

Abstract
We have recently reported upon the development of crosslinked urethane-doped polyester (CUPE) network elastomers, which was motivated by the desire to overcome the drawbacks presented by crosslinked network polyesters and biodegradable polyurethanes for soft tissue engineering applications. Although the effect of the isocyanate content and post-polymerization conditions on the material structure-property relationship was examined in detail, the ability of the diol component to modulate the material properties was only studied briefly. Herein, we present a detailed report on the development of CUPE polymers synthesized using diols 4, 6, 8, 10, or 12 methylene units in length in order to investigate what role the diol component plays on the resulting material’s physical properties, and assess their long-term biological performance in vivo. An increase in the diol length was shown to affect the physical properties of the CUPE polymers primarily through lowered polymeric crosslinking densities and elevated material hydrophobicity. The use of longer chain diols resulted in CUPE polymers with increased molecular weights resulting in higher tensile strength and elasticity, while also increasing the material hydrophobicity to lower bulk swelling and prolong the polymer degradation rates. Although the number of methylene units largely affected the physical properties of CUPE, the choice of diol did not affect the overall polymer cell/tissue-compatibility both in vitro and in vivo. In conclusion, we have established the diol component as an important parameter in controlling the structure-property relationship of the polymer in addition to diisocyanate concentration and post-polymerization conditions. Expanding the family of CUPE polymers increases the choices of biodegradable elastomers for tissue engineering applications.
Keywords: biodegradable, biomaterials, elastomers, polyesters, structure-property relations
Introduction
The application of semicrystalline biodegradable polymers such as poly(L-lactic acid) (PLLA), poly(glycolic acid) (PGA), and their co-polymers has enabled significant progress in the engineering of hard tissues.[1-7] In contrast, there has been unsatisfactory progress in the engineering of soft tissues including blood vessels, heart valves, bladder, and cartilage primarily due to the limited available soft and elastic biomaterials. When materials are placed in a mechanically demanding environment, there is a potential to cause irritation the surrounding tissues if the implant material is not mechanically compliant.[8] Ideally, biomaterials should possess unique chemical, physical, and mechanical properties to mimic the extracellular matrix of the targeted tissue being regenerated.[9-12] In addition, the importance and effects of adequate mechanical conditioning on the expression of appropriate cellular phenotypes and extracellular matrix deposition have been well documented in numerous studies,[13-15] and it has also been reported that dissected soft tissues like nerves and blood vessels are nearly 30% shorter than their in situ length,[15] which further emphasizes the need for materials specifically designed to be elastic in nature for soft tissue replacement therapy.
In order to fulfill this need of materials suited for soft tissue engineering, researchers have developed numerous biodegradable and biocompatible elastomers over the past few years.[8, 16-25] Poly(1,8-octanediol citrate) (POC) is one such biodegradable elastomer,[19, 20] which has demonstrated excellent cell/tissue-compatibility and hemocompatibility.[26] Although biphasic tubular scaffolds fabricated from POC are attractive for potential use as small diameter vascular grafts due to their elasticity and hemocompatibility,[27] poor load bearing capabilities and inadequate suture retention strengths limit the utility of POC.
To overcome the limitations of POC, we have recently reported on the synthesis and characterization of crosslinked urethane-doped polyesters (CUPE) network, a novel biodegradable and biocompatible elastomer with excellent mechanical properties.[28] The superior mechanical strength of CUPE motivated the development tubular scaffolds, and further evaluation of the graft hemocompatibility.[29] Our earlier work on CUPE focused on a single polyester soft segment composition and examined the material’s structure-property relationship as a function of the ratio of polyester to diisocyanate used in the synthesis.[28] In this current work, we have focused on investigating the effect of soft segment polymer composition on the resulting material and biological properties while keeping the ratio of polyester to diisocyanate constant. This study provides further evidence that CUPE is a highly flexible material whose properties can be controlled, and accurately matched to the target application by varying numerous reaction parameters.
Experimental Section
CUPE Synthesis
All chemicals, cell culture medium and supplements were purchased from Sigma–Aldrich (St. Louis, MO), except where mentioned otherwise and used as received. The different CUPE polymers were synthesized as per a three step procedure previously reported (Fig. 1).[28] Step 1 involves the polycondensation reaction of citric acid with different C4-C12 aliphatic diols: 1,4-butanediol (≥99%), 1,6-hexanediol (99%), 1,8-octanediol (98%), 1,10-decanediol (98%), or 1,12-dodecanediol (99%). Step 2 involves the introduction of urethane linkages to form the final CUPE pre-polymer, and step 3 involves a post-polymerization step for final thermal crosslinking. Briefly, citric acid and various diols were bulk polymerized in a three neck flask equipped with an inlet and outlet adapter, at 160-165 °C. A monomer ratio of 1:1.1 (acid to diol) was used for the all synthesis procedures. The temperature was reduced to 140 °C when the monomer mixture had melted, and the reaction was allowed to continue for 1 hour to obtain the soft segment pre-polymer. The resulting polymer was purified by dropwise precipitation in de-ionized (DI) water under constant stirring. The purified pre-polymer precipitate was carefully collected from the aqueous phase and lyophilized for 48 hours to remove traces of water. In step 2, the purified pre-polymer was dissolved in 1,4-dioxane to form a 3.0% solution (wt/wt), and the polymer solution was reacted with 1,6-hexamethylene diisocyanate (HDI) under constant stirring with stannous octoate as catalyst (0.1% wt). The reaction was carried out at 55 °C, and reaction completion was indicated by the absence of a diisocyanate peak in the FT-IR spectrum of the reaction mixture at 2267 cm−1. In step 3, the pre-CUPE synthesized in step 2 was cast in Teflon molds, air dried to remove all solvent, and then cured in an oven maintained at 80 °C for time periods ranging from 1-4 days, to obtain the final crosslinked urethane doped polyester.
