

Abstract
Purpose
To determine whether lacrimal and salivary gland nerves of an animal model of Sjögren's syndrome, the MRL/lpr mouse, are able to release acetylcholine. The second purpose was to determine whether activation of the lacrimal gland nerves of the MRL/lpr mouse leads to protein secretion.
Methods
Total saliva was collected for 10 minutes from the oral cavity of male and female MRL/lpr and MRL/+ mice, after intraperitoneal stimulation with pilocarpine and isoproterenol. Lacrimal and salivary gland lobules prepared from 18-week-old MRL/lpr and MRL/+ mice were incubated in the presence of depolarizing KCl (75 mM) solution. Acetylcholine release and peroxidase secretion (a protein secreted by the lacrimal gland) were measured using a spectrofluorometric assay.
Results
Female, but not male, MRL/lpr mouse salivary glands were hyper-responsive to in vivo injection of secretagogues. These mice produced significantly higher amounts of saliva than did age-matched MRL/+ mice. Lacrimal and salivary gland nerves from 18-week-old MRL/+ mice released acetylcholine in response to a depolarizing KCl solution. In contrast, nerves in glands from 18-week-old MRL/lpr mice did not increase acetylcholine release in response to the depolarizing solution. Moreover, lacrimal glands from 18-week-old MRL/+ mice were able to secrete peroxidase in response to a depolarizing KCl solution, whereas those from 18-week-old MRL/lpr could not. This was not due to a defect in the secretory process, because addition of an exogenous secretagogue elicited peroxidase secretion from 18-week-old MRL/lpr as well as MRL/+ mice lacrimal glands.
Conclusions
The results show that activation of nerves of lacrimal and salivary glands infiltrated with lymphocytes does not increase the release of neurotransmitters, which results in impaired secretion from these glands. (Invest Ophthalmol Vis Sci.
Lacrimal and salivary glands are exocrine tissues that have the main function of secreting tears and saliva, respectively.1–3 These glands consist of acini, ducts, nerves, myoepithelial cells, mast cells, and plasma cells. Approximately 80% of each gland is acini, which secrete electrolytes, water, and proteins.1,4 It is well established that lacrimal and salivary gland fluid secretion is under neural control.1,4,5 For example, reflexes from the ocular surface and optic nerve, as well as from higher centers of the brain, stimulate lacrimal gland secretion using parasympathetic and sympathetic efferent pathways.2,3 Parasympathetic and sympathetic nerves innervate the acinar cells, duct cells, and blood vessels of the lacrimal and salivary glands.1,6 – 8 The parasympathetic nerves contain the neurotransmitter acetylcholine, which acts through cholinergic muscarinic receptors, and vasoactive intestinal peptide. Sympathetic nerves contain norepinephrine, which acts through adrenergic receptors.
A decrease in lacrimal and salivary gland secretion is a primary cause of dry eye and dry mouth. Sjögren's syndrome is the leading cause of the aqueous tear-deficient type of dry eye.9–11 It is an autoimmune disease that occurs almost exclusively in females (>90%). This syndrome is associated with an extensive lymphocytic infiltration of the lacrimal and salivary glands and destruction of epithelial cells.9–11 To date there is no cure for this disease. Moreover, the exact cause of Sjögren's syndrome is largely unknown but may involve numerous factors including those of viral, endocrine, neural, genetic, and environmental origin.10,12,13
Several strains of mice have been proposed as models of human Sjögren's syndrome.14–17 Each of these strains has its advantages and disadvantages. Nonobese diabetic (NOD) mice were first used as a model for type I diabetes. Subsequently, it was found that in these mice, lesions develop in the salivary and lacrimal glands that resemble human Sjögren's syndrome.18 However, the incidence of the lymphocytic accumulation in lacrimal glands in these mice is more frequent and severe in male than in female mice, which is the opposite of that which occurs in humans.19 The New Zealand black (NZB) and hybrid New Zealand black/New Zealand white (NZB/NZW) mice described by Kessler20 and the MRL/l mice and congenic MRL/Mp-lpr/lpr mice first described by Murphy21 were used as models to study another autoimmune disease, systemic lupus erythematosus. Later, it was found that these animals had coexisting Sjögren's syndrome. NZB/NZW and MRL/lpr mice show spontaneous development of a mononuclear cell infiltration of the salivary and lacrimal glands and other organs. The disease in both animals occurs almost exclusively in females (.90%) and develops in an age-dependent manner.14,15,19 MRL/lpr mice, compared with NZB/NZW mice, have more pronounced and destructive mononuclear infiltrates in the lacrimal and salivary glands.16,19
The precise mechanism(s) responsible for the decrease in tear and saliva secretion in Sjögren's syndrome is unknown.10,11,13 It is believed that the dry eye and dry mouth in this disease are due to a progressive lymphocytic infiltration of the lacrimal and salivary glands, an immune-mediated destruction of the epithelial cells, and a consequent decline in tear and saliva production.10,11,13 However, adjacent to the immuno pathologic lesions in Sjögren's syndrome, the lacrimal and salivary gland tissues appear to contain normal acinar and ductal epithelia that should be able to secrete enough tears and saliva. One hypothesis could be that a decrease in lacrimal gland innervation and/or an alteration of the signaling pathways of the remaining epithelia accounts for the decreased function of the lacrimal and salivary glands associated with Sjögren's syndrome.
