

SUMMARY
The intestinal microbiota is important for induction of inflammatory bowel disease (IBD). IBD is associated with complex shifts in microbiota composition, but it is unclear whether specific bacterial subsets induce IBD and, if so, whether their proportions in the microbiota are altered during disease. Here we fulfilled Koch’s postulates in host-genotype-specific fashion using a mouse model of IBD with human-relevant disease-susceptibility mutations. From screening experiments we isolated common commensal Bacteroides species, introduced them into antibiotic-pretreated mice, and quantitatively re-isolated them in culture. The bacteria colonized IBD-susceptible and non-susceptible mice equivalently, but induced disease exclusively in susceptible animals. Conversely, commensal Enterobacteriaceae were >100-fold enriched during spontaneous disease but an Enterobacteriaceae isolate failed to induce disease in antibiotic-pretreated mice despite robust colonization. We thus demonstrate that IBD-associated microbiota alterations do not necessarily reflect underlying disease etiology. These findings establish important experimental criteria and a conceptual framework for understanding microbial contributions to IBD.
INTRODUCTION
Inflammatory bowel disease (IBD) arises from complex interactions of genetic, environmental, and microbial factors (Xavier and Podolsky, 2007). IBD is a spectrum of chronic, non-communicable diseases, all of which exhibit a primary defining feature of spontaneous intestinal inflammation. The two major subtypes, ulcerative colitis and Crohn’s disease, are defined by both distinct and overlapping clinical and pathologic features (Stenson et al., 2009). Recent progress in understanding the host genetics of IBD has provided a critical new framework with which to evaluate how microbial factors contribute its pathogenesis (Cadwell et al., 2010). An important challenge in this field is to understand the role of commensal bacteria in the context of specific host genotypes.
Host genetics clearly influence IBD susceptibility (Xavier and Podolsky, 2007). The most current meta-analyses of genome wide association studies identified approximately 100 IBD-susceptibility loci, some specifically associated with either ulcerative colitis or Crohn’s disease and others associated with both (Franke et al., 2010; McGovern et al., 2010). A subset of these susceptibility loci are associated with signaling pathways for IL-10 and TGFβ, two important immunoregulatory cytokines that play prominent roles in intestinal homeostasis (Li et al., 2006; Ouyang et al., 2011). These susceptibility alleles, like most others, are all common gene variants that modestly elevate disease risk. Importantly, investigators also recently showed that rare recessive deficiencies in the IL- 10 receptor result in fulminant, rapid-onset IBD enterocolitis within one year of age (Glocker et al., 2009).
The intestinal microbiota also plays an important role in IBD pathogenesis (Xavier and Podolsky, 2007). The microbiota consists of a large and diverse community containing hundreds of commensal bacterial species, many of which have only been detected by culture-independent 16S rRNA gene sequencing (Eckburg et al., 2005; Qin et al., 2010). Both clinical and laboratory data implicate the microbiota in IBD induction. Medically, antibiotics provide therapeutic benefits for some manifestations of IBD (Ohkusa et al., 2010; Sartor, 2008). In patients with Crohn’s disease, surgical diversion of fecal flow produces remission in inflamed bowel segments with disease recurrence upon flow restoration (Janowitz et al., 1998). Experimental introduction of small bowel effluent into the surgically excluded bowel segments also re-induced disease, but a sterile ultrafiltrate of bowel effluent did not (Harper et al., 1985). In most spontaneous animal models of IBD, disease can be blocked by antibiotics or re-derivation into a germ-free state, and IBD-susceptible germ-free animals develop disease upon exposure to commensal microbiota from conventionally raised hosts (Sartor, 2008). Mass sequencing studies of the intestinal microbiota have revealed complex disease-associated shifts in microbiota composition, but disease is thus far not consistently associated with presence or absence of a specific microbe (Frank et al., 2007; Packey and Sartor, 2009; Qin et al., 2010).
Thus, two related and fundamentally important questions about the role of the microbiota in IBD remain unresolved: in genetically susceptible hosts, do specific subsets of commensal bacteria induce IBD (Strober, 2010; Takaishi et al., 2008) and, if so, can these subsets be identified based on disease-associated alterations in levels of colonization (Tannock, 2008)? Functional tests of the ability of specific microbes to induce IBD have relied primarily on germ-free IBD-susceptible animals. These studies demonstrate that select commensal bacterial species can induce disease in some models (Sartor, 2008). However, translating these findings to conventionally raised animal models, which more closely mimic IBD patients, has proven to be challenging. Developing experimental approaches using conventionally raised animals to functionally assess the roles of specific commensal bacteria and test the hypotheses generated by culture-independent profiling studies of the microbiota in IBD is thus a critical priority.
Here, we address these questions using non-germ-free methods in an antibiotic-responsive mouse model with IBD-relevant deficiencies in IL-10 and TGFβ signaling. We fulfill host-genotype-specific Koch’s postulates by isolating commensal bacteria, introducing them into antibiotic-pretreated mice, assessing disease development, and confirming host colonization by quantitative re-isolation of the experimentally introduced bacteria in culture. Using this approach, we identify distinct commensal bacterial subsets with and without disease-inducing potential in susceptible hosts and show that these subsets would not have been predicted based on disease-associated alterations in microbiota composition.
RESULTS
Establishment of a non-germ-free system to screen for disease-inducing microbes using dnKO mice
We evaluated the colitogenic potential of intestinal microbes in the dnKO mouse model of IBD (Kang et al., 2008). These mice contain a complete knock out of Il10r2 (Spencer et al., 1998) and express a transgene encoding dominant negative Tgfbr2 restricted to T-cells (Gorelik and Flavell, 2000). Both mutations are closely linked to pathways implicated in human IBD: IL10 and IL10RB (Franke et al., 2010; Glocker et al., 2009; McGovern et al., 2010) and SMAD3, a direct downstream target of TGFBR2 (Franke et al., 2010). We previously showed that dnKO mice develop spontaneous, severe, and rapid intestinal inflammation that is 100% penetrant (Kang et al., 2008). Co-housed Il10r2+/− littermate controls did not develop colitis, suggesting the microbial triggers of disease in dnKO mice were innocuous in non-susceptible hosts (Figure 1A). We confirmed that colitis in dnKO mice was highly responsive to treatment with metronidazole and ciprofloxacin (Figure 1A). We quantified colitis severity in several anatomically defined regions of the colon using multiple independent metrics of gross and anatomic pathology previously validated in this model (Kang et al., 2008). Intestinal whole mounts were scored for gross pathology in a blinded fashion (see Figure S1). Histologic mucosal inflammation was quantified by measuring heights and widths of well-oriented colonic crypts, which we previously showed to be sensitive metrics of colitis severity (Kang et al., 2008). By these criteria, antibiotic treatment completely blocked dnKO colitis (Figures 1B through 1G).
Figure 1. Antibiotic treatment quantitatively prevents colitis development in dnKO mice.

(A) Representative images of H&E stained rectal histology of 4-week-old untreated and antibiotic-treated Il10r2+/− and dnKO mice (distal 0.5 cm of colon). Abx = antibiotics (metronidazole + ciprofloxacin in drinking water). Scale bar = 100 μm.
(B and C) Cecum and transverse colon pathology scores of untreated and antibiotic-treated Il10r2+/− and dnKO mice as described in Figure 1A. Intestinal whole mounts were scored for gross pathology in a blinded fashion by an anatomic pathologist (T.S.S.) according to a validated system: ranging from 0 (no pathology) to 3 (severe pathology; see Figure S1 for details). Individual (squares) and median (bars) pathology scores are displayed. Kruskal-Wallis test: (B) H3 = 13.75, p = 0.0033; (C) H3 = 13.75, p = 0.0033. (D to G) Transverse colon and rectum crypt heights and crypt widths of mice in described in Figures 1B and 1C displayed as mean +/− SEM. 1-way ANOVA with post-hoc Tukey’s test: (D) F3,11 = 40.87, p < 0.0001; (E) F3,11 = 10.71, p = 0.0014; (F) F3,11 = 33.25, p < 0.0001; (G) F3,11 = 9.501, p = 0.0022. All statistically significant pairwise comparisons are displayed: **, p < 0.01, ***, p < 0.005.
