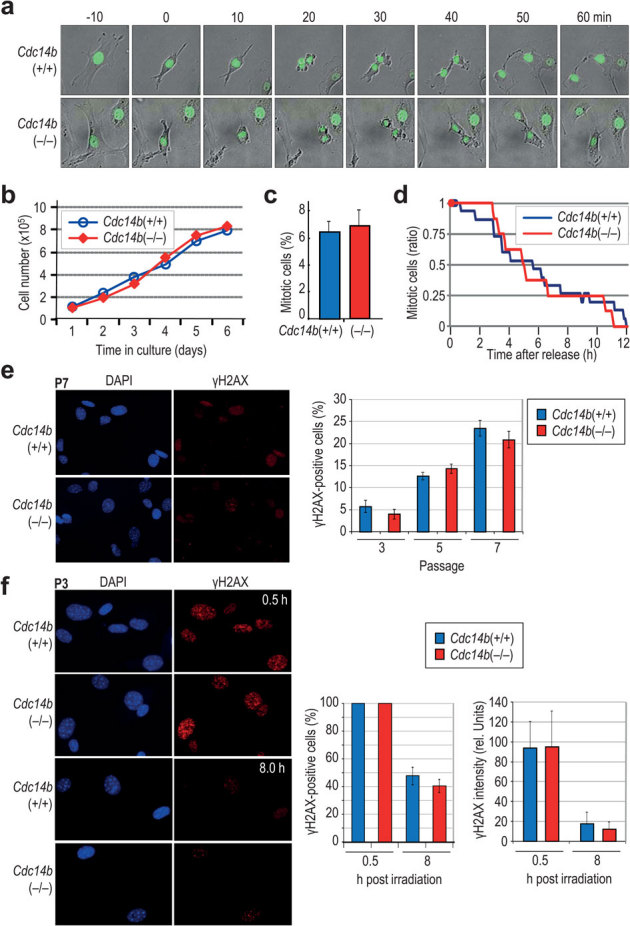
(a) Primary MEFs were transduced with lentiviruses expressing Histone H2B-GFP and cells were recorded by videomicroscopy. Time 0 indicates mitotic entry. No differences are found in the duration of mitosis in wild-type or Cdc14b-null cells. (b) Growth curve of Cdc14b-null and control primary MEFs during 6 days. (c) Percentage of mitotic cells in these asynchronous cultures. (d) Mitotic exit kinetics in Cdc14b-null (N = 84) and control (N = 67) cells in the presence of nocodazole. Cells were arrested in prometaphase with nocodazole and the percentage of mitotic cells, as observed by videomicroscopy, is scored showing no differences between these two genotypes during mitotic slippage. All these graphs show representative results from at least three separate experiments. (e) Immunofluorescence staining of passage 7 (P7) primary MEFs and quantification of the percentage of γH2AX positive cells in primary MEFs at passages 3, 5 and 7. (f) Immunofluorescence staining of passage 3 (P3) primary MEFs after 10 Gy of γ−irradiation. Quantification of γH2AX-positive cells and the average γH2AX intensity per cell shows no significant differences between wild-type and Cdc14b-null cultures. All these graphs show results from three separate experiments.
Despite the lack of obvious defects in asynchronous cultures, a detailed analysis of mitotic entry by videomicroscopy revealed that Cdc14b(−/−) MEFs entered earlier in mitosis (24.59 ± 3.07 h after serum stimulation vs. 28.90 ± 3.49 h in wild-type cells; p<0.003; Fig. 2a,b) when released from quiescence. We initially checked the expression of several mitotic regulators such as cyclin B1, Cdk1 or Aurora B and found a general upregulation of these proteins during early time points. This correlated with earlier phosphorylation of histone H3 (an Aurora-B substrate) or Cdk substrates (Fig. 2c). These data could be in agreement with a role for Cdc14 phosphatases in the control of Cdk1 activation during G2 as suggested previously8,14,15. However, this putative function does not explain the upregulation of some of these proteins at early time points (20–23 h) at which most wild-type cells have not completed S phase (Fig. 3a). Thus, we wondered whether a similar alteration in expression levels was found in G1/S regulators. Both cyclin E and cyclin A, but not cyclin D2, were upregulated in Cdc14b(−/−) cells, and these alterations correlated with increased levels in the phosphorylation of the retinoblastoma protein (pRb), a Cdk substrate whose phosphorylation induces the G1/S transition (Fig. 3b). Indeed, Cdc14b(−/−) cells entered S-phase with a faster kinetics as determined by the incorporation of nucleotide analogs (Fig. 3a). Thus, although we cannot discard a direct function for Cdc14b in Cdk1 activation, these data suggest a specific deregulation of the levels of G1/S regulators in the absence of Cdc14b.