In previous studies, using the MRL/lpr mice, we found that the lymphocytic infiltration of the lacrimal and salivary glands did not alter the parasympathetic, sympathetic, and sensory innervation of the remaining epithelial cells in these tissues.22 We also found that acinar cells isolated from lacrimal and salivary glands of diseased MRL/lpr animals were hyper-responsive to exogenous cholinergic and adrenergic stimulation when compared with age-matched control MRL/+ animals.23 We hypothesized that the exocrine tissues from diseased animals behave as denervated ones—that is, the remaining nerves are not able to release their neurotransmitters, leading to increased responsiveness of these tissues to exogenous stimulation. Another consequence of the impaired release of neurotransmitters is a loss of tear and saliva secretion from lacrimal and salivary glands that leads to dry eye and dry mouth. The purpose of the present studies was to test for this hypothesis.
Materials and Methods
Chemicals
Pilocarpine, isoproterenol, phenylephrine, neostigmine, atropine, choline oxidase, acetylcholinesterase, and horseradish peroxidase were from Sigma (St. Louis, MO). An acetylcholine assay kit (Amplex Red) was obtained from Molecular Probes (Eugene, OR).
Animals
Female and male MRL/MpJ-Fas-lpr/lpr (MRL/lpr) and MRL/MpJ+/+ (MRL/+) were purchased from Jackson Laboratories (Bar Harbor, ME). They were maintained in constant-temperature rooms with fixed light-dark intervals of 12 hours' length and were fed ad libitum. All experiments were in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Schepens Eye Research Institute Animal Care and Use Committee.
Histopathology
Lacrimal and submandibular glands were removed from euthanatized mice and fixed in 4% methanol-free formaldehyde in phosphate-buffered saline (PBS, containing in millimolar: 137 NaCl, 2.7 KCl, 6.5 Na2HPO4, and 1.5 KH2PO4) for 24 hours at 4°C. After thorough rinsing with PBS, the tissue was dehydrated in graded alcohol to 95%. The tissue was then infiltrated and embedded in resin (Historesin; Leica, Deerfield, IL). Sections (3 μm) were placed on slides and allowed to dry at 60°C. Slides were then stained with hematoxylin and eosin to check for lymphocytic infiltration of the lacrimal and salivary glands.
Measurement of Salivary Flow
Total saliva was collected from 4-, 9-, 14-, and 18-week-old MRL/lpr and MRL/+ mice, mildly anesthetized with a diluted mixture of ketamine and xylazine, as described by Hu et al.24 Secretion of saliva was stimulated by intraperitoneal injection (0.1 ml) of a cocktail containing 0.20 mg isoproterenol (a β-adrenergic agonist) plus 0.05 mg pilocarpine (a cholinergic agonist) per 100 g body weight. The doses of isoproterenol and pilocarpine used are optimal for stimulating saliva secretion.24 Five minutes after the injection of the drugs, saliva was collected continuously over a 10-minute period from the oral cavity by a micropipette and placed in microcentrifuge tubes. Care was taken to collect saliva from the same place in the oral cavity. After a brief centrifugation to remove bubbles, the volume of saliva was determined using a micropipette. In some experiments, total salivary protein was determined by the method of Bradford,25 using bovine serum albumin as the standard.
Potassium-Evoked Release of Acetylcholine and Secretion of Peroxidase
Lacrimal and submandibular glands were removed from 18-week-old MRL/lpr and MRL/+ mice. Tissue was cut into small lobules (~2 mm in diameter), placed in cell strainers, and incubated at 37°C in Krebs-Ringer bicarbonate buffer (KRB, containing in millimolar: 120 NaCl, 5 KCl, 1 CaCl2, 1.2 MgCl2, 1.2 KH2PO4, and 25 NaHCO3) supplemented with 10 mM HEPES and 5.5 mM glucose (pH 7.4). The cell strainers containing lobules were transferred into fresh KRB solution every 20 minutes for a total of 60 minutes. The lobules were then incubated for 20 minutes in a total volume of 0.8 ml in normal KRB (referred to as spontaneous release) then in depolarizing KRB (evoked release) solution where the concentration of KCl was increased to 75 mM and that of NaCl was decreased to 55 mM to maintain isotonicity. Both normal and depolarizing KRB solutions contained neostigmine bromide (0.1 mM), an inhibitor of acetylcholine esterase, to allow the accumulation of acetylcholine in the media. In some experiments, lacrimal gland lobules were further incubated for 20 minutes in 0.8 ml of normal KRB containing phenylephrine (an α1-adrenergic agonist, 10−4 M). After incubation, the media were collected and centrifuged to remove debris. The lobules were homogenized in 10 mM Tris-HCl (pH 7.5). The amounts of acetylcholine, choline, and peroxidase in the media and tissue homogenate were determined using a spectrofluorometric assay.