See also Figure S1
Since the combination of antibiotics we used does not sterilize the gut (Heimesaat et al., 2006), we hypothesized that disease in dnKO mice would depend on a subset of commensal bacteria which are eliminated by antibiotic treatment. The intestinal microbiota is normally dominated by members of two bacterial phyla: Firmicutes (Gram +) and Bacteroidetes (Gram ) (Eckburg et al., 2005). To assess the effects of antibiotic treatment on the microbiota, we collected fecal samples from antibiotic-treated and untreated mice and performed quantitative PCR using 16S rRNA gene primer sets specific for the Bacteroidetes families Bacteroidaceae-Porphyromonadaceae-Prevotellaceae (BPP) and for the Firmicutes families Enterococcaceae, Lactobacillaceae, and Lachnospiraceae-Ruminococcaceae (Nava et al., 2010). We found antibiotic-treated mice showed a decrease in BPP and Lachnospiraceae-Ruminococcaceae whereas Enterococcaceae and Lactobacillaceae were increased (Figure S2). These data supported the hypothesis that antibiotic treatment prevented disease by selectively altering subsets of commensal bacteria within the microbiota.
To test this hypothesis, we therefore developed a non-germ-free screen for colitogenic activity of commensal bacteria taking advantage of the dnKO model’s rapid disease onset and antibiotic-responsiveness. We housed mice in a specific pathogen-free facility using stringent precautions to prevent cage-to-cage contamination. We treated dnKO and control mice at weaning with metronidazole and ciprofloxacin for ≥3 weeks. Two days after halting antibiotic treatment, mice were orogastrically gavaged a single time with experimental inocula or sterile PBS as a control (Figure 2A). Using this experimental design, we found that antibiotic-pretreated dnKO mice gavaged with sterile PBS remained disease free (Figures 2B through 2H). To demonstrate that we could induce disease in this system, we gavaged antibiotic-pretreated dnKO mice with intestinal contents harvested from untreated control animals from our colony. We used a standardized frozen stock of intestinal contents for our experiments to control for effects that might occur due to variation between individual donors (Weinstein et al., 1974). Mice gavaged with freshly thawed aliquots of intestinal contents developed severe colitis by three weeks post inoculation (Figures 2B through 2H). Gavage with freshly harvested intestinal contents or cohousing with untreated mice produced similar outcomes (not shown). Total organ surveys showed that the predominate site of pathology was the colon with minor more variable effects in the lung and liver that were similar to the pathology that developed in post weaning untreated dnKO mice (Gorelik and Flavell, 2000; Kang et al., 2008). We therefore relied on colon-specific metrics of colitis rather than weight loss or serum cytokines to ensure results were not confounded by extra-intestinal processes. These results demonstrated the feasibility of screening for colitogenic bacteria using this method.
Figure 2. A non-germ-free system for colitis induction in antibiotic-pretreated dnKO mice.

(A) Experimental timeline. Antibiotic treatment began at weaning (age 3 weeks); mice were antibiotic-treated for ≥3 weeks and gavaged 2 days after treatment cessation (Abx = antibiotics).
(B) Representative images of H&E stained rectal histology of antibiotic-pretreated dnKO mice orogastrically gavaged with sterile PBS or with a standardized frozen stock of intestinal contents from untreated animals. Gavage dose = 2×107 total cfu/mouse on Anaerobic Reducible Blood (ANB) agar (non-selective anaerobic culture media; cfu = colony-forming unit). Scale bar = 100 μm.
(C and D) Cecum and transverse colon pathology scores of antibiotic-pretreated dnKO mice gavaged with sterile PBS or intestinal contents from untreated donors as described in Figure 2B displayed as individual (symbols) and median (bars) scores. Mann-Whitney U-test.
(E to F) Transverse colon and rectum crypt heights and crypt widths of mice described in Figure 2C and 2D displayed as mean +/− SEM. Unpaired t-test.
See also Figure S2
Mixed cultures of intestinal microbes induce disease in antibiotic-pretreated mice
We next asked whether cultivable intestinal bacteria could induce colitis in this system. Because of the rapid and widespread colitis in dnKO mice, we hypothesized that common, abundant members of the intestinal microbiota would likely induce disease. As an initial screen, we therefore serially diluted stock intestinal contents from untreated mice (see above) and cultured the dilutions in parallel on non-selective and selective bacterial growth media (Figure 3A). We harvested anaerobic mixed cultures from a dilution at which ~1500 colonies grew, ensuring the culture contained only relatively abundant (approximately ≥0.07% abundance) intestinal bacterial cultivable on that media type (Table S1). For colonization experiments, the anaerobic culture was mixed in equal proportion with a culture grown aerobically on non-selective media and gavaged into antibiotic-pretreated dnKO mice. The gavaged mice developed severe colitis relative to PBS-gavaged controls (Figures 3B through 3E). These results demonstrated that colitis-inducing bacteria could be grown in culture.
Figure 3. Mixed cultures of intestinal contents induce disease in antibiotic-pretreated dnKO mice.

(A) Diagram of preparation of mixed cultures of intestinal contents. An aliquot of frozen intestinal contents (see Figure 1C) was thawed, serially diluted, and cultured on the indicated media types. 3-day mixed cultures were harvested from the indicated culture plates (in squares) and frozen in 20% glycerol. Aliquots of each culture were subsequently thawed and adjusted to standardized doses for gavage.
(B and C) Cecum and transverse colon pathology scores of antibiotic-pretreated dnKO mice gavaged with sterile PBS or with the indicated anaerobic mixed cultures of intestinal contents mixed 1:1 with aerobic cultures grown on chocolate agar at 0 dilution. Individual (symbols) and median (bars) pathology scores are displayed. Gavage doses: ANB = 6.4×107 total cfu/mouse; LKV = 7.3×107 total cfu/mouse; CNA = 5.4×107 total cfu/mouse. Statistical significance relative to PBS determined by Dunn’s multiple comparison test: n.s., p > 0.05; *, p < 0.05; **, p < 0.01.
(D and E) Transverse colon and rectum crypt heights of antibiotic-pretreated dnKO mice described in Figure 3B and 3C, displayed as mean +/− SEM. Statistical significance relative to PBS determined by Dunnett’s multiple comparison test. The screen was unrepeated.
See also Table S1 and S2
To refine potential pools of colitogenic microbes, we also tested cultures grown on more selective media types. Anaerobic cultures were grown in parallel on media selective for Gram-negative obligate anaerobes (LKV agar) or media inhibitory towards Gram-negative bacilli and enriched for growth of Gram-positive anaerobes (CNA agar) (Figure 3A). Gavage mixtures were prepared as above. Mice receiving the CNA culture showed pathology that did not significantly differ from PBS-gavaged controls (Figures 3B through 3E). To evaluate why the CNA mixed culture did not induce disease, we isolated common bacteria from the feces of antibiotic-treated (non-colitic) dnKO mice using non-selective ANB agar, extracted genomic DNA, and obtained partial sequences of the 16S rRNA gene. Based on sequence analysis, we identified the isolates as belonging to a variety of families within the phylum Firmicutes including Enterococcaceae, Lactobacillaceae, Bacillaceae, and Lachnospiraceae (Table S2). We confirmed that all of these isolates grew on CNA agar. Additionally, we performed targeted 16S rRNA gene sequencing of bacterial DNA isolated from the CNA gavage inoculum and found sequences belonging to the Firmicutes species listed in Table S2. We therefore concluded that these species could colonize antibiotic-treated dnKO mice without inducing disease. In contrast, antibiotic-pretreated dnKO mice gavaged with the LKV culture developed robust disease similar in severity to mice given the non-selective ANB culture (Figures 3B through 3E). These results suggested that abundant Gram-negative obligate anaerobes would be sufficient to induce disease in dnKO mice.
Commensal Bacteroides isolates induce disease in IBD-susceptible but not non-susceptible mice
We therefore isolated and identified abundant Gram-negative, obligate anaerobes from the stock of pooled intestinal contents described above. Based on unique-appearing colony morphology, we isolated and propagated 17 Gram-negative isolates on LKV agar. Sequence analysis of the 16S rRNA gene showed that several of the isolates were redundant, but they included six unique species from the phylum Bacteroidetes (Table S3). Isolation of unique-appearing colonies from intestinal contents grown in parallel on Bacteroides bile esculin (BBE) agar produced nine additional Bacteroidetes isolates, all of which were identical to isolates obtained using LKV agar. Sequences from all isolates were ≥99% identical to 16S rRNA sequences previously detected in culture-independent studies of rodent commensal intestinal microbiota as determined using BLAST and the Ribosomal Database Project (Altschul et al., 1990; Cole et al., 2007; Cole et al., 2009). Collectively, these results confirmed the isolates were common, abundant members of the commensal microbiota.
