

Abstract
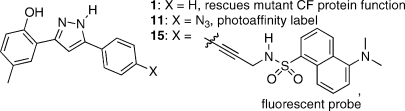
Cystic fibrosis is a genetic disease caused by mutations in the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. In vitro experiments have demonstrated that 4-methyl-2-(5-phenyl-1H-pyrazol-3-yl)phenol (VRT-532, 1) is able to partially restore the function of mutant CFTR proteins. To help elucidate the nature of the interactions between 1 and mutant CFTR, molecular probes based on the structure of 1 have been prepared. These include a photoreactive aryl azide derivative 11 and a fluorescent dansyl sulfonamide 15. Additionally, a method for hydrogen isotope exchange on 1 has been developed, which could be used for the incorporation of radioactive tritium. Using iodide efflux assays, the probe molecules have been demonstrated to modulate the activity of mutant CFTR in the same manner as 1. These probe molecules enable a number of biochemical experiments aimed at understanding how 1 rescues the function of mutant CFTR. This understanding can in turn aid in the design and development of more efficacious compounds which may serve as therapeutic agents in the treatment of cystic fibrosis.
Introduction
Cystic fibrosis (CF) is one of the most common genetic diseases among Caucasians. The illness is best known for affecting the respiratory system, where the lungs become obstructed with thick sticky mucus, resulting in difficulty breathing and increased susceptibility to bacterial infections. The disease also affects the pancreas, liver, intestines, and male reproductive system.
CF is caused by mutations in the gene for the cystic fibrosis transmembrane conductance regulator protein (CFTR). In healthy individuals, CFTR acts as a phosphorylation and nucleotide regulated channel which mediates the flux of chloride across the apical membrane of epithelial cells.(1) In CF patients, mutant CFTR fails to correctly mediate chloride flux, and as a consequence, transepithelial chloride, salt, and water transport is impaired. This transport defect leads to dehydration of the airway surface fluid, mucus desiccation, and obstruction with recurrent episodes of inflammation and infection. Currently, CF is treated through careful control of diet, physiotherapy, antibiotics and, when lung function is severely degraded, lung transplantation.(2) The average life expectancy for a CF patient is 47 years.(3) There are currently no pharmacological agents approved for use in the clinic that address the basic molecular defect underlying the disease.
The most common mutation in CFTR, occurring on at least one allele in 70% of CF patients, is the deletion of a phenylalanine residue at position 508 in the amino acid sequence (F508del-CFTR).(4) This defect results in protein misfolding, retention in the endoplasmic reticulum, and its failure to reach the cell membrane.(5) In cell culture expression systems, this trafficking defect can be partially overcome by incubating cells at 27 °C,(6) however the protein’s ability to function as a chloride channel at the cell surface remains impaired relative to that of the wild-type CFTR.(7) Another less common mutation: G551D-CFTR undergoes normal biogenesis and trafficking to the cell surface(5) yet it exhibits defective channel activation at that location.(8) Thus, two classes of small molecules have been identified, which may prove to be useful in the pharmacological treatment of CF.9,10 “Correctors” are those molecules which, through either direct interactions with mutant CFTR or perhaps any of the various chaperone proteins, rescue the cell’s ability to correctly traffic the protein to the cell membrane. “Potentiators” are those molecules which result in a restoration of normal channel activity in the mutant protein once it appears in the cell membrane.
In 2006, a group from Vertex Pharmaceuticals reported that 4-methyl-2-(5-phenyl-1H-pyrazol-3-yl)phenol (VRT-532, 1) was identified
in a high-throughput screen for the ability to potentiate channel
activity in mouse NIH 3T3 cells expressing wild-type CFTR or F508del-CFTR
after low-temperature correction of the trafficking defect.(11) Subsequent reports have shown that 1 also partially promotes proper protein trafficking of F508del-CFTR
from the endoplasmic reticulum (ER) to the cell membrane.(12) Thus, 1 is both a potentiator and
a corrector. It has been suggested on the basis of limited proteolysis
studies that this molecule may modify the conformation of F508del-CFTR
such that it more closely resembles that of the wild-type protein,
and that this interaction is sufficient to partially rescue both protein
biogenesis and channel activity.(13) Understanding
the molecular basis for this interaction will provide insight into
the mechanism of action of 1 as a modulator of CFTR and
provide a template for development of therapeutically efficacious
compounds.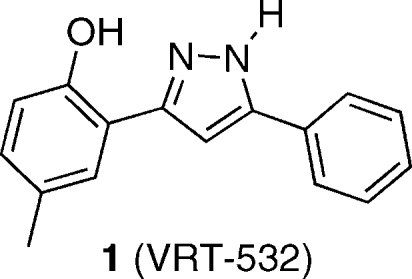
To help elucidate the nature of the interaction between 1 and mutant CFTR proteins, we have designed and prepared derivatives of 1, modified with functional groups useful in biochemical studies. These derivatives include a photoaffinity label and a fluorescent probe. We have also developed a protocol for hydrogen isotope exchange (deuteration by D2O, as a model for tritiation). We show in functional assays of the channel activity of the mutant CFTR proteins that these derivatives retain the potentiator activity of the parent compound and, hence, have the potential to define the molecular basis for this activity.
Results and Discussion
Synthesis of 3,5-Diarylpyrazoles and a Photoaffinity Label
A photoaffinity label is a molecule that contains a recognition element for specific interactions with a site on a target protein as well as a photoactivatible functional group that reacts to form a covalent bond to the protein upon photoirradiation. The labeled site can then be identified using any of a number of biochemical techniques, for instance limited proteolysis followed by gel electrophoresis and/or mass spectrometry to suggest the location of the binding site for the unlabeled ligand.(14)
Our synthetic efforts to prepare derivatives of 1 including the photoaffinity label, aryl azide 11 are illustrated in Scheme 1. Reaction of 2′-hydroxy-5′-methylacetophenone 2 with 4-iodobenzoyl chloride 3 afforded the expected ester 6 in 86% yield. The ester was subjected to the Baker–Venkataraman rearrangement by heating with t-BuOK in THF, intending to isolate diketone 8.(15) The crude product of this reaction was a yellow solid which by 1H NMR spectroscopy suggested the presence of numerous species. Repeated attempts at recrystallization failed to yield any material with apparent improved purity. Reasoning that perhaps the additional signals arise from a mixture of keto–enol tautomers, which ought to be competent in the subsequent pyrazole formation, it was decided to carry the crude yellow solid through the next step. Treatment with hydrazine in acetic acid afforded the desired pyrazole 9 in a serviceable 68% yield (from ester 6). Exchange of the iodide for azide using a Cu/proline catalyzed methodology reported by Ma(16) was hampered by the fact that the desired azide 11 and starting material iodide 9 have identical chromatographic properties on silica gel. It was therefore impossible to gauge reaction progress by TLC and impossible to isolate 11 from the reaction mixture by column chromatography. In fact, it was only by 1H NMR analysis of what appeared by TLC analysis to be clean recovered 9 that we were able to observe that the material was actually a nearly 1:1 mixture of 9 and another compound with the same NMR splitting pattern. After this discovery, TLC analysis of this material using a variety of common eluents failed to resolve the two compounds. Treatment of a portion of the mixture with triphenylphosphine in wet THF (Staudinger reaction conditions)(17) led to the formation of the amine compound 12, confirming that the second component of the 1:1 mixture was in fact the azide 11, formed in only about 50% conversion in the iodide-for-azide exchange reaction. Given this poor conversion, as well as the inability to separate 11 from 9 by any practical means, we opted instead to pursue a different route to the azide. Nitro compound 10 was prepared by the same sequence as described above. Stannous chloride reduction of 10 provided amine 12 in 71% yield. Diazotization of 12 by treatment with t-butyl nitrite, followed by treatment with azidotrimethylsilane,(18) provided the desired azide 11 in excellent 94% yield.