Spectrofluorometric Assay of Acetylcholine, Choline, and Peroxidase
The amount of acetylcholine, choline, and peroxidase were measured using the acetylcholine assay kit (Amplex Red; Molecular Probes). This kit measures the amount of hydrogen peroxide (which in the presence of horseradish peroxidase leads to the oxidation of Amplex Red) produced through the oxidation of choline. For the measurement of acetylcholine and choline, 0.1 ml of media and tissue homogenate were spotted in duplicate onto 96-well microplates. An acetylcholine and choline standard curve was used in each experiment. In each well, 0.1 ml of assay buffer (50 mM Tris-HCl, pH 7.5) containing 0.2 M Amplex Red reagent, 2 U/ml horseradish peroxidase, 0.2 U/ml choline oxidase, and 10 U/ml acetylcholinesterase was added. To measure the amount of choline, acetylcholinesterase was omitted from the assay buffer. After incubation, the fluorescence was determined in a fluorescence microplate reader (model FL600; Bio-Tek, Winooski, VT) using 530 nm excitation wavelength and 590 nm emission wavelength. The concentration of choline and acetylcholine was determined using the software provided by the manufacturer (KC4; Bio-Tek).
For the measurement of peroxidase, 0.1 ml of media and 0.01 ml of tissue homogenate were spotted in duplicate onto 96-well microplates. To each well was added 0.1 ml of assay buffer containing 0.2 M Amplex Red reagent and 0.2 M hydrogen peroxide. After incubation, the fluorescence was determined as described. The amount of secreted peroxidase was expressed as a percentage of the total: (peroxidase in media/peroxidase in media + peroxidase in tissue) × 100.
Data Presentation and Statistical Analysis
Data are expressed as means ± SEM. The data were statistically analyzed using Student's t-test for paired and unpaired values. Values of P < 0.05 were considered to be significant.
Results
Effect of Disease on Salivary Flow
To determine whether salivary glands of MRL/lpr mice are hyper-responsive in vivo to exogenous stimulation, secretion of saliva was stimulated by intraperitoneal injection of isoproterenol and pilocarpine. Saliva was then collected over a 10-minute period from 4-, 9-, 14-, or 18-week-old female MRL/lpr and MRL/+ mice. As shown in Figure 1, the volume of saliva collected from 4-week-old MRL/lpr mice was not different from that collected from 4-week-old MRL/+ mice. In contrast, the amount of saliva collected from 9-, 14-, and 18-week-old MRL/lpr was significantly higher compared with age-matched MRL/+ mice (Fig. 1). Figure 2 shows that the amount of total protein was also significantly higher in saliva collected from 18-week-old MRL/lpr mice compared with age-matched MRL/+ mice.
Figure 1.

Effect of disease on salivary flow. Total saliva was collected from mildly anesthetized 4-, 9-, 14-, and 18-week-old female MRL/lpr and MRL/+ mice. Secretion of saliva was stimulated by intraperitoneal injection of 0.20 mg isoproterenol plus 0.05 mg pilocarpine/100 g body weight and saliva was collected over a 10-minute period. Data are means ± SEM, n = 5 except for 18-week-old MRL/lpr where n = 3. *Significant difference in salivary flow between MRL/+ and MRL/lpr mice.
Figure 2.
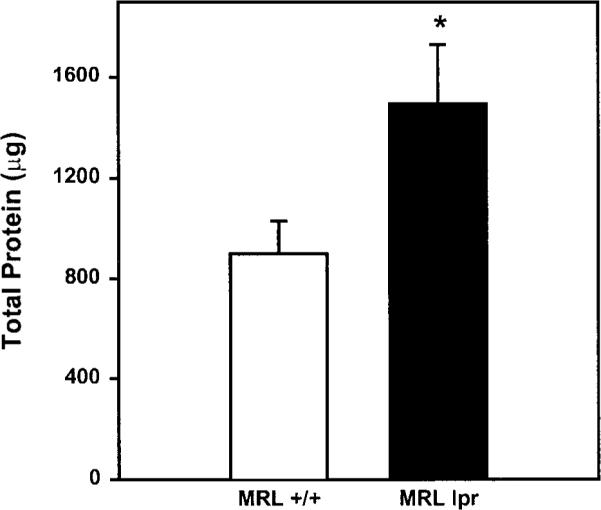
Effect of disease on total salivary protein. Total salivary protein in saliva collected from 18-week-old MRL/lpr and MRL/+ mice was determined by the method of Bradford, using bovine serum albumin as the standard. Data are means ± SEM, n = 3 to 5. *Significant difference in total salivary protein between MRL/+ and MRL/lpr mice.
To rule out the possibility that this increased salivation was due to the lpr mutation rather than inflammation of the salivary glands, we collected saliva from 16-week-old male and female MRL/lpr and MRL/+ mice, because Toda et al.19 had shown that inflammation of the exocrine glands occurs only in female but not male MRL/lpr mice. The rationale behind these experiments is that if the increased salivation observed in female MRL/lpr mice is solely due to the lpr mutation, then male MRL/lpr mice should produce more saliva than age-matched male MRL/+ mice. As shown in Figure 3, the amount of saliva secreted by MRL/lpr male mice was not significantly different from that secreted by MRL/+ male mice. It is important to note that consistent with the data reported in Figure 1, female MRL/lpr mice produced significantly higher amounts of saliva than did female MRL/+ mice (Fig. 3). Thus, the increased salivation observed in female MRL/lpr mice is not due to the lpr mutation but rather seems to correlate with the inflammation of the exocrine glands.