Figure 1.

Representative CUPE synthesis schematic. The monomers citric acid and various aliphatic diols (4, 6, 8, 10, or 12 methylene units in length) underwent condensation polymerization to produce hydroxyl group capped pre-poly(diol citrates). Next, 1,6-hexamethylene diisocyanate was used to extend the pre-poly(diol citrates) to produce pre-CUPE. Pre-CUPE can be post-polymerized to obtain a crosslinked polyester network.
The different crosslinked urethane doped polyesters obtained were designated as CUPEX, where X denotes the number of carbon atoms in the diol used as the monomer for the reaction with citric acid. For example, CUPE10 indicates that 1,10-decanediol was the monomer used in the reaction. For all the different polymers obtained, the soft segment prepolymer: diisocyanate ratio of 1:1.2 was maintained.
Polymer Characterization
A Nicolet 6700 FT-IR spectrometer (Thermo Fisher Scientific) was used to obtain Fourier transform infrared (FT-IR) spectra of the different polymers synthesized. Briefly, a dilute solution (3.0% wt/wt) of the CUPE pre-polymer dissolved in 1,4-dioxane was cast onto a clean potassium bromide (KBr) crystal, and allowed to dry in vacuum for 6 hours prior to spectrum acquisition.
The glass transition temperature (Tg) of the different CUPE polymers synthesized was determined by differential scanning calorimetry (DSC) on a DSC2010 Differential Scanning Calorimeter (TA Instruments, New Castle, DE). The different polymer samples were subjected to two heating cycles. In the first cycle, the polymers were scanned to 150 °C, under nitrogen atmosphere with a step size of 10 °C/min and then cooled rapidly to −60 °C at a cooling rate of −40 °C/min. The second cycle comprised of heating the sample to 230 °C with a step size of 10 °C/min. All readings were collected during the second heating scan. The Tg value was determined from the middle of any step changes in the heat capacity from the second scan.
All mechanical testing was conducted on a MTS Insight 2 mechanical tester equipped with a 500 N load cell (Eden Prairie, MN). Mechanical testing was carried out to determine the effect of the different diols and different polymerization conditions on CUPE tensile properties. All films were cut into dog bone shape as per ASTM D412a standard (25 × 6 × 1.5mm, length × width × thickness). The samples were stretched at a deflection rate of 500 mm/min until break. The initial modulus was derived from the gradient of the curve at 10% elongation in the stress strain curve. A minimum sample size of n = 5 was maintained for all the films tested.
A density measurement kit (Mettler Toledo, Columbus, OH) was used to determine the density of the different CUPE polymers synthesized. The auxiliary fluid used in the density measurement experiments was DI water.
In order to determine the molecular weight between crosslinks and the crosslink density, the theory of rubber elasticity was used (Equation 1)[18-20, 24]:
| (1) |
where η is the number of moles of active network chains per unit volume, E0 is the initial modulus of the polymer, R is the universal gas constant, T is the absolute temperature, ρ is the density of the polymer and Mc is the relative molar mass between crosslinks.
Contact angle measurements were made on the different CUPE thin films using the sessile drop method. A KSV101 Optical Contact Angle and Surface Tension meter (KSV Instruments Inc, Helsinki, Finland) was used for obtaining the contact angle values. Thin films of the different CUPE pre-polymers were made by smearing 1 ml of a dilute polymer solution in 1,4-dioxane (3.0% wt/wt) on a clean glass slide. All readings were collected within 10 s after drop elution. A minimum of 10 readings was collected from different regions of the different thin films from each sample.
In Vitro Degradation
An accelerated in vitro degradation study of the different CUPE polymers was carried out in 0.01M sodium hydroxide (NaOH) solutions. Briefly, the polymer films were cut into 7 mm discs using a cork borer and the initial weights of these discs was recorded. The discs were transferred to clean test tubes containing 10 ml of 0.01M NaOH and incubated at 37 °C for pre-determined time intervals of 3, 6, 9 and 12 hours. A minimum of 8 specimens per time point was present for each polymer being tested. At each time interval, first the NaOH was aspirated out of the test tube and the polymer samples were washed thoroughly with DI water twice to remove any traces of NaOH. The polymer samples were then lyophilized for 72 hours to remove traces of water and weighed to obtain the final degraded weight corresponding to that time interval. The initial weight of the specimen and the degraded weight at a particular time interval were used to determine the rate of degradation of the polymer corresponding to that time interval using Equation (2):
| (2) |
where W0 = initial weight of polymer disc and Wt = final weight of the degrading polymer disc. A long-term degradation study was also conducted as per the aforementioned method using PBS as the degrading agent and CUPE8 as the representative polymer. The degradation of the polymer discs was monitored over a total period of 8 months.
In Vitro Cell Culture
The biocompatibility of the different CUPE polymers was evaluated by seeding the polymer films with two different cell lines and observing the morphology and proliferation of the cultured cells after 3 days post seeding. Human aortic smooth muscle cells (HASMCs) and the NIH 3T3 fibroblasts were selected as the model cells lines for this assessment. Briefly, the polymer samples were cut into 10 mm diameter discs using a cork borer. These discs were sterilized by a two-step process comprising of ultraviolet (UV) light treatment for 1 hour followed by immersion in 70% ethanol for 30 minutes. The sterilized discs were washed thoroughly with sterile PBS to remove any traces of alcohol and then allowed to dry completely prior to cell seeding. Cells were cultured in 75 cm2 culture flasks with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. For seeding, the cells were trypsinized, centrifuged and re-suspended in fresh complete culture medium. The volume of culture medium used to re-suspend the cell pellet was adjusted to obtain a seeding density of 3×105 cells/ml. 200μl of the cell suspension was evenly spread over the surface of each of the CUPE films being tested. After 1 hour of incubation at 37 °C, 5ml of complete culture medium was added to each of the petri dishes containing the cell seeded films. The cells were cultured on the films for 3 days during which the media was changed daily.