Scheme 1.

Reagents and conditions: (i) pyridine, 0 °C, 1 h; (ii) KOt-Bu, THF, 50 °C, 30 min; (iii) H2N–NH2·xH2O, AcOH, 65 °C, 16 h; (iv) NaN3, CuI, l-proline, NaOH, DMSO, 100 °C, 16 h; (v) PPh3, H2O, THF; (vi) SnCl2·2H2O, EtOH, 78 °C, 16 h; (vii) t-BuONO, TMS-N3, MeCN, 0 °C to rt, 1 h.
Subsequent to these experiments, we discovered that the entire synthesis of the pyrazole core from acetophenone 2 and benzoyl chloride 5 could be accomplished in one pot, using pyridine as a solvent, at 50 °C. With monitoring by TLC, additional 5 was added until 2 was completely converted. At this point, t-BuOK was added, again using TLC to gauge complete consumption of the intermediate ester. Finally, aqueous hydrazine was added, followed by acetic acid. Thus, in less than one day’s reaction time, in one pot, and with only a single, final chromatographic purification step, 1 was isolated in 65% yield from 2. Using the same one-pot sequence, 10 has also been prepared from 2 and 4 in 24% yield.
Under UV irradiation, aryl azides extrude a molecule of N2, transiently forming a reactive nitrene intermediate, which rearranges to an electrophilic cyclic ketenimine.(14) In photoaffinity labeling experiments, this occurs within a protein binding site, and the ketenimine is quenched by a nucleophilic functional group from the protein, thus forming a covalent linkage between the label and the protein (Scheme 2a). To demonstrate that azide 11 is capable of undergoing this chemistry, a benzene solution of 11, containing an excess of diethylamine, was irradiated with an unfiltered (full spectrum) mercury lamp (Scheme 2b). After workup and chromatographic purification, diethylamine adduct 13 was isolated in 40% yield. Thus, 11 exhibits the necessary photoreactivity for photoaffinity labeling experiments.
Scheme 2.

(a) Typical photoreaction of aryl azide photoaffinity labels. Upon irradiation, the azide decomposes to the reactive nitrene, which rearranges to the cyclic ketenimine, which is ultimately quenched by a nucleophilic functional group from the protein target. (b) Photoreaction of 11, with trapping by diethylamine.
Synthesis and Properties of a Fluorescent Conjugate
The amino group on compound 12 was envisaged to be a handle for further functionalization, for instance by reaction with fluorescent electrophiles like 4-chloro-7-nitrobenzofurazan (NBD chloride)(19) or 5-(dimethylamino)naphthalene-1-sulfonyl chloride (dansyl chloride),(20) however no reaction was observed on treatment of 12 with either of these reagents under their prescribed reaction conditions. Rather than attempt to optimize reaction with these costly reagents, we reasoned that incorporation of a glycine amide might provide a more reactive aliphatic amino group for further derivatization. Unfortunately, attempts to acylate 12 with Boc-glycine using a variety of standard peptide coupling agents also failed. Ultimately, we decided to abandon this approach, instead choosing to focus on palladium-catalyzed cross-coupling chemistry to install a fluorophore. The N-propargyl dansyl amide 14 was prepared(21) and found to react cleanly with iodide 9 under Sonogashira reaction(22) conditions to afford 15 in 47% yield (Scheme 3). Given the success of this reaction, Boc-propargylamine 16 was also prepared(23) and reacted with 9, affording 17 in 69% yield. Upon Boc-deprotection, this compound will afford a nucleophilic primary aliphatic amine, which can be readily derivatized with electrophiles, should the need arise to prepare new derivatives in the future.
Scheme 3.

Reagents and conditions: (i) PdCl2·dppf·CH2Cl2, CuI, NEt3, THF, 45 °C, 15 h.
Dansyl sulfonamides typically display fluorescence that is sensitive to their local environments. Figure 1 shows the emission spectrum of 15, recorded in solvents of different polarity. When dissolved in chloroform, fluorescence emission is maximal and centered at about 504 nm. In the most polar of the media tested, an aqueous buffer solution, the emission maximum was red-shifted by 37 nm to 541 nm, with an intensity 32% that of the emission in chloroform. In the organic solvents dimethyl sulfoxide and methanol, which are increasingly more polar than chloroform, but less so than water, fluorescent emissions maxima were intermediate (529 nm in both) and intensity decreased with increasing polarity. Probe 15 contains the recognition element of 1, conjugated to a fluorescent dansyl group through a propargylamine linker and, as such, is expected to be a useful probe for the binding interaction between 1 and CFTR. The direct binding of probe 15 to CFTR, and/or the conformational changes which mediate protein folding and function, may be detected in fluorescence anisotropy measurements.(24) The probe’s fluorescence intensity or emission maximum may change upon binding to a hydrophobic pocket of CFTR, which would be monitored by fluorescence experiments. Alternatively, as the emission for the tryptophan residues in CFTR overlaps with the excitation of this dansyl derivative, there may be measurable resonance energy transfer upon binding to the protein.(25) Lastly, we anticipate that competition experiments with unlabeled drugs will identify other small molecules that bind directly to CFTR and permit quantification of their relative binding constants.
Figure 1.

Fluorescence emission spectra of 20 μM solution of 15 in CHCl3 (black solid line), DMSO (black dotted line), MeOH (gray solid line), and aqueous buffer containing 20 mM MOPS, 75 mM KI, 1 mM n-dodecyl-β-d-maltoside (gray dotted line).
Hydrogen Isotope Exchange
A radiolabeled version of 1 would also be useful for quantitative studies of its binding properties to CFTR. Radioligands have shown to be invaluable tools for the understanding of the mechanism of action of membrane proteins, receptors, transporters, and channels.(26) To date, binding of CFTR modulators has been detected using functional assays which report the consequences of binding rather than binding per se.11,27,28 Hence, the development of radiolabeled 1 would enable direct assay of the relative affinities for binding to CFTR and mutant CFTR proteins, thereby providing insight into potential genotype-specific structural differences in the binding site. Further, this tool would enable comparison of the binding sites for various CFTR modulators as they are identified.