To assess whether any of the isolates could induce disease, we screened five of them in antibiotic-pretreated dnKO mice. We found that pure cultures of each induced colitis to varying degrees. Bacteroides vulgatus and Bacteroides thetaiotaomicron induced severe ulcerative disease while the Bacteroides sp. TP5 induced milder disease consisting of lymphocytic infiltrate in the mucosa (Figures 4A and 4B).
Figure 4. Commensal Bacteroides induce disease in antibiotic-pretreated dnKO but not Il10r2+/− mice.

(A) Screen for colitis induction by Bacteroidetes isolates. Cecum gross pathology scores of antibiotic-pretreated dnKO mice gavaged with 1×108 cfu/mouse of pure cultures of the indicated primary bacterial isolates (see Table S3). Not repeated for B. uniformis and P. goldsteinii.
(B) Representative H&E stained cecal histology of mice gavaged with pure cultures of the indicated bacterial isolate as described in Figure 4A. Scale bar = 200 μm. Cecal histology of a non-colitic Il10r2+/− mouse gavaged with B. thetaiotaomicron is shown for comparison.
(C and D) Cecum and transverse colon pathology scores of antibiotic-pretreated mice of the indicated genotypes gavaged with PBS or a pure culture of the Bacteroides thetaiotaomicron isolate (7×107 cfu/mouse). Kruskal-Wallis test with post-hoc Dunn’s test. (C) H2 = 12.26, p = 0.0022; (D) H2 = 11.36, p = 0.0034. All significant pairwise comparisons are displayed: *, p < 0.05.
(E to H) Transverse colon and rectum crypt heights and crypt widths of mice described in Figure 4C and 4D displayed as mean +/− SEM. 1-way ANOVA with post-hoc Tukey’s test: (E) F2,10 = 8.596, p = 0.0067; (F) F2,10 = 12.55, p = 0.0019; (G) F2,10 = 2.057, p = 0.1786; (H) F2,10 = 12.05, p = 0.0022. All significant pairwise comparisons are displayed if the omnibus p-value by ANOVA was significant: *, p < 0.05; **, p < 0.01.
(I) Graph of the number of neutrophils per 50 well-oriented crypts located within the intestinal epithelium/crypt lumen in colons of the mice described in 4C and 4D. Kruskal-Wallis test: H2 = 6.529, p = 0.0382.
(J) Percent of CD4+ T-cells expressing Ifnγ, determined using validated procedures (Kang et al., 2008), in the colonic lamina propria of antibiotic-treated Il10r2+/− (n=2) and dnKO (n=4) mice or antibiotic-pretreated Il10r2+/− (n=3) and dnKO (n=3) mice gavaged with 108 cfu/mouse of B. thetaiotaomicron and sacrificed 3 weeks post-gavage. 1-way ANOVA with post-hoc Tukey’s test: F3,8 = 27.93, p = 0.0001. All significant pairwise comparisons are displayed: **, p < 0.01; ***, p < 0.005.
(K) Immunoblots of B. thetaiotaomicron lysate probed with a 1:200 dilution of pooled serum collected from antibiotic-pretreated Il10r2+/− (n=3) or dnKO (n=8) mice immediately prior to gavage with 108 cfu/mouse of B. thetaiotaomicron (pre-gavage) or at sacrifice 3 weeks later (post-gavage). Secondary antibody is α-mouse IgG. Equal amounts of bacterial lysate were separated by SDS-PAGE, transferred to nitrocellulose membranes, blotted, and exposed in parallel for all groups
Importantly, we confirmed in additional experiments that B. thetaiotaomicron, a well-characterized symbiotic species (Goodman et al., 2009) potently induced colitis in dnKO mice but was innocuous in Il10r2+/− controls (Figures 4C to 4H and S3). The pathology included a significant colonic neutrophil infiltration with trans-epithelial migration (Figure 4I) and a significant increase in the percentage of interferon producing CD4+ T cells isolated from the lamina propria (Figure 4J). These findings were similar to untreated, spontaneously colitic dnKO mice (Kang et al., 2008).
Previous studies have demonstrated serum antibody responses to commensal bacterial antigens in IBD, but assessing the relationship of these antibody responses to disease induction and etiology has been experimentally challenging (Brandwein et al., 1997; Lodes et al., 2004). We found that antibiotic-pretreated mice exhibited no serum IgG response to lysates of cultured Bacteroidetes species prior to gavage. By contrast, dnKO mice given either B. thetaiotaomicron or B. vulgatus developed robust IgG responses to multiple antigens of the respective isolate post-gavage whereas Il10r2+/− controls given the same bacteria did not (Figure 4K and Figure S3B). Taken together, these results demonstrate that colitis induced by commensal Bacteroides species involves activation of both innate and adaptive immune responses in a host-genotype-specific fashion.
Antibiotic treatment eliminates cultivable intestinal Bacteroides from the fecal microbiota
Since diverse intestinal Bacteroidetes species induced colitis in dnKO mice, we reasoned that antibiotic treatment should eradicate them from the microbiota. To test this hypothesis, we performed quantitative fecal cultures on Bacteroides-selective media. Fecal samples from antibiotic-treated dnKO mice exhibited no bacterial growth when quantitatively cultured on Bacteroides bile esculin (BBE) agar at a detection limit of 17 colony-forming units (cfu) per sample (~103 cfu/g of feces). By contrast, we observed high bacterial titers (~109 cfu/g of feces) on BBE agar in samples from untreated dnKO mice (Figure 5A). While antibiotic treatment decreased the total bacterial load as assessed by qPCR for the bacterial rpoB gene (Nava et al., 2010), fecal titers on ANB media did not differ with antibiotic treatment (Figures 5A and S4A). Collectively, these data further confirmed that antibiotics prevented disease by selectively altering microbiota composition rather than by sterilizing the gut.
Figure 5. Intestinal Bacteroides are eliminated by antibiotics and stably re-colonize gavaged, antibiotic-pretreated mice independently of host genotype.
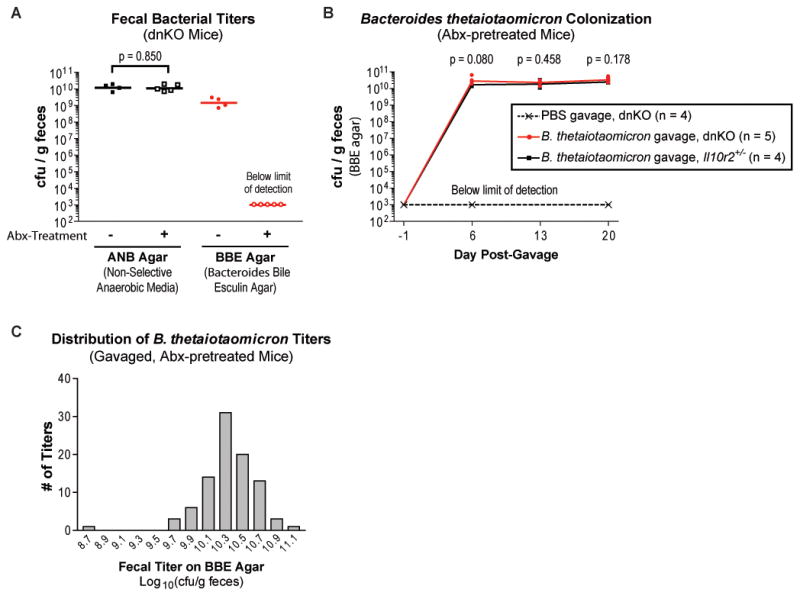
(A) Plot of fecal titers from untreated and antibiotic-treated dnKO mice grown in parallel anaerobically on non-selective (ANB) agar and Bacteroides bile esculin (BBE) agar. Titers from individual mice (symbols) and means of log10-transformed titers (bars) are displayed. Unpaired t-test (log10-transformed titers).