Figure 2. Early mitotic entry in synchronous Cdc14b-deficient cells.
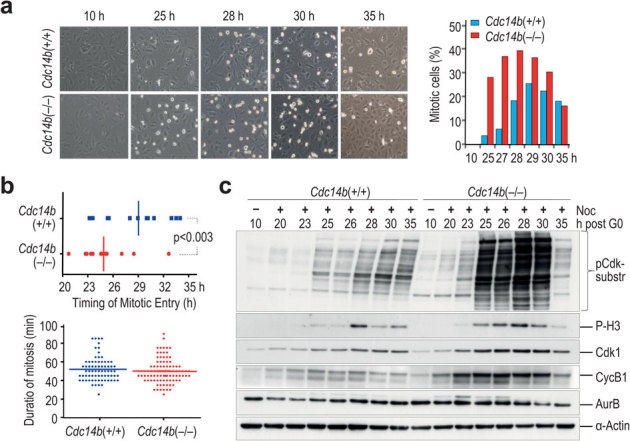
(a) Primary MEFs were serum starved for 72 h and forced to enter into cell cycle after stimulation with 20% fetal bovine serum (FBS). Micrographs show an earlier mitotic entry in Cdc14b(−/−) than in Cdc14b(+/+) cells. The percentage of mitotic cells was scored after immunofluorescence with DAPI and phospho-histone H3 at different time points after serum stimulation. These graphs show representative results from at least three separate experiments. (b) Cdc14b(−/−) and Cdc14b(+/+) MEFs expressing H2B-GFP were recorded by videomicroscopy during 35 h after serum stimulation. Cdc14b(−/−) cells enter earlier in mitosis but no differences were found in the duration of mitosis (N = 70–90 cells per genotype). (c) Immunodetection of the indicated mitotic proteins in total lysates from Cdc14b(+/+) and Cdc14b(−/−) MEFs at the indicated time-points after serum stimulation. These blots are representative from at least three different experiments.
Figure 3. Earlier entry into S-phase in Cdc14b-null cells.
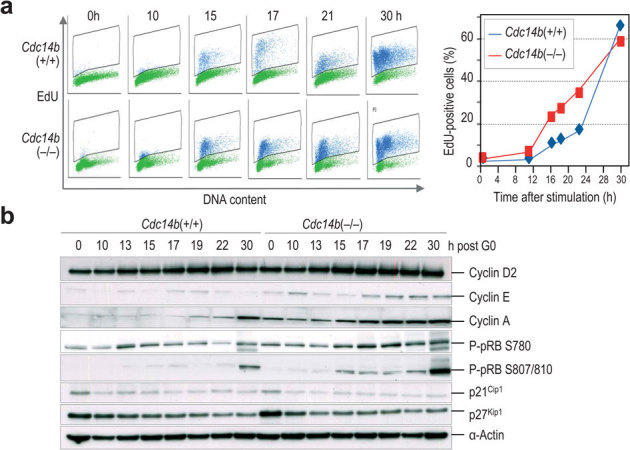
(a) MEFs were pulsed with EdU after serum stimulation and the percentage of EdU-positive cells was scored at different time-points. (b) Immunodetection of the indicated cell-cycle proteins in total lysates from Cdc14b(+/+) and Cdc14b(−/−) MEFs at the indicated time-points after serum stimulation. These results show representative data from at least three separate experiments with different MEFs.