While it would be possible to incorporate a radioactive electrophile onto the propargylamino group accessible from 17, a method that would be much less disturbing to the original structure of 1 would be to replace a “non-exchangeable” hydrogen atom for a tritium atom. As a model for this, a study was conducted on the incorporation of less expensive and more safely handled nonradioactive deuterium. A small sample of 1 was dissolved in 1 M NaOH in D2O and heated at 90 °C. Under these conditions, in addition to the classically exchangeable phenolic O–H and pyrazole N–H, the pyrazole C4–H also gradually exchanged for deuterium (Figure 2a). The reaction progress was readily monitored by 1H NMR spectroscopy. The singlet at 6.7 ppm, arising from the pyrazole C4–H, was observed to gradually decrease in integration, reaching ∼50% deuterium incorporation over 24 h, decreasing to ∼20% (i.e., ∼80% deuterium incorporation) after 48 h. Parts b and c of Figure 2 show the aromatic region of the 1H NMR spectrum of 1 at the beginning (t = 0) and end (t = 48 h) of the experiment, respectively. After workup (including aqueous acid to re-exchange the phenolic O–H and the pyrazole N–H for protons) and TLC purification, 1 was reisolated in 88% yield and determined to have 80% deuterium incorporation at pyrazole C4. If these conditions were to be repeated using commercially available tritiated water (radioactivity level 185 GBq/g),(29) they would be expected to provide [3H]-1 at a level of 1.33 × 1012 Bq/mol,(30) without the need for multiple synthetic steps or purification of radioactive materials.
Figure 2.

(a) Experimental conditions for hydrogen isotope exchange. (b) Aromatic region of the 1H NMR spectrum of 1 in 1 M NaOH/D2O, prior to any heating. The signal from the proton on pyrazole C4 is indicated by the arrow. (c) The same region of the spectrum after heating at 90 °C for 48 h, wherein ∼80% of the pyrazole C4 protons have been exchanged for deuterium.
Biological Activity
For these molecular probes to be of use in biochemical experiments, it must be demonstrated that the substitutions, in every case at the 4-position of the phenyl ring of the parent structure 1, do not interfere with the compound’s ability to interact with the protein. To determine this, the effects of 9–12 and 15 on CFTR channel function were assessed using patch clamp electrophysiology.11,31,32 In our version of this assay,27,28 baby hamster kidney (BHK) cells expressing F508del-CFTR at the cell membrane (after biosynthetic rescue by low temperature culture conditions)(6) are loaded with NaI, stimulated using forskolin (10 μM), and then treated acutely with the compounds at a final concentration of 10 μM at approximately the 500 s mark. The potentiation of channel function is observed as an increased rate of iodide efflux from the cells, measured using an iodide-sensitive electrode positioned in the cell bath. After sufficient data is collected (around 800 s), cells are lysed, releasing all remaining iodide. Figure 3a depicts the measured level of iodide in the cell bath over time, after acute treatment with either 1 or 11 at a final concentration of 10 μM or DMSO control. The experiment was also repeated with addition of 9, 10, 12, and 15 (traces not shown). The channel potentiating effects of the compounds (measured as the maximum slope of the line prior to addition subtracted from the maximum slope after addition of the compound) are depicted in figure 3b. The DMSO control showed negligible change in halide efflux post forskolin stimulation (1.4 ± 0.4 nM/s, n = 4) whereas acute addition of 1 displayed a marked increase in efflux post forskolin stimulation (9.9 ± 2.3 nM/s, n = 7). Analogues 9–12 were found to be biologically active, with efflux rates ranging from 5.1 to 7.5 nM/s. In this assay, fluorescent analogue 15 proved to be inactive (1.7 ± 1.2 nM/s, n = 6). The effects of 1 and 9–12 were all determined to be significantly different from DMSO control, as assessed using analysis of variance (ANOVA) statistics. Importantly, the rates of efflux promoted by 9–12 were not significantly different than the rate observed for the parent compound 1, thus suggesting that substitution of a small functional group at the 4-position of the phenyl ring is not detrimental to the function of the molecule as a potentiator of CFTR channel function.
Figure 3.

(a) Typical iodide efflux traces for 1 (black line), 11 (dark-gray), and DMSO control (light-gray). Cells were activated with forskolin at 120 s (arrow i), treated with compound at 500 s (arrow ii), and completely lysed at 800 s (arrow iii). (b) Potentiating effects of all compounds tested (measured as the maximum slope of the iodide efflux trace prior to addition of the compound, subtracted from the maximum slope post addition).
The reduced activity in the iodide efflux assay of dansyl derivative 15 may be attributable to the compound’s reduced ability to permeate the cell membrane rather than some hindered interaction with CFTR. To test this possibility, we examined the effect of this molecule in a less complex reconstitution system, which allows the direct interactions of small molecules with isolated CFTR to be studied. G551D-CFTR, a clinically relevant mutant which exhibits more severely defective channel activity than F508del-CFTR, was reconstituted into liposomes of egg phosphatidylcholine. In this reconstituted system, a population of protein molecules are oriented inside out within the liposome (i.e., domains that are normally intracellular in live cells face out of the liposome, and vice versa). Therefore a compound does not need to cross the membrane in order to interact with the intracellular domains of the protein. If 15 retains the ability to interact with CFTR, it should potentiate channel activity in this population of protein molecules.
The liposomes were loaded with potassium iodide and suspended in a 10 μM solution of the compound to be tested in buffer lacking iodide. The concentration of iodide outside the liposomes is continuously measured using an iodide sensitive probe, and CFTR activity is observed after addition of protein kinase A and ATP to activate CFTR, and valinomycin to prevent charge build-up across the membrane due to CFTR-mediated iodide efflux which would slow and eventually stop further efflux. Figure 4a shows the traces of iodide concentration vs time for compound 1 (black line) and 15 (dark-gray) as well as a DMSO control (light-gray). Figure 4b shows the effect of each compound, measured as the maximum slope of the iodide efflux trace following valinomycin activation. Under these conditions, compound 15 proved to be an effective potentiator of channel activity, with an iodide efflux rate 11.18 ± 2.08 nM/s, n = 3, although with slightly diminished activity compared to 1 (16.85 ± 1.26 nM/s, n = 5). These data suggest that 15 maintains the ability of the parent compound 1 to interact with CFTR and modulate its activity but has a diminished ability to cross the cell membrane. Therefore 15 should be a useful fluorescent probe in systems of purified and reconstituted protein and other in vitro experiments not involving whole live cells.
Figure 4.

(a) Iodide efflux assay on purified G551D-CFTR, reconstituted in proteoliposomes for compound 1 (black line), fluorescent derivative 15 (dark-gray) and DMSO control (light-gray). (b) The effect of 1 and 15 on iodide efflux in this system, measured as the maximum slope of the iodide efflux trace after valinomycin activation.