(B) Plot of serial fecal titers on BBE agar from antibiotic-pretreated mice gavaged on Day 0 with sterile PBS or B. thetaiotaomicron (7×107 cfu/mouse). Individual titers (symbols) and means of log10-transformed titers (lines) are displayed. Unpaired t-tests comparing log10-transformed fecal titers from B. thetaiotaomicron-gavaged dnKO mice to Il10r2+/− mice.
(C) Frequency histogram of all 92 B. thetaiotaomicron fecal titer measurements (log10-transformed) we have performed on individual, single-isolate-gavaged, antibiotic-pretreated mice regardless of mouse genotype. Fecal titers were performed between 6 and 20 days post-gavage; data are compiled from multiple independent experiments with 4–9 mice per experiment. Gavage doses ranged from 6.6×107 to 8.6×108 cfu/mouse. Excluding the single outlier value (8.78), the titers are normally distributed as assessed by the D’Agostino & Pearson K2 omnibus normality test: p = 0.5894, K2 = 1.057, skewness = −0.153, kurtosis = 0.3345. Statistical analysis performed using GraphPad Prism v5.01 (GraphPad Software).
See also Figure S4
Commensal Bacteroides species fulfill Koch’s postulates in host-genotype-specific fashion
We could thus fulfill Koch’s postulates for commensal Bacteroides species by performing serial, quantitative fecal cultures from single-isolate-gavaged dnKO mice on BBE agar. Antibiotic-pretreated mice gavaged with PBS remained free of cultivable Bacteroides for up to 28 weeks, confirming absence of cage-to-cage microbial contamination (Figures 5B and S4B). By contrast, mice given pure cultures of B. thetaiotaomicron, B. vulgatus, or Bacteroides species TP5 became stably colonized at ~1010 cfu/g of feces (Figures 5B, 5C, S4B, and not shown). Bacterial identity was determined based on characteristic colony size, morphology, and pigmentation pattern and was confirmed by 16S rRNA gene sequencing of representative colonies (Figures S4C and S4D). Colonization levels were consistent from animal to animal and experiment to experiment (Figure 5C). Antibiotic-pretreated Il10r2+/− and dnKO mice became stably colonized with the gavaged bacteria at equivalent levels (Figures 5B, 5C, and not shown). These results demonstrated that disease induction was host-genotype-specific.
Commensal Enterobacteriaceae but not Bacteroides are strikingly enriched in the microbiota during spontaneous disease
Despite our finding that commensal Bacteroides induced colitis in dnKO mice, studies of Bacteroides dynamics in the intestinal microbiota of both IBD patients and animal IBD models have produced variable results, with several studies detecting no significant disease-associated enrichment (Dicksved et al., 2008; Frank et al., 2007; Onderdonk et al., 1998; Ott et al., 2008; Takaishi et al., 2008). We therefore examined fecal Bacteroides levels using both quantitative titers on Bacteroides bile esculin agar and qPCR species-specific primers for the B. thetaiotaomicron the 16S rRNA gene (Figures 6A, 6B, 6D, 6F, and S5). We found no significant difference between dnKO (spontaneously colitic) and Il10r2+/− (non-colitic) mice by either of these methods. By contrast, many previous studies have documented IBD-associated enrichment of Enterobacteriaceae, which are Gram-negative, facultative anaerobes (including the species Escherichia coli) from the phylum Proteobacteria that can be selectively cultured on MacConkey agar (Burke, 1997; Darfeuille-Michaud et al., 1998; Frank et al., 2007; Onderdonk et al., 1998). In agreement with findings from these studies, we observed that commensal Enterobacteriaceae were strikingly enriched in spontaneous disease in dnKO mice, accounting for ~50% of total cultivable fecal bacteria compared with <0.5% of cultivable bacteria in co-housed Il10r2+/− controls (Figures 6A, 6C, 6E, and S5).
Figure 6. Commensal Enterobacteriaceae are enriched in spontaneous colitis.
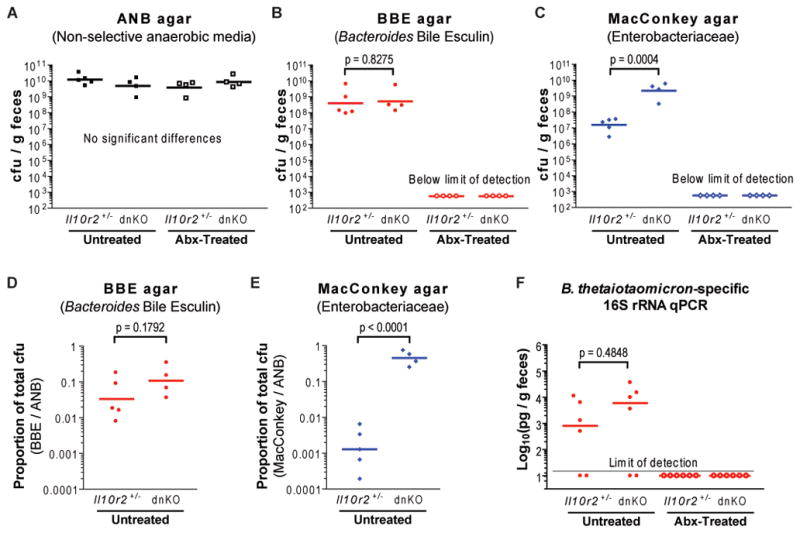
(A to C) Plot of fecal titers from untreated or antibiotic-treated mice of the indicated genotypes grown in parallel anaerobically on ANB and BBE agar and aerobically on MacConkey agar (selective for Enterobacteriaceae). Il10r2+/− and dnKO mice were co-housed; data compiled from ≥2 cages/group. 1-way ANOVA (ANB titers: F3,13 = 1.344, p = 0.3031) or unpaired t-test (log10-transformed titers).
(D and E) Proportions of total cultivable bacteria in Figure 6A to 6C that grew on BBE agar and MacConkey agar, calculated by dividing BBE and MacConkey titers by ANB titers. Data is displayed as individual (symbols) and means of log10-transformed proportions (bars). Unpaired t-test (log10-transformed proportions).
(F) Plots of fecal samples from untreated and antibiotic-treated dnKO and Il10r2+/− mice (n = 6 per group) analyzed by quantitative PCR using primers specific for the B. thetaiotaomicron 16S rRNA gene. Il10r2+/− and dnKO mice within each treatment group were co-housed; data are compiled from ≥3 cages/group. Results of qPCR assays were normalized to sample weight. Mann-Whitney U-test (log10-transformed values).
See also Figure S5
Commensal Enterobacteriaceae are not sufficient for disease induction
The observation that Enterobacteriaceae are often elevated in IBD has led to the hypothesis that these bacteria may play a pathogenic role in disease (Burke, 1997; Frank et al., 2007; Onderdonk et al., 1998). However, it is unclear whether Enterobacteriaceae enrichment is a cause or effect of disease because enrichment also occurs during non-IBD intestinal inflammation experimentally induced by chemicals or microbial pathogens (Heimesaat et al., 2006; Lupp et al., 2007; Stecher et al., 2007). We found that commensal Enterobacteriaceae were eliminated from antibiotic-treated mice (Figure 6C), allowing us to directly test their colitogenic activity in dnKO mice. We isolated an Enterobacteriaceae species that was highly enriched in the feces of an untreated, colitic dnKO mouse (~33% of total cultivable bacteria) and identified it as E. coli by 16S rRNA sequence analysis and phenotypic characterization (Figure S6). To test the isolate’s disease-inducing potential, we gavaged it into antibiotic-pretreated dnKO mice. Other groups of dnKO mice were gavaged with B. thetaiotaomicron or sterile PBS as positive and negative controls. We quantified bacterial colonization by performing serial fecal titers on Bacteroides bile esculin and MacConkey agar (Figures 7A and 7B). E. coli stably colonized antibiotic-pretreated animals at higher levels than those observed in untreated dnKO mice (compare Figures 6C and 7B). However E. coli colonization did not induce significant disease relative to PBS, whereas B. thetaiotaomicron induced significant disease relative to both E. coli and PBS (Figures 7C to 7H).
Figure 7. A colitis-enriched commensal Enterobacteriaceae isolate robustly colonizes antibiotic-pretreated dnKO mice without disease induction.