At the end of the culture period, the cells on the polymer discs were fixed by addition of 5 ml of 2.5% (wt/vol) gluteraldehyde-PBS solution. All samples were kept in the fixating medium for at least 2 hours. After fixation, the cells were sequentially dehydrated in a graded series of ethanol (50, 75, 90 and 100%) and lyophilized to remove any minute traces of water. The dehydrated cell seeded polymer discs were mounted on stubs, sputter coated with silver for 2 minutes and observed under a Hitachi S300N (Hitachi Corp, Tokyo, Japan) scanning electron microscope (SEM). Different images of cellular morphology were captured at different magnifications to gauge the pattern of cellular growth and proliferation on the polymer surfaces.
Foreign Body Response
CUPE8 was used as the representative polymer to study the extent of foreign body response incited by the presence of the polymer for a medium term implantation period of 8 weeks and a long-term implantation period of 6 months. CUPE8 scaffolds and PLLA control scaffolds were prepared by salt leaching with a salt size of 150-250 μm. The scaffolds were cut into discs (10mm × 1mm, diameter × thickness) using a cork borer and implanted subcutaneously in the back of healthy, female Sprague Dawley (SD) rats (Harlan Sprague Dawley, Inc., Indianapolis, IN). Animals were cared for in compliance with regulations of the animal care and use committee of the University of Texas at Arlington. Prior to implantation, the discs were sterilized by treatment with 75% ethanol for 1 hour followed by 1 hour of UV light treatment. A total of 8 rats were used for this study; 4 rats each for each time point of 8 weeks and 6 months. The rats were anesthetized using an isofluorane-oxygen mixture and the test samples were implanted in the upper back by blunt dissection.
At the end of each time period the rats tagged for that time period were sacrificed by CO2 asphyxiation. The implant and the surrounding tissue was collected and frozen in OCT embedding media (Polysciences Inc., Warrington, PA), at −80 °C for further histological analysis. The tissue blocks were sectioned into 10 μm sections using a cryostat and stained with hematoxylin and eosin (H&E) to examine the tissue responses. Stained section images were acquired at 10x magnifications using a Leica DMLP microscope fitted with a Nikon E500 camera (Nikon Corp., Japan). A minimum of three images was obtained from each section for analysis, and three sections were examined per animal sacrificed. Image J software was used to determine the fibrous capsule thickness in each of the analyses performed. In order to determine an average fibrous response, at least 25 readings of capsule thickness were obtained from different parts of the section images obtained and averaged.
Statistical Analysis
All data obtained are presented as the mean ± standard deviation. The statistical significance between independent data sets was calculated using Student’s two-tail t-test. A p < 0.05 value was used as a measure of significant difference.
Results and Discussion
Although elastomeric biomaterial development has been largely driven by the prerequisites of most soft tissues, elastic tissues vary greatly and cover a wide range of mechanical properties. For example, the mechanical properties of bladder tissue[30] vary greatly when compared to that of aortic tissue[31] or skin.[32] It has been well established that for successful tissue engineering one of the main requirements that need to be fulfilled is a similarity between the properties of the extracellular matrix of the target tissue and the replacement scaffold.[23, 33-36] Being able to control the properties of a polymer through multiple modalities is especially important to improve the utility of a bio-polymer for diverse soft tissue engineering applications.[37]
In our previous work,[28] we successfully synthesized and characterized the physical and biological properties of crosslinked urethane doped polyesters. From the results obtained, we were able to establish CUPE as a biodegradable, biocompatible, and strong yet soft elastomer. Although we concentrated primarily on the effects of different molar ratios of isocyanate to pre-polymer, we briefly demonstrated that the polymer properties could be manipulated by varying the diol component as well. The focus on controlling the material performance through various diols motivated us to explore the development of a family of such elastomers, whose diols varied in their methylene content.
FT-IR spectra obtained for all the synthesized polymers are depicted in Figure 2. The absence of an isocyanate peak at 2267 cm−1 was characteristic of all the spectra obtained, which indicated that the synthesis of the different CUPE polymers consumed all the diisocyanate in the reaction process. The peaks centered at 1733 cm−1 were assigned to carbonyl groups of the free carboxylic acid functional groups on the citric acid backbone and carbonyl groups of the ester bonds in the polymer backbone. Peaks between 2931 cm−1 and 2919 cm−1 were assigned to the methylene units in the polymer backbone.[19, 38] FT-IR also confirmed the incorporation of the isocyanate in the polymer backbone by urethane bond formation. All the CUPE polymers had sharp peaks at 1670 cm−1 and 1560 cm−1 which were attributed to amide I and amide II vibrations, respectively. A narrow shoulder peak at 3350 cm−1 also suggests the presence of urethane linkages in the polymer chains.[39]
Figure 2.