Conclusion
The interaction between 1 and mutants of CFTR imbues the protein with properties more like the wild-type, including improved protein maturation and halide channel function (the two key deficiencies which lead to the symptoms of cystic fibrosis). However, little is known about the exact nature of this interaction. As tools to help elucidate these details, useful molecular probes based on the structure of 1 have been synthesized and characterized. Compound 11 bears an azide group substituted at the 4-position of the parent compound’s phenyl ring. The compound (and other derivatives bearing small substitutions at the same position) maintains the ability to potentiate channel function in live cells expressing the major mutant F508del-CFTR and also undergoes a photoreaction generating a strong electrophile. These properties combined suggest that 11 will be useful in photoaffinity labeling experiments. Sonogashira coupling between iodide 9 and propargylamine derivatives provided further useful compounds, including the fluorescent dansyl sulfonamide 15. This compound is active as a potentiator in a purified proteoliposome assay, however exhibits almost no activity in live cells, suggesting that 15 maintains the ability to interact with CFTR mutants but that the larger substituent abrogates the ability to cross the cell membrane. The environment-dependent fluorescence properties of 15 will make it a useful probe molecule for experiments in purified protein and/or cell lysates. Finally, it has been demonstrated that the pyrazole C4–H in 1 undergoes gradual exchange in hot alkaline water, and a protocol to exploit this property to incorporate radioactive tritium has been developed. These molecular probes enable an array of biochemical experiments to determine how and where 1 interacts with mutant CFTR, which will in turn enable the design of new, more potent and specific compounds as potential treatments for cystic fibrosis. These experiments are currently underway, and their results will be reported in due course.
Experimental Section
General (Chemistry)
All reagents and solvents were used as received from commercial suppliers, except for tetrahydrofuran, which was dispensed under nitrogen from an Mbraun solvent purification system immediately prior to use as a reaction solvent. Propargylamine derivatives 14(21) and 16(23) were prepared according to literature methods. TLC was carried out on silica gel 60 F254 aluminum backed plates, supplied by EMD chemicals, eluting with the solvent system indicated, and visualizing with UV light. Column chromatography was carried out on Silicycle SiliaFlash P60, 40–63 μm silica gel, eluting with the solvent system indicated below for each compound. Nuclear magnetic resonance (1H NMR, 400 MHz, and 13C NMR, 100 MHz) spectroscopy was carried out on a Bruker Avance 400 instrument in the deuterated solvents indicated below for each compound, and spectra were referenced to the solvent residual proton and 13C peaks. High-resolution mass spectrometry was performed using ESI-TOF or DART-TOF as indicated. UV/Visible spectroscopy was performed using a Perkin-Elmer Lambda 20 instrument. Melting points were determined in open air and are uncorrected. The purity of all compounds subjected to biological assays was determined to be ≥95% by HPLC analysis, using an Agilent Zorbax Rx-C8 4.6 mm × 150 mm column, eluting at 0.6 mL/min with a binary gradient of 100% H2O to 100% MeCN over the course of 1 h, except for compound 15, for which the gradient was 100% H2O to 50% MeCN over the course of 1 h.
2′-(4-Iodobenzoyloxy)-5′-methylacetophenone (6)
Solid 2′-hydroxy-5′-methylacetophenone (2, 3.948 g, 26.3 mmol) was added to an ice-bath cooled suspension of 4-iodobenzoyl chloride (3, 7.008 g, 26.3 mmol) in pyridine (140 mL). The resultant clear yellow solution was stirred over an ice-bath for one hour, after which time it was quenched by pouring into an ice/HCl mixture (2M, 600 mL). This aqueous phase was extracted three times with CH2Cl2 (200 mL each time). The combined organic extracts were washed aqueous HCl (1M, 200 mL), then NaHCO3 (saturated, 200 mL), and finally NaCl (saturated, 200 mL), before drying over Na2SO4 and evaporating to afford the title ester 6 as a yellow solid, which was used without further purification. Yield: 86% (8.566 g). 1H NMR (400 MHz, (CD3)2SO): δ 7.99–7.95 (m, 2H), 7.84–7.81 (m, 2H), 7.76 (d, J = 1.5 Hz, 1H), 7.45 (ddd, J = 8.5 Hz, J = 2.0 Hz, J = 0.5 Hz, 1H), 7.22 (d, J = 8.5 Hz, 1H), 2.46 (s, 3H), 2.38 (s, 3H). 13C NMR (100 MHz, (CD3)2SO) δ 196.87, 163.89, 145.74, 137.41, 135.39, 133.53, 130.92, 130.27, 129.58, 127.97, 123.19, 102.14, 28.83, 19.81.
2′-(4-Nitrobenzoyloxy)-5′-methylacetophenone (7)
Prepared as described above for 6, but using 2 (2.508 g, 16.7 mmol) and 4-nitrobenzoyl chloride (4, 4.324 g, 23.3 mmol). As above for 6, this compound was used after extractive workup without further purification. Yield 98% (4.898 g); mp 274–276 °C. 1H NMR (400 MHz, CDCl3): δ 8.38–8.36 (m, 4H), 7.68 (d, J = 2 Hz, 1H), 7.42 (ddd, J = 12 Hz, J = 2 Hz, J = 1 Hz, 1H), 7.13 (d, J = 8 Hz, 1H), 2.54 (s, 3H), 2.45 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 196.79, 163.47, 150.91, 146.75, 136.45, 135.19, 134.02, 131.32, 131.03, 130.22, 123.66, 123.55, 28.65, 19.97.
4-Methyl-2-(5-(4-iodophenyl)-1H-pyrazol-3-yl)phenol (9)
Potassium tert-butoxide (613 mg, 5.46 mmol) was dissolved in THF (25 mL). The solution was warmed to 50 °C, and ester 6 (2.000 g, 5.26 mmol) was added, upon which a yellow precipitate was observed to form. The mixture was stirred at 50 °C for 30 min, then cooled in an ice-bath and quenched by addition of aqueous acetic acid (10% v/v, 20 mL). The aqueous mixture was extracted three times with CH2Cl2 (50 mL each time). The combined organic extracts were dried over Na2SO4 and evaporated to afford a yellow solid. This solid was taken up in glacial acetic acid (30 mL) and cooled in an ice-bath. Hydrazine hydrate (“50–60%”, 5.7 mL, ∼100 mmol) was added dropwise to the mixture. The ice bath was removed, and the mixture was warmed to 65 °C for 16 h. The mixture was then cooled to room temperature and quenched with water (30 mL) and then extracted three times with CH2Cl2 (50 mL each time). The combined organic extracts were dried over Na2SO4 and evaporated. The crude product was purified by silica gel column chromatography, eluting with a gradient of 1:10 to 1:1 ethyl acetate/hexanes, to afford the title compound 9 as a yellow solid. Yield 68% (1.346 g); mp 211–213 °C. 1H NMR (400 MHz, (CD3)2CO) δ 13.03 (bs, 1H), 10.64 (bs, 1H), 7.86 (d, J = 8 Hz, 2H), 7.68 (d, J = 8 Hz, 2H), 7.58 (s, 1H), 7.28 (s, 1H), 7.01 (d, J = 8 Hz, 1H), 6.84 (d, J = 8 Hz, 1 H), 2.29 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 177.09, 138.16, 135.28, 135.11, 129.74, 127.91, 127.44, 127.03, 124.44, 123.56, 118.14, 116.49, 116.23, 19.77. ESI-TOF MS: m/z = 377.0140 (M + H) calculated for C16H14N2OI: 377.0145. HPLC rt = 25.2 min.