(A and B) Plot of serial fecal titers grown on BBE and MacConkey agar from antibiotic-pretreated dnKO mice (5–6 per group) gavaged with PBS, B. thetaiotaomicron (1×108 cfu/mouse), or Escherichia coli (2×108 cfu/mouse). Bacterial identity was determined by colony characteristics, confirmed by sequencing the 16S rRNA gene of representative colonies.
(C and D) Cecum and transverse colon pathology scores of mice described in Figures 7A and 7B displayed as individual (symbols) and median (bars) scores. Kruskall-Wallis test with post-hoc Dunn’s tests: (C) H2 = 11.49, p = 0.0032; (D) H2 = 12.02, p = 0.0025. All significant pairwise comparisons are displayed: *, p < 0.05; **, p < 0.01.
(E to H) Transverse colon and rectum crypt heights and crypt widths from mice described in Figures 7A and 7B displayed as mean +/− SEM. 1-way ANOVA with post-hoc Tukey’s test: (E) F2,14 = 2.580, p = 0.1112; (F) F2,14 = 5.434; p = 0.0179; (G) F2,14 = 6.438, p = 0.0104; (H) F2,14 = 7.268, p = 0.0068. All significant pairwise comparisons are displayed if omnibus p-value by ANOVA was significant: *, p < 0.05.
See also Figure S6.
DISCUSSION
The commensal microbiota influences the pathogenesis of many diseases including IBD, but role of specific microbes is incompletely understood. Two central related question are whether specific commensal bacterial subsets induce IBD and, if so, whether their proportions in the microbiota are altered during disease (Strober, 2010; Takaishi et al., 2008; Tannock, 2008). To address these questions, we utilized an antibiotic-responsive genetic mouse model with defects in IL-10 and TGFβ signaling, both of which are associated with human IBD (Franke et al., 2010; Glocker et al., 2009; McGovern et al., 2010). Using these mice, we developed a non-germ-free approach to screen for specific disease-inducing commensal bacteria. Based on the results of this screen, we fulfilled host-genotype-specific Koch’s postulates for commensal Bacteroides species. We isolated Bacteroides species that were common to susceptible and non-susceptible hosts and were eliminated by disease-blocking antibiotics. We administered pure cultures of the isolates to antibiotic-pretreated mice and found that they induced colitis with features of adaptive and innate immunopathology in susceptible hosts but not in non-susceptible hosts. Finally, we showed that the Bacteroides species could be quantitatively re-isolated from experimentally colonized animals at equivalent levels regardless of host genotype and disease status. These results suggest the genotype-dependent disparity in disease induction was attributable to differences in host response rather than altered colonization susceptibility. Conversely, we found that commensal Enterobacteriaceae were strikingly enriched in the microbiota during spontaneous disease but a colitis-enriched Enterobacteriaceae isolate was not sufficient to induce disease despite robust colonization. We further found that antibiotic-treated, non-colitic dnKO mice were colonized at high levels with bacteria belonging to several families of Firmicutes. We thus identified distinct subsets of commensal bacteria with and without disease-inducing potential and demonstrated that the colitogenicity of these subsets would not have been predicted based on disease-associated shifts in colonization.
Given the important role of the intestinal microbiota in IBD pathogenesis, identification of specific disease-inducing commensal microbes has been a longstanding goal (Burke, 1997; Sartor, 2008). Many studies have employed germ-free animals because germ-free re-derivation can provide a source of healthy, yet susceptible hosts for experimental colonization with defined microbial populations. Specifically measuring colonization levels of introduced isolates in ex-germ-free animals is relatively uncomplicated, and the absence of related, indigenous commensal organisms increases confidence that host effects can be directly attributed to the introduced bacteria (Hapfelmeier et al., 2010). But while studies in germ-free animals have been crucial in demonstrating that the microbiota plays a role in disease induction, there are important caveats for using them to identify specific disease-inducing bacteria. Importantly, the innate and adaptive immune system of germ-free animals is immature (Ivanov et al., 2009; Lee and Mazmanian, 2010) and bacteria known to induce chronic inflammation or cancer in conventionally raised animals can produce dramatically different outcomes in germ-free hosts ranging from no disease induction to rapid death (Garrett et al., 2010; Lofgren et al., 2010; Rhee et al., 2009). These disparate results highlight the need for additional strategies to identify and characterize disease-inducing bacteria. The non-germ-free approach we used here generates healthy, IBD-susceptible mice by pre-treating conventionally raised animals with antibiotics that have been widely studied in human IBD (Elahi et al., 2009; Ohkusa et al., 2010; Rahimi et al., 2006). We showed that antibiotic treatment eradicates certain bacterial subsets from the microbiota, enabling us to experimentally colonize antibiotic-pretreated animals with primary isolates of commensal bacteria indigenous to our mouse colony and specifically quantify colonization using culture- and molecular-based techniques. Interestingly, the isolates colonized antibiotic-pretreated mice at levels even higher than those observed in untreated animals, suggesting antibiotics produce an altered microbial ecosystem highly susceptible to expansion of individually re-introduced commensal species. This approach should be broadly applicable to a wide range of animal models and disease processes.
We found that a range of common, commensal Bacteroides species were sufficient for disease induction in antibiotic-pretreated dnKO mice, with the two most potent disease-inducing isolates belonging to the species B. thetaiotaomicron and B. vulgatus. Intestinal Bacteroides are among the most abundant and well-studied members of the mammalian commensal microbiota. They provide many beneficial effects to the host including breakdown of complex dietary carbohydrates and modulation of mucosal glycosylation, gene expression, angiogenesis, and immune maturation (Comstock, 2009; Hooper et al., 2001; Round and Mazmanian, 2010; Stappenbeck et al., 2002; Xu et al., 2007). To our knowledge, B. thetaiotaomicron has not previously been implicated in IBD pathogenesis. Conflicting data exist on the role of B. vulgatus in the pathogenesis of IBD. Germ-free rats transgenically expressing HLA-B27, a human MHC class I allele linked to spondyloarthritis-associated IBD (Brakenhoff et al., 2010), developed T-cell dependent colitis when colonized with B. vulgatus but not with various other bacteria including E. coli (Hoentjen et al., 2007; Rath et al., 1999). Interestingly, E. coli but not Bacteroides species were enriched in the intestines of conventionally raised, spontaneously colitic transgenic rats (Onderdonk et al., 1998). Similarly, B. vulgatus colonization was sufficient for disease induction in germ-free guinea pigs treated with carrageenan (Onderdonk et al., 1983). However B. vulgatus did not induce disease in germ-free Il10−/− mice and appeared to be protective in germ-free Il2−/− mice, whereas some strains of E. coli were colitogenic (Kim et al., 2007; Sellon et al., 1998; Waidmann et al., 2003). The Il10−/− and Il2−/− mice used in those studies were on different genetic backgrounds than the dnKO mice we used here, making it unclear whether the disparity in results was due to host genetic differences or to the fact that our studies were performed in conventionally raised rather than germ-free animals. Given the human-relevant genetic features of dnKO mice and the use of a non-germ-free approach, we believe our findings suggest commensal Bacteroides species may have specific colitogenic effects in at least a subset of human IBD. Mazmanian and colleagues recently coined the term “pathobiont” to describe commensal or symbiotic microbes that induce disease only in certain genetic or environmental contexts (Chow and Mazmanian, 2010; Mazmanian et al., 2008). We propose that fulfillment of host-genotype-specific Koch’s postulates in dnKO mice demonstrates that commensal Bacteroides species act as pathobionts in this genetic context.