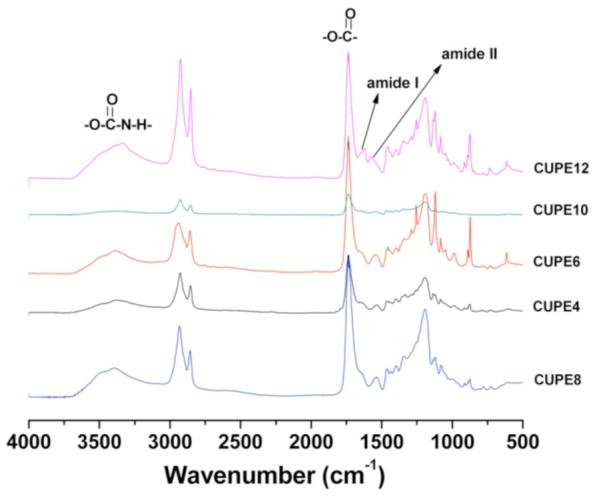
FT-IR spectra of the different crosslinked urethane doped polyester pre-polymers synthesized with different diols. The pre-polymers include pre-CUPE4, pre-CUPE6, pre-CUPE8, pre-CUPE10 and pre-CUPE12.
Figure 3 depicts the glass transition temperature of the different CUPE polymers, as determined by DSC. No obvious hard segment transitions were observed in the DSC thermograms indicating a low degree of microphase separation in all the CUPE polymers examined.[40-42] Furthermore, all thermograms lacked any crystallization and melting peaks, indicating the absence of hard segment crystallization.[43] The glass transition temperature of the polymers varied as an inverse function of the length of the diol component. CUPE12, which had the longest diol chain, had the lowest glass transition temperature (Tg = −3.22 °C), whereas CUPE4, which had the smallest diol component, demonstrated a higher value of Tg (27.47 °C). CUPE8 which had a Tg value of 3.06 °C[28] also supports this trend. This increase in glass transition temperature with decreasing diol length may be explained by the segmented architecture of urethanes wherein strong hydrogen bonding between the polymer chains may function as physical crosslinks.[44] Increasing the length of the diol may serve to reduce the crosslink density by increasing the separation between the urethane groups in the polymer backbone, thus increasing the polymer chain mobility and subsequently lowering Tg. A decrease in the molecular weight of the soft segment would also promote an increase in the hard segment effect on the chain mobility as a result of phase mixing,[45] thus increasing the glass transition temperature of the CUPEs synthesized with smaller diols.
Figure 3.
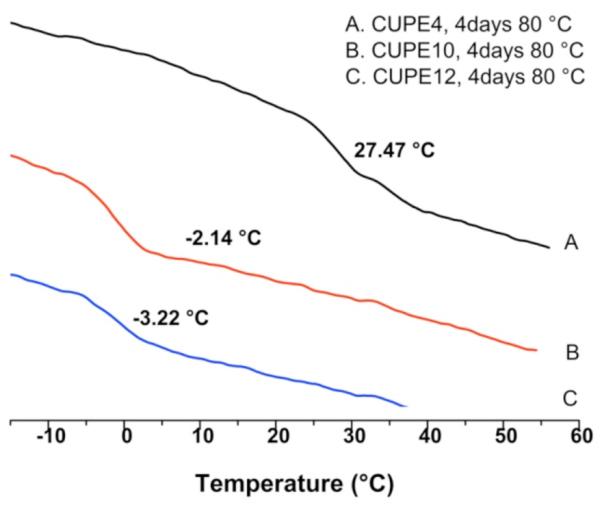
Effect of different diols on the DSC thermograms of the CUPE polymers. All samples tested were post-polymerized for 4 days at 80 °C prior to analysis. A two-step heating cycle was used to generate DSC thermograms for the different polymers.
Assuming the different polymers recovered completely after deformation, the effect of diol length on the crosslink density of the crosslinked polyester network was further examined using the theory of rubber elasticity, which has been used in previous studies to evaluate other biodegradable polyester elastomers.[18-20, 24] Data represented in Table 1 indicate that formulations with increasing diol length did indeed reduce the ester bond crosslink density allowed for by the citric acid component of the CUPE polymer. Under similar post-polymerization conditions (2 days at 80 °C) CUPE6 had the highest crosslink density of all the polymers evaluated (η = 1142.12 ± 120.79 mol/m3) while CUPE10 had the lowest crosslink density (η = 783.07 ± 167.52 mol/m3). Increasing the post-polymerization duration to 4 days, also led to an increase in crosslink density due to the formation of more inter-chain ester bond crosslinking (CUPE10_4D η = 978.31 ± 157.26 mol/m3). As a result of the growing length of methylene units in the polymer backbone with the incorporation of longer diols and subsequent increase in the molecular weight between crosslinks, the specific density of the different CUPE polymers decreased with increasing diol size (Table 1).[19]
Table 1.