4-Methyl-2-(5-(4-nitrophenyl)-1H-pyrazol-3-yl)phenol (10)
Prepared as described above for 9 but beginning with ester 7 (3.500 g, 11.7 mmol) and driving the rearrangement step with 17.6 mmol of sodium tert-butoxide. The rest of the procedure was scaled up directly. The crude product was purified by silica gel column chromatography, eluting with a 3:17 ethyl acetate/hexanes, to afford the title compound 10 as an orange solid. Yield 67% (2.313 g); mp 105–106 °C. 1H NMR (400 MHz, (CD3)2CO) δ 12.94 (bs, 1H), 10.81 (bs, 1H), 8.35 (d, J = 1.5 Hz, 2H), 8.17 (d, J = 9 Hz. 2H), 7.62 (d, J = 1.5 Hz, 1H), 7.46 (s, 1H), 7.04 (dd, J = 8.5 Hz, J = 1.5 Hz, 1H), 6.89 (d, J = 6 Hz, 1H), 2.30 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 161.24, 148.25, 136.27, 130.83, 129.35, 128.17, 127.12, 125.08, 124.46, 119.15, 117.37, 116.78, 102.01, 20.55. ESI-TOF MS: m/z = 296.1024 (M + H) calculated for C16H14N3O3: 296.1029. HPLC rt = 31.2 min.
4-Methyl-2-(5-(4-aminophenyl)-1H-pyrazol-3-yl)phenol (12)
Stannous chloride dihydrate (3.850 g, 17 mmol) was added to a solution of 10 (1 g, 3.4 mmol) in 95% ethanol (30 mL). The mixture was heated to reflux for 16 h. The mixture was cooled to room temperature and poured into aqueous NaOH (1 M, 150 mL). The mixture was extracted twice with ethyl acetate (75 mL each time). The combined organic extracts were washed with aqueous NaCl (saturated, 75 mL), then dried over Na2SO4 and evaporated. The crude product was purified by silica gel column chromatography, eluting with a 1:4 ethyl acetate/hexanes, to afford the title compound to afford the title compound 12 as a pale-yellow solid. Yield 71% (638 mg); mp: 200–202 °C. 1H NMR (400 MHz, (CD3)2CO) δ 13.2 (s, 1H), 10.9 (s, 1H), 7.54 (s, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.03 (s, 1H), 6.97 (d, J = 8 Hz, 1H), 6.79 (d, J = 8 Hz, 1H), 6.63 (d, J = 8 Hz, 2H), 5.42 (s, 2H), 2.27 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 154.17, 149.38, 129.54, 129.34, 127.66, 126.77, 126.69, 126.32, 120.08, 116.72, 116.35, 114.36, 97.06, 19.70. ESI-TOF MS: m/z = 266.1288 (M + H) calculated for C16H16N3O: 266.1287. HPLC rt = 29.0 min.
4-Methyl-2-(5-(4-azidophenyl)-1H-pyrazol-3-yl)phenol (11)
Conversion of the amine to the azide was conducted using the method reported by Moses.(18) Amine 12 (265 mg, 1 mmol) was dissolved in acetonitrile (10 mL) and cooled in an ice-bath. To this was added tert-butyl nitrite (178 μL, 154 mg, 1.5 mmol), followed by azidotrimethylsilane (158 μL, 138 mg, 1.2 mmol). The mixture was warmed to room temperature and stirred for one hour, after which the solvent was evaporated and the crude product was purified by silica gel column chromatography, eluting with a gradient of 1:10 to 1:1 ethyl acetate/hexanes, to afford the title compound 11 as a yellow solid. Yield 94% (273 mg); mp 153–154 °C. 1H NMR (400 MHz, (CD3)2CO) δ 12.82 (bs, 1H), 10.70 (bs, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 2 Hz, 1 H), 7.28–7.22 (m, 3H), 7.02 (dd, J = 8.5 Hz, J = 2 Hz, 1H), 6.83 (d, J = 8 Hz, 1H), 2.30 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 153.89, 143.72, 140.18, 129.68, 127.99, 127.14, 126.97, 126.38, 125.50, 119.63, 116.49, 116.37, 99.14, 19.76. ESI-TOF MS: m/z = 291.1196 (M + H) calculated for C16H14N5O: 292.1192. HPLC rt = 33.8 min.
4-Methyl-2-(5-phenyl-1H-pyrazol-3-yl)phenol (1) (One-Pot Method)
Benzoyl chloride (5, 510 μL, 617 mg, 4.4 mmol) was added to a solution of 2 (601 mg, 4 mmol) in pyridine (10 mL), and the mixture was warmed to 50 °C. At 15 min intervals, more 5 (50 μL, 61 mg, 0.4 mmol) was added, until TLC analysis (1:3, ethyl acetate/hexanes) indicated complete conversion of 2 (Rf = 0.8) into the ester (Rf = 0.7) (three aliquots were added over 45 min, for a total of 660 μL, 800 mg, 5.7 mmol benzoyl chloride in the reaction). While stirring at 50 °C, solid potassium tert-butoxide (987 mg, 8.8 mmol) was gradually added to the reaction mixture. After 15 min, TLC analysis (1:3, ethyl acetate/hexanes) indicated substantial but incomplete conversion of the ester (Rf = 0.7) to the rearranged “dione” (under these conditions, the TLC of this material was a streak extending from the baseline to a major spot with Rf = 0.6). More potassium tert-butoxide (258 mg, 2.3 mmol) was added, and after an additional 30 min of stirring, TLC analysis indicated complete consumption of the ester. Hydrazine hydrate (“50–60%”, 2.5 mL, ∼44 mmol) was added over the course of one minute, followed by glacial acetic acid (2.0 mL, 2.1 g, 35 mmol). After 1.5 h, TLC analysis indicated completed conversion to the desired product (1:3, ethyl acetate/hexanes, Rf = 0.5). The reaction mixture was poured into an ice/HCl mixture (1M, 250 mL), which was then extracted three times with ethyl acetate (100 mL each time). The combined organic extracts were washed with NaHCO3 (saturated, 100 mL) and NaCl (saturated, 100 mL) and then dried over MgSO4 and evaporated. The crude product was purified by silica gel column chromatography, eluting with a 1:9 ethyl acetate/hexanes, to afford the title compound 1 as a yellow solid. Yield 65% (648 mg); mp 160–161 °C (lit.(33) 156–157 °C). 1H NMR (400 MHz, (CD3)2CO) δ 12.78 (bs, 1H), 10.65 (bs, 1H), 7.908–7.876 (m, 2H), 7.606 (d, J = 2 Hz, 1H), 7.542–7.488 (m, 2H), 7.454–7.397 (m, 1H), 7.274 (s, 1H), 7.022 (ddd, J = 8.5 Hz, J = 2 Hz, J = 0.5, 1H), 6.838 (d, J = 7.5 Hz, 1H), 2.303 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 154.82, 152.95, 145.16, 130.50, 130.28, 129.92, 129.57, 128.79, 127.79, 126.48, 117.33, 117.28, 100.04, 20.59. DART-TOF MS: m/z = 251.1175 (M + H) calculated for C16H15N2O: 251.1184. HPLC rt = 34.3 min. The synthesis of 1 has been previously reported,11,33 however no characterization data aside from melting point have been published.