Many studies have observed IBD-associated enrichment or depletion of specific commensal bacterial subsets, including most notably enrichment of Enterobacteriaceae and other Proteobacteria (Burke, 1997; Darfeuille-Michaud et al., 1998; Frank et al., 2007; Takaishi et al., 2008). Our finding that a colitis-enriched E. coli isolate was not sufficient to induce disease in antibiotic-pre-treated dnKO mice adds to a growing body of evidence that intestinal inflammation provides both commensal and pathogenic Enterobacteriaceae with a selective colonization advantage regardless of inflammation etiology (Heimesaat et al., 2006; Lupp et al., 2007; Stecher et al., 2007; Winter et al., 2010). Interestingly, Proteobacteria enrichment also occurs in the lung microbiota of asthma patients (Hilty et al., 2010) and the gastric microbiota of patients with Helicobacter pylori-induced chronic atrophic gastritis (Maldonado-Contreras et al., 2010). The pathogenic Enterobacteriaceae species Salmonella typhimurium directly exploits intestinal inflammation by using tetrathionate a compound generated in the presence of host-derived reactive oxygen species as a respiratory electron acceptor to outcompete the commensal bacteria (Winter et al., 2010). We speculate that analogous mechanisms for exploiting mucosal inflammation may account for Enterobacteriaceae enrichment in IBD and might also be related to the frequency with which non-pathogenic Enterobacteriaceae strains have independently and convergently evolved pathogenic phenotypes (Pupo et al., 1997; Wirth et al., 2006). Thus, although colitis-enriched bacteria, including some Enterobacteriaceae, may play pro-colitic roles in certain circumstances (Darfeuille-Michaud et al., 1998; Garrett et al., 2010), our findings underscore the importance of functionally assessing a microbe’s disease-inducing potential rather than simply extrapolating from disease-associated shifts in microbiota composition.
In summary, our results provide important insights into the intestinal microbiota’s role in IBD induction. This work does not support models in which a single microbial species is both necessary and sufficient for disease to occur, nor can we exclude the possibility that additional commensal species unexamined in our study may also be capable of disease induction. Nonetheless, we provide evidence that specific bacterial subsets can induce IBD whereas other subsets cannot. We demonstrate that the identification and validation of these subsets is experimentally feasible. As new spontaneous animal models of IBD based on other human susceptibility mutations become available, the experimental criteria and conceptual framework developed here will allow the contributions of commensal bacteria to be assessed in additional genetic contexts.
EXPERIMENTAL PROCEDURES
Mouse Care
Animal protocols were approved by Washington University’s animal studies committee. Generation of dnKO mice was previously described (Kang et al., 2008). dnKO mice are CD4-dnTgfβrII × Il10r2−/−. Il10r2+/− littermates were used as controls. Mice were housed in a specific pathogen-free facility in cages that were autoclaved after assembly and opened only in a laminar flow cabinet after thorough disinfection of all outer surfaces with a 1:8:1 preparation of Clidox®S (Pharmacal Research Laboratories, Inc.). Il10r2+/− and dnKO mice were co-housed. Antibiotic treatment consisting of 0.66 mg/ml ciprofloxacin, 2.5 mg/ml metronidazole (Sigma), and 20 mg/ml sugar-sweetened grape Kool-Aid Mix (Kraft Foods) in drinking water (Kang et al., 2008) was begun at weaning (3 weeks old); mice were transferred to fresh sterile cages 2–3 times per week. For gavage experiments, mice were antibiotic-pretreated for ≥3 weeks, gavaged 2 days after treatment cessation, and euthanized 3 weeks post-gavage or upon loss of ≥30% of maximum body weight. Data are representative of at least 2 independent experiments per group unless otherwise indicated.
Tissue harvest, fixation, and preparation for histology
Mice were euthanized and ceca and colons were immediately removed, flushed with PBS and Bouin’s fixative, opened longitudinally with scissors (ceca were opened from the ileocecal junction to the cecal tip along the greater curvature), pinned open mucosa-upward in square Petri dishes filled with wax (Carolina Biological), fixed in Bouin’s fixative for 4–8 hours at 4°C, washed and stored in 70% ethanol, and processed for histology as previously described (Kang et al., 2008).
Gross pathology scoring, histology, and histologic quantification of mucosal inflammation
Whole mount images of fixed intestines were taken at 7X (cecum only), 20X, 40X, and 90X magnification using an Olympus SZX12 dissecting microscope with an Olympus DP70 Digital Microscope Camera at 1360×1024 image size (Diagnostic Instruments). Images were scored for gross pathology according to a previously validated scoring system (Figure S1) (Kang et al., 2008) by an anatomic pathologist (TSS) blinded to the identity of the samples. Representative images of rectal histology (≤0.5 cm from the anorectal junction) were taken using the same camera on an Olympus BX51 microscope. Blinded microscopic analysis of HE-stained histologic samples for height and width of well-oriented crypts in the transverse colon, and distal 0.5 cm of the rectum was performed at 400X magnification as previously described (Kang et al., 2008). Counts of neutrophil trans-epithelial migration per 50 crypts were performed on non-ulcerated segments of colon by TSS.
DNA extraction and PCR amplifications
Fecal pellets were collected from untreated mice or mice treated with antibiotics for ≥3 weeks and frozen. Bacterial genomic DNA was extracted using a bead-beating system (Matrix E and Bio101 FastPrep cell disrupter, MP Biomedicals; Solon, OH) and a commercial kit (QIAamp DNA Stool Mini Kit, Qiagen, Valencia, CA) following the manufacturer recommendations. Extracted genomic DNA was used as a template for PCR amplification and quantification (pg of DNA/μl) of different bacterial groups. Bacterial DNA concentrations were estimated by quantitative PCR (qPCR) assays using primers and protocols previously validated in our laboratory (Nava et al., 2010). Total bacterial load was estimated based on amplification of the bacterial rpoB gene (Nava et al., 2010). 16S rRNA gene primers specific for bacterial families were used to quantify the groups Bacteroidaceae-Porphyromonadaceae-Prevotellaceae, Lachnospiraceae-Ruminococcaceae, Enterococcaceae (Rinttila et al., 2004) and Lactobacillaceae (Barman et al., 2008).
Bacteroides thetaiotaomicron was quantified using established species-specific primers targeting the 16S rRNA gene (Miyamoto et al., 2002). Specificity of this primer set was verified using the Probe Match function at the Ribosomal Database Project (Cole et al., 2009) and by PCR assays using DNA from the Bacteroides thetaiotaomicron, Bacteroides sp. TP5, B. uniformis, and Parabacteroides goldsteinii isolates described in Table S3. The cycling protocol was: denaturation at 94°C (3 min), 33 cycles of 94°C (45 sec), 61.6°C (30 sec), 72°C (45 sec), followed by one cycle of 72°C (7 min). Specificity was confirmed by visualizing single and expected-size PCR amplicons via agarose gel electrophoresis.
All qPCR assays were conducted in a Mastercycler ep realplex real-time PCR machine (Eppendorf, Westbury, NY) using SYBR Green PCR technology (Clontech Laboratories, Mountain View, CA). Each 17 μl qPCR mixture consisted of 8.5 ul of 2X SYBR Green Master Mix (Clontech Laboratories), 1.7 ul of BSA (100 μg/ml), 0.5 ul of each primer (10 μM), 2 ul of extracted genomic DNA and 3.10 ul PCR-grade water. Denaturation curves were determined from 60°C to 95°C for all products for quality assurance. DNA concentrations (rpoB and 16S rRNA genes) in each fecal sample were determined using the absolute quantification method. Standard curves were constructed with 5-fold dilutions of genomic DNA templates of known concentration. DNA extracted from mouse intestinal contents was used as a template for total bacteria and family-specific qPCR, whereas DNA extracted from B. thetaiotaomicron was used for species-specific qPCR. Concentrations of DNA used in the standard curves ranged from 100 ng to 1.3 pg/μl. For each qPCR assay, standard curves were amplified at the same time as fecal samples. PCR amplifications were performed in triplicate. Bacterial group-specific qPCR signals were normalized to total Bacteria (rpoB gene) qPCR signal. Then, bacterial DNA densities were estimated by gram of fecal pellet. Detection limit for family-specific and B. thetaiotaomicron qPCR assays were around 6.4 /μl and 1.28 /μl, respectively.
Intestinal content and mixed culture preparation
All anaerobic bacteriology and manipulation of uncultured intestinal contents was performed in a Bactron IV Anaerobic Chamber (Sheldon Manufacturing) in an atmosphere of 5% hydrogen/5% carbon dioxide/90% nitrogen (Airgas-Mid America). Anaerobic cultures were incubated at 35–36°C while aerobic cultures were incubated at 37°C in ambient air.
To develop a standardized source of intestinal contents, ceca were harvested from untreated littermates of dnKO mice and were then transferred to the anaerobic chamber where contents were removed, suspended in sterile, pre-reduced PBS + 20% glycerol, and frozen at −80°C in single-use aliquots in 1.8 mL CryoTube Vials (377267, Nunc).