Cdc14 phosphatases modulate phosphorylation of RNA polymerase II
Transcriptional control during the cell cycle is mediated by so-called “cell cycle” and well as “transcriptional” Cdks1. Whereas the first group (Cdk1-4,6) phosphorylates pRb to inactivate its repressor functions, the second group (Cdk7-9) phosphorylates the C-terminal domain (CTD) of the largest subunit (Rpb1) of RNA polymerase II (RNAPII) thus activating the transcriptional machinery. Since Cdc14 phosphatases have preference for Cdk substrates we then tested whether these two major Cdk substrates, pRb and RNAPII, are dephosphorylated by Cdc14 mammalian phosphatases. As shown in Fig. 4a, Cdc14a, but not Cdc14b, was able to efficiently remove phosphates added by Cdk1-cyclin B1 complexes on pRb. However, both Cdc14a and Cdc14b were able to dephosphorylate the RNAPII-CTD and, interestingly, whereas Cdc14a was able to decrease the phosphor-signal in Ser2 and Ser5, Cdc14b displayed certain preference for Ser5 (Fig. 4a).
Figure 4. Cdc14 proteins dephosphorylate RNA polymerase II and repress transcription.
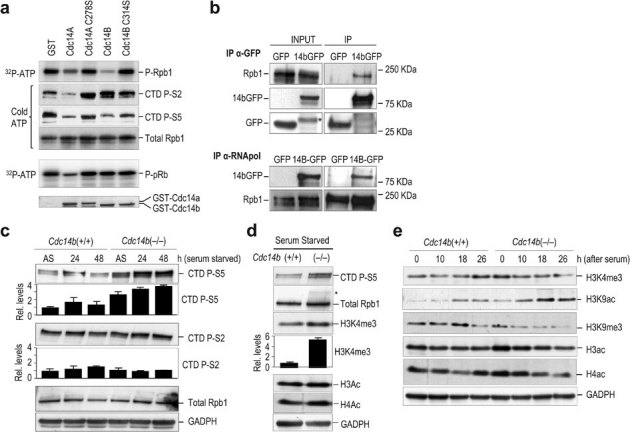
(a) The C-terminal domain (CTD) fragment of RNA polymerase II and pRb (amino acids 773–928) were used as substrates of Cdk1-cyclin B complexes in the presence of 32P-ATP or non-radiactive ATP and then incubated with GST-Cdc14a, GST-Cdc14b, phosphatase-dead mutants or GST alone. Samples were resolved by SDS-PAGE and detected by autoradiography or immunoblot with antibodies against Rpb1 or specific for phospho-Ser2 (P-S2) and phospho-Ser5 (P-S5) forms of the CTD. (b) Immunoblots of cell lysates from 293 cells expressing Cdc14b-GFP or GFP alone, before (INPUT) or after immunoprecipitation (IP) with either GFP or Rpb1 antibodies. The endogenous Rpb1 was detected using specific antibodies whereas exogenous Cdc14b-GFP (14bGFP) was detected using specific antibodies against GFP (* indicates non-specific bands). (c) Immunodetection of phospho-Ser2 (P-S2) and phospho-Ser5 (P-S5) forms of the CTD in asynchronous (AS) MEFs or in cells serum-deprived for 24 or 48 h. The relative levels of these marks versus asynchronous wild-type cells are indicated in the histograms. (d) Immunodetection of phospho-Ser5 CTD and the indicated epigenetic marks during serum starvation in wild-type or Cdc14b-null cells. (e) Immunodetection of the indicated epigenetic marks during cell cycle entry after stimulation of quiescent cells (serum-starved for 48 h) with serum. Samples were analyzed 0, 10, 18 and 26 h. after stimulation with serum. GADPH was used as a loading control in all these immunodetection studies.
To further test the molecular interactions between Cdc14b and RNAPII we then analyzed whether these proteins can be found as a complex in mammalian cells. Since current available antibodies do not detect endogenous Cdc14b proteins, we instead used a Cdc14b-GFP fusion protein. As observed in Fig. 4b, the endogenous Rpb1 was detected in Cdc14B-GFP immunoprecipitates, but not in lysates from cells expressing GFP alone. In addition, immunoprecipitation of the endogenous Rpb1 was able to pull down the Cdc14b-GFP fusion protein but not GFP alone, suggesting specific interaction between Cdc14b and RNAPII complexes.