4-Methyl-2-(5-(4-nitrophenyl)-1H-pyrazol-3-yl)phenol (10) (One-Pot Method)
Prepared by a method analogous to 1 above, beginning with 1 g (6.7 mmol) of 2 and employing a total of 1.781 g (9.6 mmol) of 4-nitrobenzoyl chloride 4. Yield: 24% (475 mg). Characterization data were the same as reported above for this compound.
Photoreaction of 11
A solution of 11 (40 mg, 0.14 mmol) and diethylamine (0.4 mL) in C6D6 (1.5 mL) in a quartz NMR tube was irradiated with a 140 W Hanovia Utility ultraviolet quartz lamp. The tube was held ∼10 cm from the light bulb, and a stream of air was blown over the tube to maintain the contents at room temperature. After 5 h of exposure, the mixture was evaporated under reduced pressure, and the crude product was purified by silica gel column chromatography, eluting with 3:7 ethyl acetate/hexanes to afford the diethylamine adduct 13. Yield 40% (17.3 mg). 1H NMR (400 MHz, (CD3)2CO) δ 10.75 (bs, 1H), 7.54 (s, 1H), 7.21 (d, J = 8.0 Hz, 1H), 6.98 (dd, J = 8.5 Hz, J = 1.5 Hz, 1H), 6.92 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.04 (d, J = 8.0 Hz, 1H), 5.73 (t, J = 7.5 Hz, 1H), 3.46 (q, J = 7.0 Hz, 4H), 2.28 (s, 3H), 1.29 (bs, 2H), 1.26–1.01(m, 6H). 13C NMR (100 MHz, (CD3)2CO) δ 154.05, 152.09, 146.01, 144.21, 142.88, 129.44, 129.40, 127.74, 126.79, 116.56, 116.35, 109.33, 106.77, 99.40, 42.95, 30.97, 19.66, 13.46.
5-Dimethylamino-N-(3-(4-(3-(2-hydroxy-5-methylphenyl)-1H-pyrazol-5-yl)phenyl)-2-propynyl)-1-naphthalenesulfonamide (15)
A 10 mL Schlenck tube was fitted with a magnetic stirring bar and a glass stopper and flushed with argon gas. The tube was then charged with 9 (203 mg, 0.54 mmol), 14(21) (312 mg, 1.08 mmol), and PdCl2·dppf·CH2Cl2 (22 mg, 0.027 mmol). The tube was evacuated and backfilled with argon. Under a stream of argon, THF (4 mL), triethylamine (0.12 mL, 0.8 mmol), and finally CuI (4 mg, 0.02 mmol) were added. The tube was sealed, and the mixture was heated to 45 °C for 15 h and then cooled back to room temperature. The mixture was diluted with ethyl acetate (25 mL) and extracted with 0.1 M HCl (10 mL), then brine (10 mL), then dried over anhydrous sodium sulfate and evaporated under reduced pressure. The crude material was purified by silica gel column chromatography, eluting with 1:4 ethyl acetate/hexanes, to afford the title compound 15 as a yellow solid. Yield 47% (135 mg); mp 216–218 °C. 1H NMR (400 MHz, (CD3)2CO) δ 12.82 (bs, 1H), 10.62 (bs, 1H), 8.51 (d, J = 8.5 Hz, 1H), 8.42 (d, J = 8.5, 1H), 8.33 (dd, J = 7.5 Hz, J = 1.0 Hz, 1H), 7.72 (d, J = 8 Hz, 2H), 7.66 – 7.57 (m, 3H), 7.29 – 7.23 (m, 3H), 7.02 (dd, J = 8 Hz, J = 2 Hz, 1H), 6.94 (d, J = 8 Hz, 2H), 6.84 (d, J = 8 Hz, 1 H), 4.12 (d, J = 5 Hz, 2H), 2.81 (s, 6H), 2.30 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 152.85, 137.24, 137.21, 132.72, 130.98, 130.90, 130.75, 130.59, 130.29, 130.27, 128.93, 128.73, 127.86, 126.08, 124.27, 123.27, 120.40, 117.36, 117.28, 117.12, 115.97, 100.47, 86.47, 83.50, 45.59, 33.77, 20.57 (one 13C signal was not observed after 15000 scans). DART-TOF MS: m/z = 537.1958 (M + H) calculated for C31H29N4O3S: 537.1960. HPLC rt = 25.2 min.
O-t-Butyl-N-(3-(4-(3-(2-hydroxy-5-methylphenyl)-1H-pyrazol-5-yl)phenyl)-2-propynyl)carbamate (17)
Prepared by a method analogous to 15 above, but using 9 (610 mg, 1.62 mmol), 16(23) (500 mg, 3.24 mmol), PdCl2·dppf·CH2Cl2 (66 mg, 0.084 mmol), THF (12 mL), triethylamine (0.35 mL, 2.5 mmol), and CuI (11 mg, 0.06 mmol). Title compound 17 was isolated as an orange/brown solid. Yield 69% (445 mg); mp 185–187 °C. 1H NMR (400 MHz, (CD3)2CO) δ 12.86 (bs, 1H), 10.66 (bs, 1H), 7.87 (d, J = 8 Hz, 2H), 7.60 (d. J = 1.5 Hz, 1H), 7.54 (d, J = 7 Hz, 2H), 7.31 (s, 1H), 7.03 (dd, J = 8.5 Hz, J = 2 Hz, 1H), 6.87–6.82 (m, 1H), 6.45 (bs, 1H), 4.13 (d, J = 6 Hz, 2H), 2.30 (s, 3H), 1.45 (s, 9H). 13C NMR (100 MHz, (CD3)2CO) δ 156.32, 154.70, 136.16, 135.98, 133.02, 130.59, 128.90, 127.87, 126.46, 124.12, 119.04, 117.36, 117.16, 100.53, 89.87, 82.18, 79.38, 31.30, 28.59, 20.58. DART-TOF MS: m/z = 404.1966 (M + H) calculated for C24H26N3O3: 404.1974. HPLC rt = 19.8 min.