Figure 3.
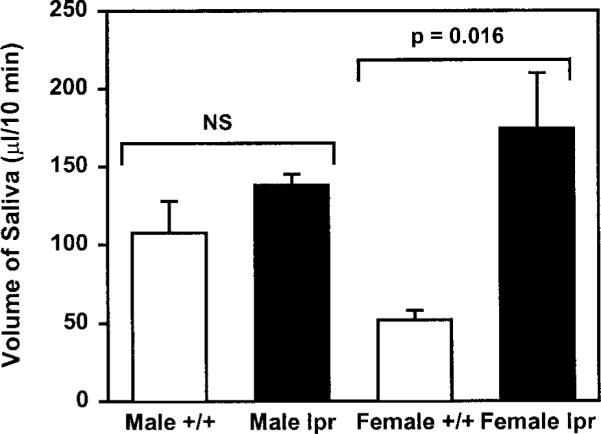
Effect of gender and disease on salivary flow. Total saliva was collected from mildly anesthetized 16-week-old male and female MRL/lpr (lpr) and MRL/+ (+/+) mice. Secretion of saliva was stimulated by intraperitoneal injection of 0.20 mg isoproterenol plus 0.05 mg pilocarpine per 100 g body weight, and saliva was collected over a 10-minute period. Data are means ± SEM, n = 4 to 5.
These results suggest that exocrine glands from MRL/lpr mice behave as denervated tissues. We hypothesize that this is due to the inability of their nerves to release neurotransmitters.
Effect of Disease on Acetylcholine Release
To directly test our hypothesis that the nerves of exocrine tissues from MRL/lpr are not able to release their neurotransmitters, we measured the spontaneous and evoked release of acetylcholine. As shown in Figure 4, high KCl induced a small but significant release of acetylcholine from submandibular and lacrimal glands of 18-week-old MRL/+ mice. In contrast, nerves of submandibular and lacrimal glands from 18-week-old MRL/lpr mice were not able to release acetylcholine in response to the depolarizing solution (Fig. 4).
Figure 4.

Effect of disease on acetylcholine release. Submandibular (top panels) and lacrimal glands (bottom panels) were removed from 18-week-old MRL/lpr and MRL/+ mice, and the release of acetylcholine was measured over a 20-minute period in buffer containing 5 mM KCl (spontaneous release) and 20 minutes in 75 mM KCl (evoked release). Data are means ± SEM, n = 3 to 5. *Significant difference from spontaneous release of acetylcholine.
One explanation for the impaired release of acetylcholine from nerve endings of exocrine tissues of MRL/lpr mice may be the loss of acetylcholine in these nerves. As shown in Figure 5, this explanation seems unlikely because, although the total amount of tissue acetylcholine was reduced in 18-week-old MRL/lpr tissues, there was a substantial amount of acetylcholine available in both lacrimal and submandibular gland nerves. However, we cannot rule out the possibility that only the releasable pool of acetylcholine was lost in nerves from diseased animals. The slight decline in acetylcholine amounts in 18-week-old MRL/lpr animals could be explained by the general loss of glandular tissue in these animals due to the extensive lymphocytic infiltration. In agreement with our previous findings, histopathologic staining of submandibular and lacrimal glands revealed that tissues from 18-week-old MRL/lpr mice showed an extensive degree of lymphocytic infiltration (Fig. 6). In contrast, tissues from 18-week-old MRL/+ mice showed a very mild inflammation (Fig. 6). As a result of the lymphocytic accumulation in MRL/lpr mice tissues, there was a substantial loss of glandular tissue.
Figure 5.
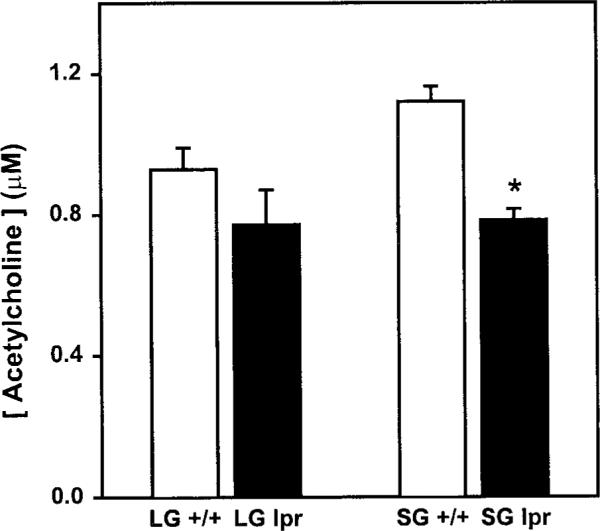
Effect of disease on tissue acetylcholine. Lacrimal (LG) and submandibular (SG) glands were removed from 18-week-old MRL/lpr (lpr) and MRL/+ (+/+) mice and the concentration of acetylcholine in tissue homogenates was measured. Data are means ±SEM, n = 3 to 5. *Significant difference from submandibular glands of MRL/+ mice.
Figure 6.