Transcriptional activation of cell cycle genes in Cdc14b-null cells
RNAPII regulation is crucial for controlling the status of gene expression and has significant consequences for cell response pathways. The phosphorylation of the CTD modulates the recruitment of chromatin modification enzymes. Thus, phosphorylation of RNAPII CTD at Ser5 promotes H3 and H4 acetylation at active promoters16,17,18 and is crucial for high levels of methylation of histone H3 lysine 4 (H3K4) at active promoters16,19,20. In agreement with the ability of Cdc14b to dephosphorylate RNAPII, Cdc14b-null cells displayed increased CTD-Ser5 and H3K4me3 signals, as well as increased acetylation of histones H3 and H4 (Fig. 4c,d). These marks of active chromatin were also increased during the early phases of the cell cycle (0–10 h after cell cycle entry from quiescence; Fig. 4e) in correlation with decreased levels of specific repression marks such as H3K9me3, a binding site for heterochromatin protein 1 (HP1) which blocks access by the transcription factors and polymerases required for gene transcription.
To further analyze the consequences of the genetic ablation of Cdc14b in transcription, we analyzed the expression profile in Cdc14b-deficient cells. Cdc14b-null cells displayed a significant (p < 0.01) induction of genes involved in three molecular routes: G1 pathway (FDR = 0.071), cell cycle regulation (FDR = 0.013), and the ALK pathway (FDR = 0.016), which triggers cell cycle progression by transducing several mitogenic signals (Fig. 5a and Table 1). As an example, Cdc14b-null cells displayed increased transcript levels of A-type and B-type cyclins (Fig. 5b), in agreement with the overexpression of the corresponding proteins (Figs. 2 and 3). In addition, ectopic expression of Cdc14b in normal fibroblasts results in a significant repression of A- and B-type cyclins. This effect is not observed upon ectopic expression of a phosphatase-dead form of Cdc14b (Fig. 5b), in agreement with a catalytic function of Cdc14b in the transcriptional repression of cell cycle regulators.
Figure 5. Altered transcriptional profiles in Cdc14b-deficient cells.
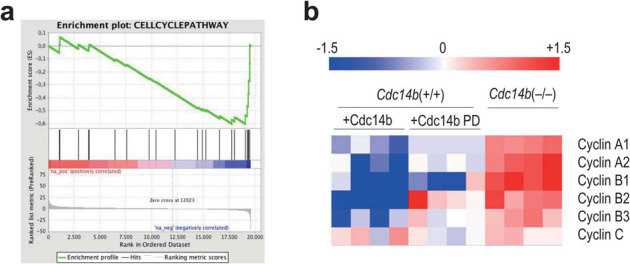
(a) Transcriptional alteration of cell cycle regulators in Cdc14b-null cells (blue side in the red-blue bar; FDR = 0.013). Gene annotations were retrieved from Biocarta. (b) Relative levels of the indicated transcripts in Cdc14b-null cells or in wild-type cells overexpressing Cdc14b or the phosphatase-dead (PD) form of Cdc14b. Fold induction is in red whereas fold repression is in blue. Cdc14b-null cells were normalized versus the levels in wild-type cells whereas overexpression of Cdc14b or Cdc14b PD was normalized versus cells transfected with the empty vector.
Table 1. Significant deregulation of Biocarta pathways in asynchronous Cdc14-null MEFs.