Hydrogen Isotope Exchange on 1
To a standard NMR tube containing 1 mL of 1 M NaOH in D2O was added 1 (25 mg, 0.1 mmol). The tube was capped and heated in a sand bath at 90 °C. Reaction progress was monitored by periodically collecting a 1H NMR spectrum and observing the relative integration of the pyrazole C4–H signal at δ 6.7 ppm. After 48 h, the reaction was cooled to room temperature and diluted with 10 mL of H2O. The mixture was cooled in an ice bath and acidified to pH ∼1 by dropwise addition of 1 M HCl. The mixture was extracted with 3 × 10 mL Et2O, concentrated under reduced pressure, and purified by preparative TLC eluting with 1:3 EtOAc/hexanes to afford 22 mg (88%) of 1C4-D with ∼80% deuteration as determined by 1H NMR integration. 1H NMR (400 MHz, (CD3)2CO) δ 12.78 (bs, 1H), 10.65 (bs, 1H), 7.91–7.88 (m, 2H), 7.61 (d, J = 2 Hz, 1H), 7.53–7.49 (m, 2H), 7.45–7.41 (m, 1H), 7.27 (s, 0.2H), 7.02 (dd, J = 8.5 Hz, J = 1.5 Hz, 1H), 6.84 (d, J = 8.5 Hz, 1H), 2.30 (s, 3H). 13C NMR (100 MHz, (CD3)2CO) δ 155.94, 154.82, 142.80, 130.49, 129.97, 129.56, 128.80, 127.82, 126.48, 117.33, 117.30, 100.06, 20.58 (one 13C signal not observed after 13686 scans).
Fluorescence Emission Spectra Determination of 15
A stock solution of 15 (5 mM in DMSO) was diluted to 20 μM in 100% chloroform, 100% methanol, 100% DMSO or buffer containing 20 mM MOPS, 75 mM KI, and 1 mM n-dodecyl-β-d-maltoside (DDM). Emission spectra were obtained with an excitation wavelength of 360 nm, and 4 nm excitation and emission bandpass, at 22 °C in a 0.5 cm micro quartz cuvette on a PTI (Photon Technology International, London, ON, Canada) Quantamaster QM/80 steady state spectrofluorimeter. Curves are an average of two measurements at 0.5 nm increments with a 1 s integration time and were corrected using a solvent blank measured under identical conditions.
Continuous Recording Cell-Based Iodide Efflux Assay
Using BHK cells stably expressing F508del-CFTR, continuous recording cell-based iodide efflux assays were performed as previously described.(27) In brief, cells were grown to approximately 90–100% confluency. Cells were loaded with NaI loading buffer (3 mM KNO3, 2 mM Ca(NO3)2, 11 mM glucose, 20 mM HEPES, and 136 mM NaI) at 37 °C for 1 h. NaI loading buffer was aspirated, and cells were washed four times in iodide-free efflux buffer (3 mM KNO3, 2 mM Ca(NO3)2, 11 mM glucose, 20 mM HEPES, and 136 mM NaNO3). Cells were scraped in 1 mL of iodide-free efflux buffer and collected by centrifugation (350 g for 5 min at 25 °C). Iodide-free efflux buffer was removed, and the cell pellet was resuspended in 400 μL of iodide-free efflux buffer. Iodide efflux was measured at room temperature using an iodide-sensitive electrode (Lazar Research Laboratories, Los Angeles, CA). F508del-CFTR at the cell surface was stimulated with 10 μM forskolin, followed by addition of 10 μM of compound (1, 9, 10, 11, 12, or 15). Five minutes after addition of each compound, Triton X-100 (Sigma) was used to lyse the cells to ensure iodide was properly loaded. The maximal iodide efflux rate was quantified over a 1 min interval associated with the largest positive slope during the 4- to 5-min time period after the addition of the activation cocktail. Traces were recorded using the Digidata 1320A data acquisition system with Clampex 8 software (Molecular Devices, Sunnyvale, CA).
G551D-CFTR Purification and Reconstitution
Mutant G551D-CFTR was purified from Sf9 cells expressing the channel with a C-terminal His10 tag from 0.5 L of culture. Cells were homogenized in the presence of protease inhibitors using an Emulsiflex C3 high-pressure homogenizer (Avenstin, Ottawa, ON. Canada), and plasma membranes were isolated on a 35% sucrose cushion by ultracentrifugation. One-fifth of the membrane pellet was solubilized by the presence of 2% fos-choline 14 (Anatrace, Maumee, OH) for 1 h with resuspension by 30 G needle, and insoluble material was removed by ultracentrifugation. The sample was bound to Ni-NTA (Qiagen Inc. Mississauga, ON, Canada), and the beads were washed with 10–50 mM imidazole buffer containing 1 mM dodecylmaltoside (DDM). G551D-CFTR was eluted with 600 mM imidazole buffer containing 1 mM DDM, and lower molecular weight contaminants and imidazole were removed by centrifugation with an Amicon Ultra centrifugal filter device (Milipore Corp. Billerica, MA), yielding up to ∼0.25 mg of pure G551D-CFTR protein per liter of culture used, in 20 mM MOPS, 75 mM KI, and 1 mM DDM, pH 7.4. G551D-CFTR was reconstituted into 5 mg of egg phosphatidylcholine (Avanti Polar Lipids, Alabaster, AB) at a protein:lipid ratio of approximately 1:300 (w/w) by incubation in the presence of 20 mM MOPS, 75 mM KI, 1 mM DDM, and lipid for 30 min, followed by passage through an Extracti-Gel D detergent-binding column (Pierce Corp. Rockford, IL).
Iodide Efflux Measurements for Purified and Reconstituted G551D-CFTR
The external iodide in the reconstituted protein sample was exchanged for 75 mM K-glutamate by Sephadex G50 gel filtration resin equilibrated with 20 mM MOPS, 75 mM K-glutamate, yielding G551D-CFTR liposomes containing 75 mM KI on the vesicle interior and 75 mM K-glutamate in the bulk solution. The increase in external iodide concentrations was monitored continuously using an iodide-selective electrode (Lazar Research Laboratories, Los Angeles, CA) interfaced to the Digidata 1320A data acquisition system and controlled by Clampex 8 software (Axon Instruments, Sunnyvale, CA), as described above for the cell-based assay. Protein kinase A (catalytic subunit; Promega Corp. Madison, WI) and Mg-ATP (200 nM and 1 mM, respectively) were added to the external solution to initiate iodide flux, and 20 nM valinomycin was added to prevent charge-buildup across the membrane that would limit iodide flux. Control liposomes were prepared and treated identically but in the absence of G551D-CFTR. Verification that sufficient iodide was trapped in the proteoliposomes was done by lysing the liposomes with 0.5% Triton X-100 at late time points. Stock solutions of 5 mM 1 or 15 were prepared in DMSO. A working solution of 20 μM was prepared from the stock in 20 mM MOPS and 75 mM K-glutamate buffer and added to the vesicle solution at a ratio of 1:1 to yield a final concentration of 10 μM, 5 min before initiation of flux by valinomycin.
Acknowledgments
These studies were supported by the Canadian Institutes of Health Research (CIHR) through Operating Grant MOP-97954 and Emerging Team Grant GPG-102171, both awarded to R.D.V. and C.E.B. P.D.W.E. was supported by postdoctoral fellowships from Cystic Fibrosis Canada and the Canadian Institutes of Health Research.