Effect of age on the lymphocytic infiltration of exocrine tissues. Lacrimal (LG) and submandibular (SMG) glands were removed from 18-week-old MRL/lpr and MRL/+ mice and processed for histopathology. Areas of lymphocytic infiltration are denoted by an asterisk. Similar results were obtained in at least three other animals.
Another possible explanation for the impaired release of acetylcholine is that the activity of the acetylcholinesterase in lacrimal and submandibular glands of MRL/lpr mice is upregulated with the onset and progression of the disease. As a consequence, highly active acetylcholinesterase degrades the putatively released acetylcholine and thus makes it undetectable. However, this possibility is unlikely because, as described in the Methods section, the release of acetylcholine was measured in the presence of neostigmine, a potent inhibitor of acetylcholinesterase.
These results show that activation, with high KCl, of nerve endings of lacrimal and salivary glands infiltrated with lymphocytes does not increase the release of acetylcholine.
Effect of Disease on Peroxidase Secretion
The main function of the lacrimal gland is to synthesize and secrete proteins, as well as water and electrolytes, onto the ocular surface. We have previously shown that secretion of peroxidase can be induced by exogenous stimulation of lacrimal gland acinar cells with cholinergic or adrenergic agonists. Figure 7A shows that KCl-induced depolarization of BALB/c mice lacrimal gland nerve endings induced a concentration-dependent secretion of peroxidase. Preincubation of lacrimal gland lobules with atropine (10−5 M), a cholinergic muscarinic antagonist, significantly reduced the amount of peroxidase secreted in response to 75 mM KCl (Fig. 7B). These results show that high KCl-induced peroxidase secretion was due to the activation of nerve endings and not to a direct effect of KCl on the lacrimal gland acinar cells. Thus peroxidase secretion can be used as an indirect index to measure KCl-induced neurotransmitter release from lacrimal gland nerve endings.
Figure 7.

Effect of KCl and atropine on peroxidase secretion. (A) Lacrimal glands from 10- to 12-week old BALB/c mice were processed to measure peroxidase secretion, over a 20-minute period, in buffer containing increasing concentrations of KCl (5–90 mM). Peroxidase secretion is expressed as a percentage of the total. Data are means ± SEM, n = 3 to 7. *Significant difference from 5 mM KCl-induced peroxidase secretion. (B) Lacrimal gland lobules from 10- to 12-week-old BALB/c mice were preincubated for 10 minutes, with or without atropine (10−5 M). Lobules were then incubated, for 20 minutes, in buffer containing 5 mM KCl (spontaneous secretion) and 20 minutes in 75 mM KCl (evoked secretion). Peroxidase secretion is expressed as a percentage of the total. Data are means ± SEM, n = 3. *Significant difference from spontaneous peroxidase secretion and atropine alone. #Significant difference from evoked peroxidase secretion in the presence of atropine.
As shown in Figure 8, high KCl induced a significant secretion of peroxidase from lacrimal glands of 18-week-old MRL/+ mice. In contrast, lacrimal glands from 18-week-old MRL/lpr mice were not able to secrete peroxidase in response to high KCl (Fig. 8). This was not due to a defect in the secretory process in MRL/lpr mice lacrimal glands because, as shown in Figure 8, exogenous addition of phenylephrine stimulated peroxidase secretion from MRL/+ as well as MRL/lpr lacrimal glands.
Figure 8.

Effect of disease on KCl-evoked peroxidase secretion. Lacrimal gland lobules were prepared from 18-week-old MRL/lpr and MRL/+ mice. To measure peroxidase secretion, lobules were incubated for 20 minutes in buffer containing 5 mM KCl (spontaneous secretion), for 20 minutes in 75 mM KCl (evoked secretion), and for 20 minutes in the presence of phenylephrine (10−4 M). Peroxidase secretion is expressed as a percentage of the total. Data are means ± SEM, n = 3 to 5. *Significant difference from spontaneous peroxidase secretion.
These results show that activation of nerve endings with high KCl does not stimulate peroxidase secretion from lacrimal glands infiltrated with lymphocytes.
Discussion
The precise mechanism(s) responsible for the decreased tear and saliva secretion in Sjögren's syndrome is unknown.9,10,13 It is believed that the dry eye and dry mouth in this disease are due to a progressive lymphocytic infiltration of the lacrimal and salivary glands, an immune-mediated destruction of the epithelial cells, and a consequent decline in tear and saliva production.9,10,12,13 Recent studies have suggested that production of autoantibodies against acinar cell surface receptors (M3 muscarinic) may be involved as well in the impaired production of tears and saliva.26,27 The mechanisms that lead to the accumulation of lymphocytes in the lacrimal and salivary glands and the production of autoantibodies are still unknown. Using NFS/sld mice thymectomized 3 days after birth, Haneji et al.28 proposed a-fodrin as a candidate autoantigen in Sjögren's syndrome.