| Biocarta pathways | Adjusted pvalue (FDR) | Genes |
|---|---|---|
| Alk Pathway | 0.016 | Gsk3B, Tcf1, Bmp7, Atf2, Apc, Tgfbr1, Bmp10, Nkx2-5, Chrd, Tgfb1, Gata4, Acvr1, Mef2C, Dvl1, Axin1, Bmpr2, Tgfbr2, Fzd1, Bmpr1a, Tgfbr3, Bmp5, Wnt1, Rfc1, Map3K7, Bmp2, Nppb, Tgfb2, Nppa, Nog, Tgfb3, Bmp4. |
| G1 pathway | 0.071 | Gsk3B, Tp53, Cdkn1B, E2F1, Atm, Ccna1, Cdk4, Atr, Cdk6, Dhfr, Tgfb1, Abl1, Cdk2, Tfdp1, Rb1, Cdc25a, Skp2, Ccne1, Hdac1, Tgfb2, Cdkn1a, Cdkn2B, Cdkn2a, Tgfb3, Ccnd1. |
| Cell Cycle | 0.135 | Cdkn1B, Rbl1, Ccnb1, Ccnh, E2F1, Ccna1, Cdk4, Cdkn2D, Cdk6, Cdk2, Tfdp1, Cdkn2C, Rb1, Cdc25a, Ccne1, Cdk7, Cdkn1a, Ccnd3, Ccnd2, Cdkn2B, Cdkn2a, Ccnd1. |
Discussion
Whereas the relevance of yeast Cdc14 during mitotic exit has been well established2, other phosphatases such as PP1 or PP2A/B55 seem to drive mitotic exit in mammals6,7,21. Mammalian Cdc14 phosphatases are able to dephosphorylate several Cdk substrates including Sirt2, Cdc25, Erk3, Skp2 or Cdh1, among others [reviewed in Ref.9]. Yet, their relevance during the cell cycle remains obscure and loss-of-function studies have shown that individual inactivation of mammalian Cdc14a or Cdc14b does not compromise cell proliferation.
By generating Cdc14b-knockout primary MEFs, we have observed a role for Cdc14b in the entry into the cell cycle from quiescence. Cdc14b-null MEFs display increased levels of multiple cell cycle regulators, such as A- or B-type cyclins, as well as Cdk1, in correlation with earlier entry into S-phase and mitosis. The alteration in the levels of these proteins could be due to an effect of Cdc14b in the degradation machinery. In fact, Cdc14 proteins may dephosphorylate the Anaphase-promoting complex (APC/C) cofactor Cdh1, thus promoting the activation of APC/C-Cdh1 complexes and the ubiquitination and subsequent degradation of several cell cycle regulators22,23. The absence of Cdc14b could therefore result in lack of activation of Cdh1 and defective degradation of several cell cycle regulators. This mechanism, however, does not explain the alteration in the levels of Cdk1 (Fig. 2c) or Cyclin E (Fig. 3b) which are not APC/C targets. On the other hand, Cdh1--phase and do not display the earlier entry into mitosis observed in Cdc14-deficient cells24. These results suggest that Cdc14b may also modulate cell cycle progression by regulating transcription. Although pRb is a major transcriptional target of Cdks during the cell cycle25, this protein does not seem to be efficiently dephosphorylated by Cdc14b, at least in vitro (Fig. 4a).
RNA polymerase II (RNAPII), which transcribes all protein-coding genes and many non-coding RNAs, is another well-known substrate of Cdks. Rpb1, the largest subunit of RNAPII, contains an unusual CTD that comprises 52 repeats of the heptapetide consensus sequence YSPTSPS. Post-translational modifications, such as phosphorylation, glycosylation and cis-trans isomerization, at any of these residues generate a complex regulatory code that dictates the functional state of the polymerase20,26. The CTD repeat contains two proline-directed serines (Ser2 and Ser5) that are major targets of co-called transcriptional Cdks (Cdk7-9)1. However, the CTD is not only phosphorylated by transcriptional Cdks since cell-cycle Cdks such as Cdk1 or Cdk2 may also phosphorylate Ser2 and Ser5 of the CTD [reviewed in Refs.16,27]. This dual role is likely to promote the necessary link between cell cycle progression and transcription1 although the molecular details have not been explored. Both Cdc14a and Cdc14b are able to dephosphorylate Cdk-dependent phosphosites in the RNAPII CTD (Fig. 4a). Whereas Cdc14a is active both in Ser2 and Ser5, Cdc14b seems to preferentially dephosphorylate Ser5. Similarly, Ser5 is preferentially phosphorylated by Cdk7-9, as well as mitogen-activated protein kinases (ERK), whereas Cdk1-2 can modify both Ser2 and Ser5 with similar selectivity27. The phosphorylation of these two sites has distinct implications during mRNA processing, and different repeats in the CTD may serve different functions, adding a further level of complexity to the CTD code. Phosphorylation of RNAPII also mediates the recruitment of chromatin remodeling enzymes. For instance, phosphorylation of Ser5 contributes to the association of methyltransferases onto the 5′-portion of actively transcribed genes shortly after initiation, thereby enhancing histone H3 K4 trimethylation (H3K4me3). Lack of Cdc14b results in increased phopho-Ser5 signal as well as increased H3K4me3, suggesting that Cdc14b deficiency may enhance RNAPII-mediated transcription. How this deregulation affects to specific genes is not clear. Cell division is normal in asynchronous Cdc14b-null cultures and the deregulation of cell cycle genes is only obvious during the re-entry into the cell cycle from quiescence. Thus, Cdc14b could participate in the dephosphorylation of RNAPII during quiescence or during the early phases of the cell cycle. Since Cdc14b is localized at the nucleolus, this may specifically affect to nucleolus-associated heterochromatic regions, although these hypothesis deserve further investigation.