| Sample | Density (g/cm3) |
Young’s Modulus (MPa) |
Tensile Strength (MPa) |
n (mol/m3) |
Mc (g/mol) |
|---|---|---|---|---|---|
| CUPE6_2D | 1.30±0.05 | 8.00±1.22 | 9.56±2.00 | 1144.12±120.79 | 1148.21±140.97 |
| CUPE8_2D | 1.24±0.01 | 7.02±0.85 | 17.38±2.34 | 951.40±123.59 | 1320.47±187.36 |
| CUPE10_2D | 1.15±0.02 | 6.02±1.07 | 24.19±2.04 | 783.07±167.52 | 1522.97±351.14 |
| CUPE10_4D | 1.17±0.02 | 7.22±0.90 | 38.36±1.69 | 978.31±157.26 | 1130.972±145.74 |
All the CUPE polymers displayed break elongations exceeding 100% and elastomeric properties: full recovery of the sample dimensions after removal of applied stress. Increasing the length of the diol resulted in stronger polymers (Fig. 4A). Under similar polymerization conditions of 2 days in an oven maintained at 80 °C, CUPE10 had the highest peak stress (38.36 ± 1.69 MPa) and CUPE6 was the weakest polymer (9.56 ± 2.00 MPa). This may be attributed to an increase in the molecular weight of the polymer due to the incorporation of higher molecular weight diols. The initial Young’s modulus of the different polymers varied from 6.02 ± 1.07 MPa to 8.00 ± 1.22 MPa, with CUPE6 being the most stiff (Fig. 4B), which could be attributed to the higher crosslink density in CUPE6 thereby necessitating higher initial stress to cause deformation.[46, 47] The higher crosslink density of polymers with lower number of methylene units in the diol also results in the polymers being more brittle, as can be seen from the increase in the final strain with increasing diol size (Fig. 4C). In addition to diol concentration, increasing polymerization conditions resulted in stronger, stiffer and more brittle polymers, which corresponds to our earlier results.[28] In order to assess the material’s elastomeric properties in detail, hysteresis cycle was performed at room temperature. As a typical example, hysteresis cycle of CUPE8 films after the 10th elongation is shown in Figure 4D: 50% elongation and back at room temperature. CUPE8 samples showed excellent recovery with no loss of energy, which could be attributed to the strong intermolecular cohesive energy between the crosslinked polyester network components.
Figure 4.
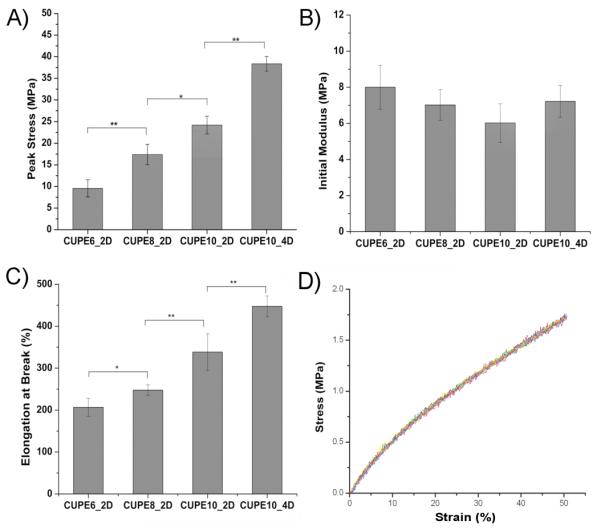
Mechanical properties of the different CUPE polymers. All the polymers shown were polymerized at 80 °C for 2 days except for CUPE10_4D which was polymerized for 4 days at 80 °C. Effect of diol component and post polymerization conditions on the peak stress (A), initial modulus (B), break strain (C), and hysteresis (D) are shown. ** corresponds to p<0.01, * denotes p<0.05. A minimum sample size of N = 5 was used for all tests.
The influence of the monomers on the surface properties of the CUPE polymers is summarized in Figure 5. As expected, using a more hydrophobic diol in the synthesis of CUPEs resulted in more hydrophobic polymers. CUPE4 had the most hydrophilic nature with an initial water in air contact angle value of 77.00 ± 1.28°, while CUPE12 was most hydrophobic, with a contact angle value of 98.91 ± 1.34°.
Figure 5.
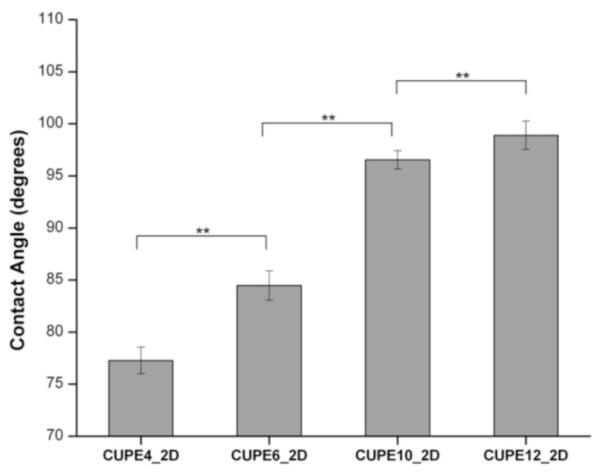
Initial contact angles of the different CUPE pre-polymers. Readings were taken for each specimen and averaged (N = 8). Contact angle was observed to increase by increasing the number of methylene units in the diol component. ** represents p<0.01.
Swelling studies indicated that the choice of monomer diol also affected the bulk properties of the CUPE family of polymers. CUPE4 and CUPE12 had the highest (168.50 ± 24.38%) and lowest (10.26 ± 2.83%) degrees of swelling in PBS, respectively, as depicted in Figure 6. The bulk swelling of the different family polymers decreased with increasing number of carbon atoms in the diol monomer used in the synthesis due to the hydrophobic nature of the larger methylene diol. Thus, one would expect that with a higher degree of crosslinking the CUPE polymers with lower number of methylene units would exhibit the lowest degree of swelling. However, the results demonstrate that the hydrophobicity of the diol plays a more predominant role than the crosslink density in determining water uptake and hence swelling, which is in agreement with published literature.[46]
Figure 6.
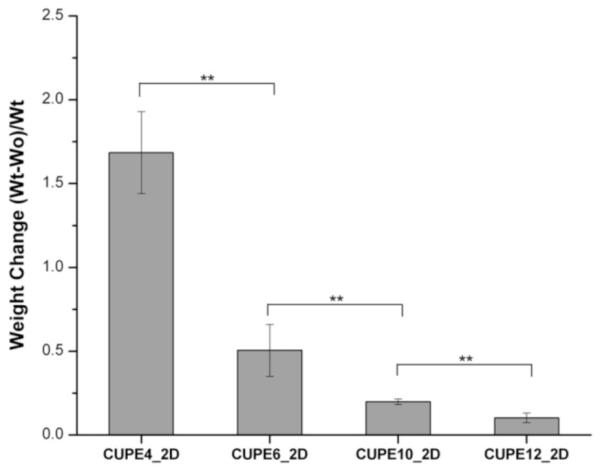
Bulk water uptake and swelling of the different CUPE pre-polymers. A minimum sample size of N = 8 was used for each polymer tested. Water uptake was found to increase with incorporation of more hydrophilic diols in polymer chain. ** denotes p<0.01.