Several studies have shown that intestinal inflammation is accompanied by changes in physiological function that reflect the actions of inflammatory mediators on epithelial cells, muscle cells, and enteric nerves.29 It was found that acetylcholine release from myenteric nerves evoked by electrical field stimulation (EFS) or high KCl solutions was significantly decreased by more than 80% in Trichinella spiralis-infected rats compared with noninfected control rats.30 Similarly, norepinephrine release induced by EFS was decreased by 66% and that induced by KCl was suppressed by 72% in infected rats compared with control noninfected rats.31 In addition to alterations in neurotransmitter release, it was found that the maximum tension generated in vitro by carbachol or serotonin was significantly greater in smooth muscle from inflamed intestine than in control noninflamed intestine,32 suggesting a denervation-like supersensitivity. Thus, it appears that inflammation of the intestine may have two consequences: a blockade of neurotransmitter release and an increased responsiveness of the inflamed tissue to neural agonists, which resembles denervation supersensitivity.
Similar to the inflamed intestine, acinar cells isolated from lacrimal and salivary glands of diseased MRL/lpr animals were previously reported to be hyper-responsive to exogenous cholinergic and adrenergic stimulation when compared with age-matched control MRL/+ animals.23 Moreover, in the present study, we found that MRL/lpr salivary glands were hyper-responsive to in vivo injection of secretagogues, because these mice produced significantly higher amounts of saliva than did age-matched MRL/+ mice. These findings combined with the fact that nerves are still present in the inflamed lacrimal and salivary glands22 led us to hypothesize that these tissues behaved as denervated ones.
In the present study, we showed that stimulation of nerves from inflamed, but not those from noninflamed, lacrimal and salivary glands with high KCl did not increase the release of acetylcholine. Furthermore, we found that, whereas activation of noninflamed lacrimal gland nerves with high KCl resulted in protein secretion, activation of those in inflamed glands did not elicit protein secretion. These findings demonstrate that, as suggested earlier by Sullivan,12 inflammation of exocrine glands in Sjögren's syndrome leads to impaired release of neurotransmitters from nerves and thus to impaired fluid secretion.
What are the mechanisms involved in inflammation-induced inhibition of neurotransmitter release? Several studies have shown that suppression of acetylcholine and norepinephrine release from myenteric nerves was mediated by proinflammatory cytokines including interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α.33–36 Moreover, in a rat model of acute colitis, an inflammatory disease of distal colon, IL-1β was implicated in blocking KCl-induced norepinephrine release from the myenteric plexus.37 IL-1β has been also shown to decrease the acetylcholine content of rat hippocampus.36 Another study showed that TNF-α alters neurotransmitter release in cultured sympathetic neurons.38 IL-1β and TNF-α were also implicated in the pathogenesis of acute graft-versus-host disease (GVHD).39 Based on these studies, inflammation-mediated production of cytokines seems to inhibit neurotransmitter release, thus altering the physiology of the inflamed tissues.
Given this abundant literature on the role of proinflammatory cytokines, we hypothesize that concomitant with inflammation of the lacrimal and salivary glands in Sjögren's syndrome, there is an increase in production of cytokines that inhibits neurotransmitter release leading to decreased tear and saliva production from the acinar and ductal cells. In support of this hypothesis, it has been shown that the levels of proinflammatory cytokines are elevated in lacrimal and salivary glands of Sjögren's syndrome patients as well as in animal models.40 – 43 Moreover, we found that the protein level of IL-1β increased, in a disease-dependent manner, in lacrimal and salivary glands of MRL/lpr, but not MRL/+ or BALB/c, mice.44
In summary, our results show that nerves of inflamed lacrimal and salivary glands of MRL/lpr mice are not able to release their neurotransmitters, which results in impaired secretion from these glands.
Acknowledgments
The authors thank David Sullivan, James Zieske, Darlene Dartt, and Robin Hodges for critical reading of the manuscript.
Supported by National Institutes of Health Grant EY12383.
Footnotes
Commercial relationships policy: N.
References