Altogether, our results suggest that mammalian Cdc14b dephosphorylates RNAPII-CTD Ser5, a modification known to contribute to the transcriptional activation of chromatin by increasing H3K4me3 and acetylation of histones H3 and H4. Lack of Cdc14b results in increased phospho-Ser5 levels, in correlation with the presence of epigenetic modifications, such as H3K4me3 or acetylated H3 and H4, that mark active chromatin. These alterations may contribute to the transcriptional induction of specific cell cycle regulators as detected by gene expression profiling. Since yeast Cdc14 also regulates transcription throughout the control of RNA polymerase II (L. Aragón, personal communication), these data indicate that the repression of cell cycle machinery is a critical, conserved function of Cdc14 phosphatases to regulate cell division.
Methods
Generation of Cdc14-null mice
The murine Cdc14b locus was modified by flanking exons 8 and 9 with loxP sequences using a routine targeting strategy24. Two out of 559 ES cell clones analyzed underwent homologous recombination and were aggregated to generate quimeras. Expression of Cre recombinase24 resulted in the excision of exons 8–9 creating the null allele [Cdc14b(−/−)]. Further details on the targeting strategy and genotyping protocols are available upon request. Animal procedures were approved by the corresponding Ethics Committee of Animal Experimentation in our institution.
Cell culture and immunofluorescence
Primary MEFs were prepared from E14.5 embryos essentially as described previously24. For cell cycle re-entry synchronization, early passage primary MEFs were starved in DMEM + 0.1% fetal bovine serum (FBS) for 72 hours and re-stimulated with 20% FBS. These cultures were treated with nocodazole (100 ng/ml; Sigma) 20 h after re-stimulation. EdU incorporation was assayed according to the manufacturer's protocol (ClickiT EdU assay kit; Molecular Probes, Invitrogen). For videomicroscopy, H2B-GFP expressing cells were recorded using a Deltavision Apparatus (Applied Precision) using 5 min frames during 35 hr. To induce DNA damage, primary MEFs at passage 3 where synchronized at G0 by serum starvation and 20 h after serum stimulation they were γ-irradiated (10 Gy) for 5 minutes. Nocodazole (100 ng/ml; Sigma) was added immediately to prevent mitotic exit. For immunostaining, cells were cultured on coverslips, fixed in 4% paraformaldehyde (PFA), permeabilized in 0.5% Triton X-100, and blocked with PBS containing 10% donor calf serum. Cells were then incubated with primary antibodies (γH2AX, Upstate biotechnology) for 2 h at room temperature and with fluor-conjugated secondary antibodies (Alexa 594) for 45 min at room temperature. Finally, cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole) to visualize DNA.