This phenomenon was also evident in the accelerated degradation studies conducted on the different CUPE polymers in 0.05 M sodium hydroxide solutions. As seen from Figure 7A, it can be noted that the intrinsic nature of the polymers composed of more hydrophobic diols degraded slower than those containing more hydrophilic monomers. CUPE4 underwent complete degradation within 9 hours, whereas other polymers were still present at the final time point of 12 hours. In contrast, CUPE12 demonstrated the slowest degradation profile, with 96.68 ± 0.97% of the polymer still remaining at the 12-hour time point. The increased water uptake of the hydrophilic CUPE polymers would provide greater access to the hydrolytically labile ester bonds in the polymer backbone resulting in faster degradation through hydrolysis to yield to yield the original monomers, which can be metabolized and eliminated from the body. In addition to NaOH degradation, a long-term degradation study was also conducted on CUPE8 in PBS (Fig. 7B). The results indicate that CUPE8 degraded steadily for 8 months, with only 37.99 ± 7.39% of the polymer remaining at the end of the last time point in the study.
Figure 7.

Degradation studies of different CUPE polymers in 0.05M NaOH solution at room temperature. All the different polymers used in the PBS degradation study were polymerized at 80 °C for 2 days and different only in their diol component (A). Long term degradation study of CUPE8 in PBS. The polymer was polymerized at 80 °C for 4days prior to study. Sample size N = 8 (B).
The initial biocompatibility of the different CUPE polymers was assessed by seeding NIH 3T3 fibroblasts and Human Aortic Smooth muscle cells (HASMCs) on the polymer films and observing the morphology of the cells post adhesion. Photomicrographs of the seeded films (Fig. 8 and 9) indicate that both the cell types used were able to adhere to the CUPE polymers and express a normal phenotype with higher cell densities on CUPE polymers synthesized from longer length diols.[19, 28]
Figure 8.
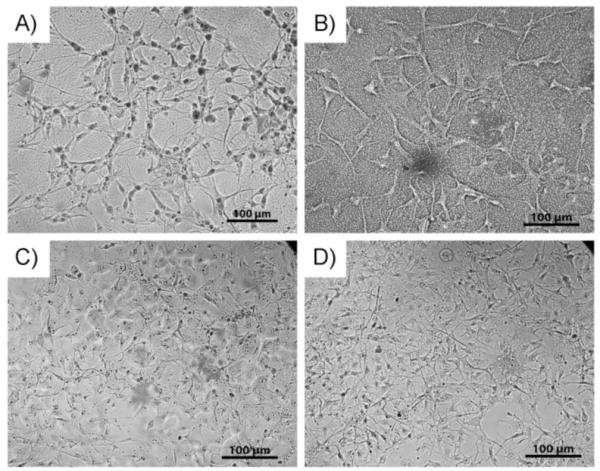
SEM images of NIH 3T3 fibroblasts growing on surface of CUPE films. Cells were allowed to grow and proliferate for 3 days post seeding. Seeding density per film = 3×105 cells. Different films used include (A) CUPE4 (B) CUPE6 (C) CUPE10 and (D) CUPE12.
Figure 9.
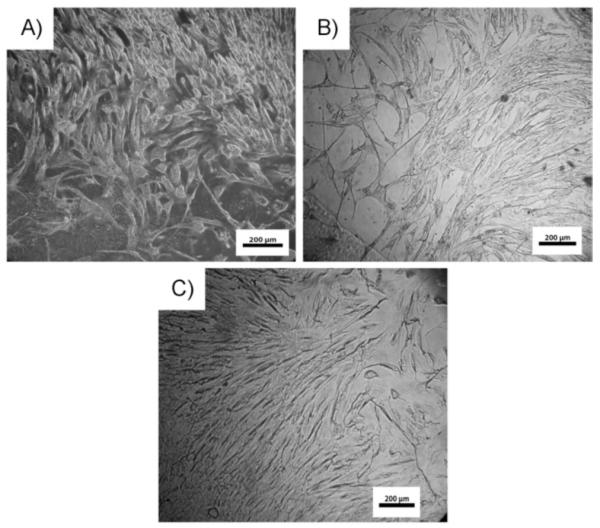
SEM images of HASMCs fibroblasts growing on surface of CUPE films. Cells were allowed to grow and proliferate for 3 days post seeding. Seeding density per film = 3×105 cells. Different films used include (A) CUPE6 (B) CUPE10 and (C) CUPE12. CUPE4 was not used because of film dissolution over long culture time.