For mixed cultures of intestinal contents, frozen intestinal contents harvested from untreated mice (see above) were thawed, transferred to the anaerobic chamber, adjusted to the concentration used for gavage in Figure 2, serially diluted in sterile, pre-reduced PBS, plated in parallel on various solid media types, and incubated either anaerobically or aerobically. Anaerobic cultures were grown on anaerobic reducible blood agar (“ANB”, R01059, Remel), anaerobic reducible blood agar with colistin and nalidixic acid (“CNA”, R01064, Remel), or anaerobic reducible laked blood agar with kanamycin and vancomycin (“LKV”, R01068, Remel). After 3 days of incubation, material was harvested from anaerobic culture plates at the indicated dilutions (Figure 3A and Table S1) using sterile swabs, suspended in sterile pre-reduced PBS, and frozen at −80 C in single-use aliquots with 20% pre-reduced glycerol. Aerobic cultures grown on chocolate agar (R01300, Remel) were harvested by the same method after 48 hours incubation (Table S1).
Bacterial isolation
To isolate bacteria from antibiotic-treated dnKO mice, fecal samples were collected in sterile microcentrifuge tubes, immediately placed on ice, weighed, transferred to an anaerobic chamber within ≤1 hr of collection, suspended in 500 μl of sterile pre-reduced PBS by repeated vigorous vortexing and disruption with a sterile pipette tip, serially diluted, and cultured on non-selective ANB agar. Unique-appearing colonies were picked from plates, sub-cultured on fresh media, and passaged to ensure purity. Genomic DNA was extracted from individual isolates, the 16S rRNA gene was PCR-amplified using primers Bact-8f and Bact-1510r (Eckburg et al., 2005; Gill et al., 2006). The resulting PCR product was directly Sanger sequenced using the original PCR primers. Sequences were edited and trimmed for quality using Trev 1.9 from the Staden Package© and assembled using SeqMan from the DNAStar© Lasergene software package, Version 8.0.2. Sequences were classified using the Ribosomal Database Project (RDP, Release 10, Update 15) Classifier and the most similar bacterial type strain sequences were identified using RDP’s SeqMatch program supplemented by BLAST (Ackermann et al., 2008; Cole et al., 2007; Cole et al., 2009; Wang et al., 2007). Sequence identity was determined by BLAST2 analysis (Table S2).
To isolate Gram-negative, anaerobic bacteria, freshly thawed aliquots of intestinal contents were diluted to the concentration used in preparation of the LKV mixed culture used for gavage (Table S1) and cultured anaerobically on LKV or BBE agar. Unique species of Gram-negative obligate anaerobes isolated from LKV agar (all of them members of the phylum Bacteroidetes) are listed in Table S3. For isolates <98% identical to the most similar type strain, the most similar cultured isolate in the RDP database was also identified. Escherichia coli (Figure S6) was isolated from feces of an untreated dnKO mouse cultured on ANB agar, and identified by 16S rRNA sequence supplemented by phenotypic identification using the API 20E strip (biocode 5144532, bioMerieux, Inc., Durham, NC) in the Barnes-Jewish Hospital Medical Microbiology Laboratory (99% confidence).
For preparation of gavage inocula from single isolates, 24-hour cultures of Bacteroidetes were grown anaerobically in standing culture in TYG broth (Goodman et al., 2009) and 16-hour cultures of E. coli were grown in Trypticase Soy Broth (#22092, Sigma) in a 37°C shaker at 225 rpm. Each culture was concentrated by centrifugation, mixed with sterile, pre-reduced PBS + glycerol to a final concentration of 20% glycerol, and frozen at −80°C in single-use aliquots.
Gavage experiments
Titers of each frozen stock were determined prior to use in gavage. To perform colonizations, aliquots were thawed in a 37°C water bath, transferred to the anaerobic chamber, volume-adjusted in sterile pre-reduced PBS, and immediately transported to the mouse facility for orogastric gavage (Goodman et al., 2009). Gavage doses were confirmed by back-titering the inocula.
Titration of Fecal Bacteria
Fecal samples from 4-week-old untreated mice, mice treated with antibiotics for ≥3 weeks, or antibiotic-pretreated mice experimentally manipulated as described were collected in sterile microcentrifuge tubes. Samples were immediately placed on ice, weighed, transferred to an anaerobic chamber within ≤1 hr of collection, suspended in 500 μl of sterile pre-reduced PBS by repeated vigorous vortexing and disruption with a sterile pipette tip, and titered by culturing 10-fold serial dilutions on ANB agar, Bacteroides bile esculin (BBE) agar (R01104, Remel), or MacConkey agar. 10 μl of each dilution was spotted in triplicate on quadrants of titer plates (see Figure S4D). MacConkey titers were counted after 12–16 hours and confirmed for negative growth at 48 hours, BBE titers were counted after 48 hours with colony characteristics confirmed under magnification, and ANB titers were counted at 48 hours and confirmed after 5 days. Identity of bacteria on BBE and MacConkey titer plates from single-isolate-gavaged mice was confirmed by 16S rRNA gene sequencing of representative colonies.
Bacterial culture images
Culture plates were photographed with backlighting using a digital camera. Representative colonies were imaged on a dissecting microscope at 12.5X magnification with both backlighting and reflected lighting at an exposure time of 1/500 s.
Serum Immunoblots
Lysates of pure cultures of B. thetaiotoaomicron or B. vulgatus were obtained by Fastprep bead lysis using 106 μm acid-washed glass beads (Sigma, Cat. # G4649). Protein was quantified using the DC protein assay (BioRAD, Cat. # 500-0111). Aliquots containing 50 μg of protein were separated using NuPAGE precast 10% polyacrylimide Bis-Tris gels and then transferred to nitrocellulose membranes. Membranes were probed at room temperature for 3 hrs with a 1:200 dilution of serum from antibiotic-pretreated Il10r2+/− or dnKO mice collected immediately prior to gavage with live cultures of the respective bacterial isolate (“pre-gavage”) or collected at sacrifice 3 weeks later (“post-gavage”). The secondary probe was HRP-conjugated goat anti-murine IgG (BioRAD, Cat. # 170-6516) diluted 1:10 000 and incubated at room temperature for 1 hour. Blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Cat. #34077). Gels were run, transferred, blotted, and exposed in parallel to ensure comparability.
Statistical Analysis
Statistical analysis was performed using Prism v3.02 and v5.01 (GraphPad Software) except for Mann-Whitney U-tests, which were calculated using SPSS 16.0 (SPSS Inc.). Significance for crypt morphometric measurements and log10-transformed fecal bacteria titers and titer proportions was determined as described in figure legends using unpaired t-test, Dunnett’s multiple comparison test, or 1-way ANOVA with post-hoc Tukey’s multiple comparison test if the omnibus p-value by ANOVA was < 0.05. F-statistics are reported as Ftreatment,residual. Significance for pathology scores, bacterial qPCR, and neutrophil trans-epithelial migration was determined by Mann-Whitney U-test, Dunn’s multiple comparison test, or Kruskal-Wallis test as indicated. Kruskal-Wallis statistics are reported as Hdegrees-of-freedom. Statistical significance was defined as p < 0.05; p-values are two-tailed. Significance of pairwise comparisons was denoted by: n.s., p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.005.
Supplementary Material
HIGHLIGHTS.
Non-germ-free screen for disease-inducing bacteria in a genetic mouse model of IBD
Common commensal Bacteroides fulfill host-genotype-specific Koch’s postulates
Commensal Enterobacteriaceae are enriched during spontaneous disease
Enterobacteriaceae not sufficient to induce disease despite robust colonization
Acknowledgments
We thank D. Kreamalmeyer S. L. Brown, and J. Y. Liu for technical support, M. E. Bloom, W. F. Stenson, and E. R. Unanue for critical reading of the manuscript, and members of the Stappenbeck laboratory for helpful discussions. Work was supported by a grant from the Broad Foundation with additional support from Pfizer and Washington University Digestive Diseases Research Core Center DK52574. S.M.B. was supported by NIH training grant 5T32AI0071632. None of the authors have a financial interest related to this work.