Biochemical analysis and phosphatase assays
For Western blotting, cells were harvested and lysed in RIPA buffer, and 50 μg of total protein was separated by SDS-PAGE and probed with antibodies against phosphoSer-CDKs substrates (Cell Signaling Technology), cyclin B1 (Chemicon International), phospho-Histone H3 (Millipore), Cdk1 (Santa Cruz), Aurora B (Abcam), Cyclin A2 (Santa Cruz), cyclin D2 (Santa Cruz), Cyclin E (Abcam), p21Cip1 (Santa Cruz), p27Kip1,(BD Transduction Laboratories), pRb P-S780 and pRb P-S807/810 (Cell signalling), H3K9ac (Abcam), H3K9me3 (Abcam), H3ac, H4ac, H3K4me3 (Upstate) and α-Actin (Sigma). For in vitro phosphatase assays, a pRb fragment (6xHis-hRb, aa773-928; Upstate) and the C-terminal domain (CTD) of human Rpb1 (purified from bacteria as a GST-CTD fusion protein using the pGEX-2TK vector) were used as substrates. These peptides were incubated with 2U of Cdk1-Cyclin B1 (New England Biolabs) in kinase buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 1 mM DTT and 50 mM ATP) in the presence of 0.15 μCi of γ(32P)ATP for 30 min at 30°C. Samples were then washed three times with 1 ml of phosphatase buffer (20 mM Tris, pH 8.3; 150 mM NaCl; 2 mM EDTA; 0.1% Triton X-100; 5 mM DTT) and incubated for 45 minutes at 30°C with GST-Cdc14a, GST-Cdc14b or their corresponding inactive forms, Cdc14a C278S and Cdc14b C314S, in the above phosphatase buffer. Approximately the same catalytic activity (as measured on the pNPP) was used for Cdc14a and Cdc14b. Reactions were stopped by the addition of loading buffer and boiling for 5 min at 95°C. Proteins were resolved by SDS-PAGE and visualized by Comassie staining and autoradiography or detected with specific antibodies: CTD phospho-Ser5 (Millipore), CTD phospho-Ser2 (Abcam), anti-RNA polymerase II (Millipore). For immunoprecipitation studies, cell lysates (1000 μg of protein) were incubated for 2 h at 4°C in a total volume of 300 μl with either 25 μl of mouse monoclonal anti-GFP (GFP; Roche) or 10 μl of mouse monoclonal anti-RNA pol II (Abcam clone AB5408) followed by incubation of protein G beads (Amersham) for an additional hour. Immunoprecipitates were collected by centrifugation, washed four times with a lysis buffer containing NaCl 0.5 M, and subjected to SDS-PAGE electrophoresis and immunoblot analysis.
Transcriptional profiling
Total RNA was prepared from cells using TRIzol reagent (Invitrogen). Gene expression arrays were purchased from Agilent and processed according to the manufacturer's recommendation. Significantly deregulated mRNAs were computed using the TM4 package (http://www.tm4.org) and the Limma package from Bioconductor (http://www.bioconductor.org). Three different clones of cells overexpressing the indicated vectors or lacking Cdc14b were compared to the corresponding controls. Gene Set Enrichment Analysis (GSEA) was carried out using limma moderated t statistic for gene ranking28. The effect of overexpression of Cdc14 or the phosphatase-dead (PD) form of Cdc14b was analyzed as reported previously10.
Author Contributions
M.G., M.C. and E.M. generated Cdc14b-null mice and characterized cell-cycle and epigenetic defects in Cdc14b-null cells, M.C. and M.S. performed kinase and phosphatase assays, G.G.-L. and D.G.P. analyzed microarray data, M.M. designed the project, analyzed the data and wrote the manuscript.
Acknowledgments
This work was funded by grants from the Association for International Cancer Research (AICR #08-0188), Foundation Ramón Areces, and the Spanish Ministry of Science and Innovation (MICINN; BFU2008-04293 to M.S.; SAF2009-07973 to M.M.). The Cell Division and Cancer Group of the CNIO is supported by the OncoCycle Programme (S-BIO-0283-2006) from the Comunidad de Madrid, the OncoBIO Consolider-Ingenio 2010 Programme (CSD2007- 00017) from the MICINN, Madrid, and the European Union Seventh Framework Programme (MitoSys project; HEALTH-F5-2010-241548).