- 1.Young JA. Salivary secretion of inorganic electrolytes. Int Rev Physiol. 1979;19:1–58. [PubMed] [Google Scholar]
- 2.Holly FJ. Tear film physiology. Int Ophthalmol Clin. 1987;27:2–6. doi: 10.1097/00004397-198702710-00002. [DOI] [PubMed] [Google Scholar]
- 3.Lamberts DW. Physiology of the tear film. In: Smolin G, Thoft RA, editors. The Cornea. Little, Brown; Boston: 1994. pp. 439–483. [Google Scholar]
- 4.Dartt DA. Signal transduction and control of lacrimal gland protein secretion: a review. Curr Eye Res. 1989;8:619–636. doi: 10.3109/02713688908995762. [DOI] [PubMed] [Google Scholar]
- 5.Schneyer LH, Young JA, Schneyer CA. Salivary secretion of electrolytes. Physiol Rev. 1972;52:720–777. doi: 10.1152/physrev.1972.52.3.720. [DOI] [PubMed] [Google Scholar]
- 6.Botelho SY, Hisada M, Fuenmayo N. Functional innervation of the lacrimal gland in the cat. Arch Ophthalmol. 1966;76:581–588. doi: 10.1001/archopht.1966.03850010583019. [DOI] [PubMed] [Google Scholar]
- 7.Sibony PA, Walcott B, McKeon C, Jakobiek FA. Vasoactive intestinal polypeptide and the innervation of the human lacrimal gland. Arch Ophthalmol. 1988;106:1085–1088. doi: 10.1001/archopht.1988.01060140241033. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto Y, Tanabe T, Ueda S, Kawata M. Immunohistochemical and enzyme histochemical studies of peptidergic, aminergic, and cholinergic innervation of the lacrimal gland of the monkey (Macaca fuscata) J Auton Nerv Syst. 1992;37:207–214. doi: 10.1016/0165-1838(92)90042-f. [DOI] [PubMed] [Google Scholar]
- 9.Talal N, Moutsopoulos HM, Kassan SS. Sjögren's Syndrome: Clinical and Immunological Aspects. Springer-Verlag; Berlin: 1987. [Google Scholar]
- 10.Fox RI. Pathogenesis of Sjögren's syndrome. Rheum Dis Clin North Am. 1992;18:517–538. [PubMed] [Google Scholar]
- 11.Fox RI, Törnwall J, Michelson P. Current issues in the diagnosis and treatment of Sjögren's syndrome. Curr Opin Rheumatol. 1999;11:364–371. doi: 10.1097/00002281-199909000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan DA. Possible mechanisms involved in the reduced tear secretion in Sjögren's syndrome. In: Homma M, Sugai S, Tojo T, Miyasaka N, Akizuki M, editors. Sjögren's Syndrome: State of the Art. Kugler; Amsterdam: 1994. pp. 13–19. [Google Scholar]
- 13.Sullivan DA, Wickham LA, Krenzer KL, Rocha EM, Toda I. Aqueous tear deficiency in Sjögren's syndrome: possible causes and potential treatment. In: Pleyer U, Hartmenn C, editors. Oculodermal Diseases. Aeolus; Buren: 1997. pp. 95–152. [Google Scholar]
- 14.Theofilopoulous AN, Dixon FJ. Etiopathogenesis of murine SLE. Immunologic Rev. 1981;55:179–216. doi: 10.1111/j.1600-065x.1981.tb00343.x. [DOI] [PubMed] [Google Scholar]
- 15.Jabs DA, Prendergast RA. Murine models of Sjögren's syndrome: immunohistologic analysis of different strains. Invest Ophthalmol Vis Sci. 1988;29:1437–1443. [PubMed] [Google Scholar]
- 16.Hoffman RW, Alspaugh MA, Waggie KS, Durham JB, Walker SE. Sjögren's syndrome in MRL/l and MRL/n mice. Arthritis Rheum. 1984;27:157–165. doi: 10.1002/art.1780270206. [DOI] [PubMed] [Google Scholar]
- 17.Haneji N, Hamano H, Yanagi K, Hayashi Y. A new animal model for primary Sjögren's syndrome in NFS/sld mutant mice. J Immunol. 1994;153:2769–2777. [PubMed] [Google Scholar]
- 18.Moore PA, Bounous DI, Kaswan RL, Humphreys-Beher MG. Histologic examination of the NOD-mouse lacrimal glands, a potential model for idiopathic autoimmune dacryoadenitis in Sjögren's syndrome. Lab Anim Sci. 1996;46:125–128. [PubMed] [Google Scholar]
- 19.Toda I, Sullivan BD, Rocha EM, et al. Impact of gender on exocrine gland inflammation in mouse models of Sjögren's syndrome. Exp Eye Res. 1999;69:355–366. doi: 10.1006/exer.1999.0715. [DOI] [PubMed] [Google Scholar]
- 20.Kessler HS. A laboratory model of Sjögren's syndrome. Am J Pathol. 1968;52:671–678. [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy ED. Lymphoproliferation (lpr) and other single-locus models for murine lupus. In: Gershwin ME, Merchant B, editors. Immunologic Defects in Laboratory Animals. Plenum; New York: 1981. pp. 143–176. [Google Scholar]
- 22.Zoukhri D, Hodges RR, Dartt DA. Lacrimal gland innervation is not altered with the onset and progression of disease in a murine model of Sjögren's syndrome. Clin Immunol Immunopathol. 1998;89:126–133. doi: 10.1006/clin.1998.4597. [DOI] [PubMed] [Google Scholar]
- 23.Zoukhri D, Hodges RR, Rawe IM, Dartt DA. Ca2+ signaling by cholinergic and α1-adrenergic agonists is up-regulated in lacrimal and submandibular glands in a murine model of Sjögren's syndrome. Clin Immunol Immunopathol. 1998;89:134–140. doi: 10.1006/clin.1998.4598. [DOI] [PubMed] [Google Scholar]