In our previous report,[28] we presented the foreign body response over a relatively short period of 1 week and 4 weeks with CUPE8 scaffolds as the representative polymer. Figure 10 represents the foreign body response to CUPE8 scaffolds over a longer time period of 8 weeks and 24 weeks with PLLA as the control. Although significant degradation could be observed in both at the later time period, CUPE and PLLA scaffolds were still present at 24 weeks. The absence of necrosis in and around the implants indicated a healthy healing response to both the materials. From Figure 10, it can be seen that even at 8 weeks 100% cellular infiltration was achieved in the CUPE and PLLA scaffolds. Both the scaffolds indicated the presence of a thin, non-uniform fibrous capsule. At 24 weeks, no tissue necrosis was observed in all test animals. The capsule could not be distinguished from the infiltrating cells. The scaffolds also lost their shape and showed reduced size compared to 8 weeks samples for both materials tested (Fig. 10). These results indicate that, in addition to demonstrating better acute inflammatory responses than PLLA, CUPE also undergoes biodegradation in vivo and does not incite a significant long-term chronic inflammatory response. Although the gross inflammatory response was presented in this work, future studies will be focused on characterizing the specific cell types as part of the inflammatory response surrounding the implant.
Figure 10.
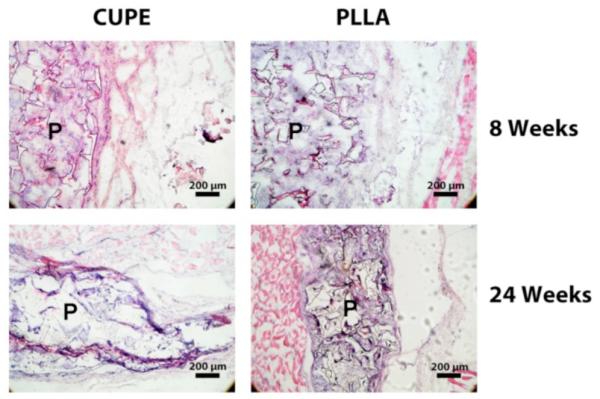
Host responses to CUPE and PLLA (control) implanted subcutaneously in Sprague-Dawley rats. Histology analysis (H&E staining) for the long-term (8 weeks and 24 weeks) implantation demonstrated that CUPE was degradable in vivo and did not elicit significant chronic inflammation. No tissue necrosis was observed in all test animals. P indicates the polymer regions.
Conclusion
A family of CUPE polymers with different diol components was synthesized and characterized. Varying the diol component was found to affect the different physical properties of the CUPE family members without significantly affecting the polymer performance with respect to cell/tissue-compatibility. We have therefore established the diol component as an important parameter in controlling the structure-property relationship of the polymer in addition to diisocyanate concentration and post-polymerization conditions. This demonstrates the versatility with which the properties of CUPEs can be varied, thereby rendering this polymer family attractive for a wide variety of tissue engineering applications.
Acknowledgements
This work was supported in part by R21 award EB009795 from the National Institute of Biomedical Imaging and Bioengineering (NIBIB) (to J.Y.), a National Science Foundation (NSF) CAREER award 0954109 (to J.Y.), and a High Impact/High Risk grant RP110412 from Cancer Prevention & Research Institute of Texas (CPRIT). The authors also would like to thank Jinming Gao at University of Texas Southwestern Medical Center for his help on DSC measurements.
References
- [1].Pineda LM, Busing M, Meinig RP, Gogolewski S. J Biomed Mater Res. 1996;31:385. doi: 10.1002/(SICI)1097-4636(199607)31:3<385::AID-JBM13>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [2].Meinig RP, Rahn B, Perren SM, Gogolewski S. J Orthop Trauma. 1996;10:178. doi: 10.1097/00005131-199604000-00006. [DOI] [PubMed] [Google Scholar]
- [3].Meinig RP, Buesing CM, Helm J, Gogolewski S. J Orthop Trauma. 1997;11:551. doi: 10.1097/00005131-199711000-00002. [DOI] [PubMed] [Google Scholar]
- [4].Zhang R, Ma PX. J Biomed Mater Res. 1999;45:285. doi: 10.1002/(sici)1097-4636(19990615)45:4<285::aid-jbm2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- [5].Zhang R, Ma PX. J Biomed Mater Res. 1999;44:446. doi: 10.1002/(sici)1097-4636(19990315)44:4<446::aid-jbm11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- [6].Burg KJ, Porter S, Kellam JF. Biomaterials. 2000;21:2347. doi: 10.1016/s0142-9612(00)00102-2. [DOI] [PubMed] [Google Scholar]
- [7].Borden M, Attawia M, Laurencin CT. J Biomed Mater Res. 2002;61:421. doi: 10.1002/jbm.10201. [DOI] [PubMed] [Google Scholar]
- [8].Yang J, Zhang Y, Gautam S, Liu L, Dey J, Chen W, Mason RP, Serrano CA, Schug KA, Tang L. Proc Natl Acad Sci U S A. 2009;106:10086. doi: 10.1073/pnas.0900004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang S, Leong KF, Du Z, Chua CK. Tissue Eng. 2001;7:679. doi: 10.1089/107632701753337645. [DOI] [PubMed] [Google Scholar]
- [10].Lukashev ME, Werb Z. Trends Cell Biol. 1998;8:437. doi: 10.1016/s0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- [11].Ingber DE. FASEB J. 2006;20:811. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- [12].Stegemann JP, Hong H, Nerem RM. Applied Physiology. 2005;98:2321. doi: 10.1152/japplphysiol.01114.2004. [DOI] [PubMed] [Google Scholar]