References
- Malumbres M. & Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci 30, 630–641 (2005). [DOI] [PubMed] [Google Scholar]
- Stegmeier F. & Amon A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet 38, 203–232 (2004). [DOI] [PubMed] [Google Scholar]
- Queralt E. & Uhlmann F. Cdk-counteracting phosphatases unlock mitotic exit. Curr Opin Cell Biol 20, 661–668 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Novelle M. D., Esteban V., Bueno A. & Sacristan M. P. Functional homology among human and fission yeast Cdc14 phosphatases. J Biol Chem 280, 29144–29150 (2005). [DOI] [PubMed] [Google Scholar]
- Li L., Ernsting B. R., Wishart M. J., Lohse D. L. & Dixon J. E. A family of putative tumor suppressors is structurally and functionally conserved in humans and yeast. J Biol Chem 272, 29403–29406 (1997). [DOI] [PubMed] [Google Scholar]
- Schmitz M. H. et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol 12, 886–893 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchado E. et al. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55alpha,delta phosphatase. Cancer Cell 18, 641–654 (2010). [DOI] [PubMed] [Google Scholar]
- Sacristan M. P., Ovejero S. & Bueno A. Human Cdc14A becomes a cell cycle gene in controlling Cdk1 activity at the G/M transition. Cell Cycle 10, 387–391 (2011). [DOI] [PubMed] [Google Scholar]
- Mocciaro A. & Schiebel E. Cdc14: a highly conserved family of phosphatases with non-conserved functions? J Cell Sci 123, 2867–2876 (2010). [DOI] [PubMed] [Google Scholar]
- Chiesa M., Guillamot M., Bueno M. J. & Malumbres M. The Cdc14B phosphatase displays oncogenic activity mediated by the Ras-Mek signaling pathway. Cell Cycle 10, 1607–1617 (2011). [DOI] [PubMed] [Google Scholar]
- Berdougo E., Nachury M. V., Jackson P. K. & Jallepalli P. V. The nucleolar phosphatase Cdc14B is dispensable for chromosome segregation and mitotic exit in human cells. Cell Cycle 7, 1184–1190 (2008). [DOI] [PubMed] [Google Scholar]
- Mocciaro A. et al. Vertebrate cells genetically deficient for Cdc14A or Cdc14B retain DNA damage checkpoint proficiency but are impaired in DNA repair. J Cell Biol 189, 631–639 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z. et al. Early-onset aging and defective DNA damage response in Cdc14b-deficient mice. Mol Cell Biol 31, 1470–1477 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Novelle M. D., Mailand N., Ovejero S., Bueno A. & Sacristan M. P. Human Cdc14A phosphatase modulates the G2/M transition through Cdc25A and Cdc25B. J Biol Chem 285, 40544–40553 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumurbaatar I., Cizmecioglu O., Hoffmann I., Grummt I. & Voit R. Human Cdc14B promotes progression through mitosis by dephosphorylating Cdc25 and regulating Cdk1/cyclin B activity. PLoS One 6, e14711 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff S. & Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet 24, 280–288 (2008). [DOI] [PubMed] [Google Scholar]
- Govind C. K., Zhang F., Qiu H., Hofmeyer K. & Hinnebusch A. G. Gcn5 promotes acetylation, eviction, and methylation of nucleosomes in transcribed coding regions. Mol Cell 25, 31–42 (2007). [DOI] [PubMed] [Google Scholar]
- Ginsburg D. S., Govind C. K. & Hinnebusch A. G. NuA4 lysine acetyltransferase Esa1 is targeted to coding regions and stimulates transcription elongation with Gcn5. Mol Cell Biol 29, 6473–6487 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell 36, 541–546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E. & Pombo A. Modifications of RNA polymerase II are pivotal in regulating gene expression states. EMBO Rep 10, 1213–1219 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. Q. et al. PP1-mediated dephosphorylation of phosphoproteins at mitotic exit is controlled by inhibitor-1 and PP1 phosphorylation. Nat Cell Biol 11, 644–651 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bembenek J. & Yu H. Regulation of the anaphase-promoting complex by the dual specificity phosphatase human Cdc14a. J Biol Chem 276, 48237–48242 (2001). [DOI] [PubMed] [Google Scholar]
- Bassermann F. et al. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134, 256–267 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I. et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol 10, 802–811 (2008). [DOI] [PubMed] [Google Scholar]
- Malumbres M. & Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1, 222–231 (2001). [DOI] [PubMed] [Google Scholar]
- Buratowski S. The CTD code. Nat Struct Biol 10, 679–680 (2003). [DOI] [PubMed] [Google Scholar]
- Palancade B. & Bensaude O. Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. Eur J Biochem 270, 3859–3870 (2003). [DOI] [PubMed] [Google Scholar]
- Subramanian A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]