References
- Ackermann M, Stecher B, Freed NE, Songhet P, Hardt WD, Doebeli M. Self-destructive cooperation mediated by phenotypic noise. Nature. 2008;454:987–990. doi: 10.1038/nature07067. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakenhoff LKPM, Heijde DMvd, Hommes DW, Huizinga TWJ, Fidder HH. The joint-gut axis in inflammatory bowel diseases. J Crohns Colitis. 2010;4:257–268. doi: 10.1016/j.crohns.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Brandwein SL, McCabe RP, Cong Y, Waites KB, Ridwan BU, Dean PA, Ohkusa T, Birkenmeier EH, Sundberg JP, Elson CO. Spontaneously colitic C3H/HeJBir mice demonstrate selective antibody reactivity to antigens of the enteric bacterial flora. J Immunol. 1997;159:44–52. [PubMed] [Google Scholar]
- Burke D. Escherichia coli and ulcerative colitis. J R Soc Med. 1997;90:612–617. doi: 10.1177/014107689709001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010;7:265–276. doi: 10.1016/j.chom.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35:D169–172. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock LE. Importance of glycans to the host-bacteroides mutualism in the mammalian intestine. Cell Host Microbe. 2009;5:522–526. doi: 10.1016/j.chom.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- Dicksved J, Halfvarson J, Rosenquist M, Jarnerot G, Tysk C, Apajalahti J, Engstrand L, Jansson JK. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. Isme J. 2008;2:716–727. doi: 10.1038/ismej.2008.37. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahi B, Nikfar S, Derakhshani S, Vafaie M, Abdollahi M. Benefit of antibiotic therapy on pouchitis after ileal pouch anal anastomosis: A systematic review and meta-analysis of clinical trials. Central European Journal of Medicine. 2009;4:164–170. [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Gallini CA, Yatsunenko T, Michaud M, Dubois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–1709. doi: 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper PH, Lee EC, Kettlewell MG, Bennett MK, Jewell DP. Role of the faecal stream in the maintenance of Crohn’s colitis. Gut. 1985;26:279–284. doi: 10.1136/gut.26.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, Jahn HK, Dunay IR, Moter A, Gescher DM, et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoentjen F, Tonkonogy SL, Qian BF, Liu B, Dieleman LA, Sartor RB. CD4+ T lymphocytes mediate colitis in HLA-B27 transgenic rats monoassociated with nonpathogenic Bacteroides vulgatus. Inflammatory Bowel Diseases. 2007;13:317–324. doi: 10.1002/ibd.20040. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowitz HD, Croen EC, Sachar DB. The role of the fecal stream in Crohn’s disease: an historical and analytic review. Inflamm Bowel Dis. 1998;4:29–39. doi: 10.1097/00054725-199802000-00006. [DOI] [PubMed] [Google Scholar]
- Kang SS, Bloom SM, Norian LA, Geske MJ, Flavell RA, Stappenbeck TS, Allen PM. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med. 2008;5:e41. doi: 10.1371/journal.pmed.0050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Tonkonogy SL, Karrasch T, Jobin C, Sartor RB. Dual-association of gnotobiotic IL-10−/− mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis. 2007;13:1457–1466. doi: 10.1002/ibd.20246. [DOI] [PubMed] [Google Scholar]
- Lee YK, Mazmanian SK. Has the Microbiota Played a Critical Role in the Evolution of the Adaptive Immune System? Science. 2010;330:1768–1773. doi: 10.1126/science.1195568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M, Hershberg RM. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, et al. Lack of Commensal Flora in Helicobacter pylori-Infected INS-GAS Mice Reduces Gastritis and Delays Intraepithelial Neoplasia. Gastroenterology. 2010;140:210–220. e214. doi: 10.1053/j.gastro.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, Brodie EL, Dominguez-Bello MG. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. Isme J. 2010 doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, Neale BM, Ong RT, Lagace C, Li C, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Watanabe K, Tanaka R, Itoh K. Distribution analysis of six predominant Bacteroides species in normal human feces using 16s rDNA-targeted species-specific primers. Microbial Ecology in Health and Disease. 2002;14:133–136. [Google Scholar]
- Nava GM, Friedrichsen HJ, Stappenbeck TS. Spatial organization of intestinal microbiota in the mouse ascending colon. 2010 doi: 10.1038/ismej.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkusa T, Kato K, Terao S, Chiba T, Mabe K, Murakami K, Mizokami Y, Sugiyama T, Yanaka A, Takeuchi Y, et al. Newly developed antibiotic combination therapy for ulcerative colitis: a double-blind placebo-controlled multicenter trial. Am J Gastroenterol. 2010;105:1820–1829. doi: 10.1038/ajg.2010.84. [DOI] [PubMed] [Google Scholar]
- Onderdonk AB, Cisneros RL, Bronson RT. Enhancement of experimental ulcerative colitis by immunization with Bacteroides vulgatus. Infect Immun. 1983;42:783–788. doi: 10.1128/iai.42.2.783-788.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onderdonk AB, Richardson JA, Hammer RE, Taurog JD. Correlation of cecal microflora of HLA-B27 transgenic rats with inflammatory bowel disease. Infect Immun. 1998;66:6022–6023. doi: 10.1128/iai.66.12.6022-6023.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott SJ, Plamondon S, Hart A, Begun A, Rehman A, Kamm MA, Schreiber S. Dynamics of the mucosa-associated flora in ulcerative colitis patients during remission and clinical relapse. J Clin Microbiol. 2008;46:3510–3513. doi: 10.1128/JCM.01512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and Functions of IL-10 Family Cytokines in Inflammation and Diseases. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 2009;22:292–301. doi: 10.1097/QCO.0b013e32832a8a5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupo GM, Karaolis DK, Lan R, Reeves PR. Evolutionary relationships among pathogenic and nonpathogenic Escherichia coli strains inferred from multilocus enzyme electrophoresis and mdh sequence studies. Infect Immun. 1997;65:2685–2692. doi: 10.1128/iai.65.7.2685-2692.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi R, Nikfar S, Rezaie A, Abdollahi M. A meta-analysis of broad-spectrum antibiotic therapy in patients with active Crohn’s disease. Clin Ther. 2006;28:1983–1988. doi: 10.1016/j.clinthera.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Rath HC, Wilson KH, Sartor RB. Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with Bacteroides vulgatus or Escherichia coli. Infect Immun. 1999;67:2969–2974. doi: 10.1128/iai.67.6.2969-2974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, Rabizadeh S, Golub J, Mathews LE, Shin J, et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun. 2009 doi: 10.1128/IAI.00814-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97:1166–1177. doi: 10.1111/j.1365-2672.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastrenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SD, Di Marco F, Hooley J, Pitts-Meek S, Bauer M, Ryan AM, Sordat B, Gibbs VC, Aguet M. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187:571–578. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A. 2002;99:15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson WF, Hanauer SB, Cohen RD. Inflammatory bowel disease. In: Yamada T, Alpers DH, Kalloo AN, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology. Hoboken, NJ: Wiley-Blackwell; 2009. pp. 1386–1472. [Google Scholar]
- Strober W. Inside the microbial and immune labyrinth: Gut microbes: friends or fiends? Nat Med. 2010;16:1195–1197. doi: 10.1038/nm1110-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaishi H, Matsuki T, Nakazawa A, Takada T, Kado S, Asahara T, Kamada N, Sakuraba A, Yajima T, Higuchi H, et al. Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int J Med Microbiol. 2008;298:463–472. doi: 10.1016/j.ijmm.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Tannock GW. The search for disease-associated compositional shifts in bowel bacterial communities of humans. Trends Microbiol. 2008;16:488–495. doi: 10.1016/j.tim.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Waidmann M, Bechtold O, Frick JS, Lehr HA, Schubert S, Dobrindt U, Loeffler J, Bohn E, Autenrieth IB. Bacteroides vulgatus protects against Escherichia coli-induced colitis in gnotobiotic interleukin-2-deficient mice. Gastroenterology. 2003;125:162–177. doi: 10.1016/s0016-5085(03)00672-3. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein WM, Onderdonk AB, Bartlett JG, Gorbach SL. Experimental intra-abdominal abscesses in rats: development of an experimental model. Infect Immun. 1974;10:1250–1255. doi: 10.1128/iai.10.6.1250-1255.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol. 2006;60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, Henrissat B, Coutinho PM, Minx P, Latreille P, et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.