- [13].Stegemann JP, Nerem RM. Annals of Biomedical Engineering. 2003;31:391. doi: 10.1114/1.1558031. [DOI] [PubMed] [Google Scholar]
- [14].MacKenna D, Summerour SR, Villarreal FJ. Cardiovasc Res. 2000;46:257. doi: 10.1016/s0008-6363(00)00030-4. [DOI] [PubMed] [Google Scholar]
- [15].Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Nat Rev Mol Cell Biol. 2002;3:349. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- [16].Guan J, Sacks MS, Beckman EJ, Wagner WR. J Biomed Mater Res. 2002;61:493. doi: 10.1002/jbm.10204. [DOI] [PubMed] [Google Scholar]
- [17].Guan J, Sacks MS, Beckman EJ, Wagner WR. Biomaterials. 2004;25:85. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- [18].Wang Y, Ameer GA, Sheppard BJ, Langer R. Nat Biotechnol. 2002;20:602. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- [19].Yang J, Webb AR, Pickerill SJ, Hageman G, Ameer GA. Biomaterials. 2006;27:1889. doi: 10.1016/j.biomaterials.2005.05.106. [DOI] [PubMed] [Google Scholar]
- [20].Yang J, Webb AR, Ameer GA. Advanced Materials. 2004;16:511. [Google Scholar]
- [21].Li XT, Sun J, Chen S, Chen G. Journal of Biomedical Materials Research Part A. 2008;87A:832. doi: 10.1002/jbm.a.31890. [DOI] [PubMed] [Google Scholar]
- [22].Bettinger CJ, Bruggeman JP, Borenstein JT, Langer RS. Biomaterials. 2008;29:2315. doi: 10.1016/j.biomaterials.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gyawali D, Nair P, Zhang Y, Tran RT, Zhang C, Samchukov M, Makarov M, Kim HK, Yang J. Biomaterials. 2010;31:9092. doi: 10.1016/j.biomaterials.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tran RT, Thevenot P, Gyawali D, Chiao JC, Tang L, Yang J. Soft Matter. 2010;6:2449. doi: 10.1039/C001605E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tran RT, Thevenot P, Zhang Y, Tang L, Yang J. Materials. 2010;3:1375. doi: 10.3390/ma3021375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Motlagh D, Allen J, Hoshi R, Yang J, Lui K, Ameer GA. Journal of Biomedical Materials Research Part A. 2007;82A:907. doi: 10.1002/jbm.a.31211. [DOI] [PubMed] [Google Scholar]
- [27].Yang J, Motlagh D, Webb AR, Ameer GA. Tissue Eng. 2005;11:1876. doi: 10.1089/ten.2005.11.1876. [DOI] [PubMed] [Google Scholar]
- [28].Dey J, Xu H, Shen J, Thevenot P, Gondi SR, Nguyen KT, Sumerlin BS, Tang L, Yang J. Biomaterials. 2008;29:4637. doi: 10.1016/j.biomaterials.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dey J, Xu H, Nguyen KT, Yang J. Journal of Biomedical Materials Research Part A. 2010;95A:361. doi: 10.1002/jbm.a.32846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dahms SE, Piechota HJ, Dahiya R, Lue TF, Tanagho EA. Br J Urol. 1998;82:411. doi: 10.1046/j.1464-410x.1998.00748.x. [DOI] [PubMed] [Google Scholar]
- [31].Lee MC, Haut RC. J Biomech. 1992;25:925. doi: 10.1016/0021-9290(92)90232-p. [DOI] [PubMed] [Google Scholar]
- [32].Clark JA, Cheng JC, Leung KS. Burns. 1996;22:443. doi: 10.1016/0305-4179(96)00038-1. [DOI] [PubMed] [Google Scholar]
- [33].Langer R, Vacanti JP. Science. 1993;260:920. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- [34].Sell SA, Francis MP, Garg K, McClure MJ, Simpson DG, Bowlin GL. Biomedical Materials. 2008;3:11. doi: 10.1088/1748-6041/3/4/045001. [DOI] [PubMed] [Google Scholar]
- [35].Zong X, Bien H, Chung CY, Yin L, Fang D, Hsiao BS, Chu B, Entcheva E. Biomaterials. 2005;26:5330. doi: 10.1016/j.biomaterials.2005.01.052. [DOI] [PubMed] [Google Scholar]
- [36].Berry CC, Campbell G, Spadiccino A, Robertson M, Curtis AS. Biomaterials. 2004;25:5781. doi: 10.1016/j.biomaterials.2004.01.029. [DOI] [PubMed] [Google Scholar]
- [37].Tran RT, Zhang Y, Gyawali D, Yang J. Recent Patents on Biomedical Engineering. 2009;2:216. [Google Scholar]
- [38].Jayakumar R, Lee YS, Nanjundan S. Journal of Applied Polymer Science. 2003;90:3488. [Google Scholar]
- [39].Guan J, Wagner WR. Biomacromolecules. 2005;6:2833. doi: 10.1021/bm0503322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li F, Hou J, Zhu W, Zhang X, Xu M, Luo X, Ma D, Kim K. Journal of Applied Polymer Science. 1996;62:631. [Google Scholar]
- [41].Garrett JT, Runt J. Macromolecules. 2000;33:6353. [Google Scholar]
- [42].Chu B, Gao T, Li Y, Wang J, Desper CR, Byrne CA. Macromolecules. 1992;25:5724. [Google Scholar]
- [43].Nakamae K, Nishino T, Asaoka S, Sudaryanto International Journal of Adhesion and Adhesives. 1996;16:233. [Google Scholar]
- [44].Skarja GA, Woodhouse KA. Journal of Applied Polymer Science. 2000;75:1522. [Google Scholar]
- [45].Gorna K, Gogolewski S. J Biomed Mater Res. 2002;60:592. doi: 10.1002/jbm.10100. [DOI] [PubMed] [Google Scholar]
- [46].Muggli DS, Burkoth AK, Anseth KS. J Biomed Mater Res. 1999;46:271. doi: 10.1002/(sici)1097-4636(199908)46:2<271::aid-jbm17>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- [47].Lee SY, Lee JC, Kim BK. Polymer International. 1997;42:67. [Google Scholar]