- 24.Hu Y, Nakagawa Y, Purushotham K, Humphreys-Beher M. Functional changes in salivary glands of autoimmune disease-prone NOD mice. Am J Physiol. 1992;263:E607–E614. doi: 10.1152/ajpendo.1992.263.4.E607. [DOI] [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Robinson CP, Brayer J, Yamchika S, et al. Transfer of human serum IgG to nonobese diabetic Igmnull mice reveals a role for autoantibodies in the loss of secretory function of exocrine tissues in Sjögren's syndrome. Proc Natl Acad Sci USA. 1998;95:7538–7543. doi: 10.1073/pnas.95.13.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bacman S, Leiros CP, Sterin-Borda L, et al. Autoantibodies against lacrimal gland M3 muscarinic acetylcholine receptors in patients with primary Sjögren's syndrome. Invest Ophthalmol Vis Sci. 1998;39:151–156. [PubMed] [Google Scholar]
- 28.Haneji N, Nakamura T, Takio K, et al. Identification of α-fodrin as a candidate autoantigen in primary Sjögren's syndrome. Science. 1997;276:604–607. doi: 10.1126/science.276.5312.604. [DOI] [PubMed] [Google Scholar]
- 29.Collins SM, Hurst SM, Main C, et al. Effect of inflammation of enteric nerves: cytokine-induced changes in neurotransmitter content and release. Ann NY Acad Sci. 1992;664:415–424. doi: 10.1111/j.1749-6632.1992.tb39780.x. [DOI] [PubMed] [Google Scholar]
- 30.Collins SM, Blennerhassett PA, Blennerhassett MG, Vermillion DL. Impaired acetylcholine release from the myenteric plexus of Trichinella-infected rats. Am J Physiol. 1989;257:G898–G903. doi: 10.1152/ajpgi.1989.257.6.G898. [DOI] [PubMed] [Google Scholar]
- 31.Swain MG, Blennerhassett PA, Collins SM. Impaired sympathetic nerve function in the inflamed rat intestine. Gastroenterology. 1991;100:675–682. doi: 10.1016/0016-5085(91)80011-w. [DOI] [PubMed] [Google Scholar]
- 32.Marzio L, Blennerhassett P, Chiverton S, et al. Altered smooth muscle function in worm-free gut regions of Trichinella-infected rats. Am J Physiol. 1990;259:G306–G313. doi: 10.1152/ajpgi.1990.259.2.G306. [DOI] [PubMed] [Google Scholar]
- 33.Hurst S, Collins SM. Interleukin-1β modulation of norepinephrine release from rat myenteric nerves. Am J Physiol. 1993;264:G30–G35. doi: 10.1152/ajpgi.1993.264.1.G30. [DOI] [PubMed] [Google Scholar]
- 34.Main C, Blennerhassett PA, Collins SM. Human recombinant interleukin 1 beta suppresses acetylcholine release from rat myenteric plexus. Gastroenterology. 1993;104:1648–1654. doi: 10.1016/0016-5085(93)90641-o. [DOI] [PubMed] [Google Scholar]
- 35.Hurst SM, Collins SM. Mechanism underlying tumour necrosis factor-α suppression of norepinephrine release from rat myenteric plexus. Am J Physiol. 1994;266:G1123–G1129. doi: 10.1152/ajpgi.1994.266.6.G1123. [DOI] [PubMed] [Google Scholar]
- 36.Rada P, Mark GP, Vitek MP, et al. Interleukin-1 beta decreases acetylcholine measured by microdialysis in the hippocampus of freely moving rats. Brain Res. 1991;550:287–290. doi: 10.1016/0006-8993(91)91330-4. [DOI] [PubMed] [Google Scholar]
- 37.Jacobson K, McHugh K, Collins SM. The mechanism of altered neural function in a rat model of acute colitis. Gastroenterology. 1997;112:156–162. doi: 10.1016/s0016-5085(97)70230-0. [DOI] [PubMed] [Google Scholar]
- 38.Soliven B, Albert J. Tumour necrosis factor modulates the inactivation of catecholamine secretion in cultured sympathetic neurons. J Neurochem. 1992;58:1073–1078. doi: 10.1111/j.1471-4159.1992.tb09364.x. [DOI] [PubMed] [Google Scholar]
- 39.Antin JH, Ferrara JLM. Cytokine dysregulation and acute graft-versus-host disease. Blood. 1992;80:294–298. [PubMed] [Google Scholar]
- 40.Saito I, Terauchi K, Shimuta M, et al. Expression of cell adhesion molecules in the salivary and lacrimal glands of Sjögren's syndrome. J Clin Lab Anal. 1993;7:180–187. doi: 10.1002/jcla.1860070309. [DOI] [PubMed] [Google Scholar]
- 41.Hamano H, Saito I, Haneji N, et al. Expression of cytokine genes during development of autoimmune sialadenitis in MRL/lpr mice. Eur J Immunol. 1993;23:2387–2391. doi: 10.1002/eji.1830231002. [DOI] [PubMed] [Google Scholar]
- 42.Fox RI, Kang HI, Ando D, Abrams J, Pisa E. Cytokine mRNA expression in salivary gland biopsies of Sjögren's syndrome. J Immunol. 1994;152:5532–5539. [PubMed] [Google Scholar]
- 43.Koh K, Sawada S, Fox RI. High levels of IL-10 and Th1 cytokine mRNA transcript in salivary gland biopsies from Sjögren's syndrome. In: Homma M, Sugai S, Tojo T, Miyasaka N, Akizuki M, editors. Sjögren's Syndrome: State of the Art. Kugler; Amsterdam: 1994. pp. 99–102. [Google Scholar]
- 44.Zoukhri D, Hodges RR, Dartt DA. Interleukin-1 beta protein is elevated in inflamed lacrimal glands in a murine model of Sjögren's syndrome [ARVO Abstract] Invest Ophthalmol Vis Sci. 1999;40:S553. Abstract nr 2919. [Google Scholar]