Glossary
Abbreviations Used
- ANOVA
analysis of variance
- BHK
baby hamster kidney
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- DANSYL
5-(dimethylamino)naphthalene-1-sulfonyl
- DDM
n-dodecyl-β-d-maltoside
- DPPF
1,1′-bis(diphenylphosphino)ferrocene
- ER
endoplasmic reticulum
- F508del
deletion of phenylalanine at position 508
- G551D
glycine to aspartic acid missense mutation at position 551
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MOPS
3-(N-morpholino)propanesulfonic acid
- NBD chloride
4-chloro-7-nitrobenzofurazan
- NIH 3T3
mouse embryonic fibroblast cell line
- NTA
nitrilotriacetic acid
- Sf9
Spodoptera frugiperda pupal ovarian cell line
References
- Riordan J. R.; Rommens J. M.; Kerem B.-S.; Alon N.; Rozhamel R.; Grzelczak Z.; Zielenski J.; Lok S.; Plavsic N.; Chou J.-L.; Drumm M. L.; Iannuzi M. C.; Collins F.; Tsui L.-C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [DOI] [PubMed] [Google Scholar]
- Zeitlin P. L. Emerging drug treatments for cystic fibrosis. Expert Opin. Emerging Drugs 2007, 12, 329–336. [DOI] [PubMed] [Google Scholar]
- Canadian Cystic Fibrosis Patient Data Registry Report; 2008, 9. Available online at http://www.cysticfibrosis.ca/assets/files/pdf/CPDR_ReportE.pdf.
- Kerem B.-S.; Rommens J. M.; Buchanan J. A.; Markiewicz D.; Cox T. K.; Chakravarti A.; Buchwald M.; Tsui L.-C. Identification of the cystic fibrosis gene: genetic analysis. Science 1989, 245, 1073–1080. [DOI] [PubMed] [Google Scholar]
- Cheng S. H.; Gregory R. J.; Marshall J.; Paul S.; Souza D. W.; White G. A.; O’Riordan C. R.; Smith A. E. Defective Intracellular Transport and Processing of CFTR Is the Molecular Basis of Most Cystic Fibrosis. Cell 1990, 63, 827–834. [DOI] [PubMed] [Google Scholar]
- Denning G. M.; Anderson M. P.; Amara J. F.; Marshall J.; Smith A. E.; Welsh M. J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [DOI] [PubMed] [Google Scholar]
- Wang F.; Zeltwanger S.; Hu S.; Hwang T.-C. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J. Physiol. 2000, 524, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory R. J.; Rich D. P.; Cheng S. H.; Souza D. W.; Paul S.; Manavalan P.; Anderson M. P.; Welsh M. J.; Smith A. E. Maturation and Function of Cystic Fibrosis Transmembrane Conductance Regulator Variants Bearing Mutations in Putative Nucleotide-Binding Domains 1 and 2. Mol. Cell. Biol. 1991, 11, 3886–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman A. S.; Galietta L. J. V. Chloride channels as drug targets. Nature Rev. Drug Discovery 2009, 8, 153–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadida S.; Van Goor F.; Grootenhuis P. D. J. CFTR Modulators for the Treatment of Cystic Fibrosis. Annu. Rep. Med. Chem. 2010, 45, 157–173. [Google Scholar]
- Van Goor F.; Straley K. S.; Cao D.; Gonzalez J.; Hadida S.; Hazlewood A.; Joubran J.; Knapp T.; Makings L. R.; Miller M.; Neuberger T.; Olson E.; Panchenko V.; Rader J.; Singh A.; Stack J. H.; Tung R.; Grootenuis P. D. J.; Negulescu P. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol., Lung Cell Mol. Physiol. 2006, 290, 1117–1130. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Bartlett M. C.; Loo T. W.; Clarke D. M. Specific Rescue of Cystic Fibrosis Transmembrane Conductance Regulator Processing Mutants Using Pharmacological Chaperones. Mol. Pharmacol. 2006, 70, 297–302. [DOI] [PubMed] [Google Scholar]
- Wellhauser L.; Kim Chiaw P.; Pasyk S.; Li C.; Ramjeesingh M.; Bear C. E. A Small-Molecule Modulator Interacts Directly with ΔPhe508-CFTR to Modify Its ATPase Activity and Conformational Stability. Mol. Pharmacol. 2009, 75, 1430–1438. [DOI] [PubMed] [Google Scholar]
- Dorman G. Photoaffinity Labelling in Biological Signal Transduction. Top. Curr. Chem. 2001, 211, 169–225. [Google Scholar]
- Thool A. W.; Ghiya B. J. Synthesis of 2-hydroxy-4,6-diphenylpyrimidines and their antimicrobial activity. J. Ind. Chem. Soc. 1988, 65, 522–524. [Google Scholar]
- Zhu W.; Ma D. Synthesis of aryl and vinyl azides via proline-promoted CuI-catalyzed coupling reactions. Chem. Commun. 2004, 888–889. [DOI] [PubMed] [Google Scholar]
- Gololobov Y. G.; Kasukhin L. F. Recent Advances in the Staudinger reaction. Tetrahedron 1992, 48, 1353–1406. [Google Scholar]
- Barral K.; Moorhouse A. D.; Moses J. E. Efficient Conversion of Aromatic Amines into Azides: A One-Pot Synthesis of Triazole Linkages. Org. Lett. 2007, 9, 1809–1811. [DOI] [PubMed] [Google Scholar]
- Ghosh P. B.; Whitehouse M. W. 7-Chloro-4-nitrobenzo-2-oxa-1,3-diazole: A New Fluorigenic Reagent for Amino Acids and other Amines. Biochem. J. 1968, 108, 155–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. Polarization of the Fluorescence of Macromolecules 2. Fluorescent Conjugates of Ovalbumin and Bovine Serum Albumin. Biochem. J. 1952, 51, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolletta F.; Fabbri D.; Lombardo M.; Prodi L.; Trombini C.; Zaccheroni N. Synthesis and Photophysical Properties of Fluorescent Derivatives of Methylmercury. Organometallics 1996, 15, 2415–2417. [Google Scholar]
- Chinchilla R.; Najera C. The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]
- Casara P.; Danzin C.; Metcalf B; Jung M. Stereospecific Synthesis of (2R,5R)-Hept-6-yne-2,5-diamine: A Potent and Selective Enzyme-Activated Irreversible Inhibitor of Ornithine Decarboxylase (ODC). J. Chem. Soc., Perkin Trans. 1 1985, 2201–2207. [Google Scholar]
- Muth S.; Fries A.; Gimpl G. Cholesterol-induced conformational changes in the oxytocin receptor. Biochem. J. 2011, 437, 541–553. [DOI] [PubMed] [Google Scholar]
- Guan L.; Nurva S.; Ankeshwarapu S. P. Mechanism of Melibiose/Cation Symport of the Melibiose Permease of Salmonella typhimurium. J. Biol. Chem. 2011, 286, 6367–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Quick M.; Zhao Y.; Weinstein H.; Javitch J. A. The Mechanism of a Neurotransmitter:Sodium Symporter—Inward Release of Na+ and Substrate Is Triggered by Substrate in a Second Binding Site. Mol. Cell 2008, 30, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Chiaw P.; Wellhauser L.; Huan L. J.; Ramjeesingh M.; Bear C. E. A chemical corrector modifies the channel function of F508del-CFTR. Mol. Pharmacol. 2010, 78 (3), 411–418. [DOI] [PubMed] [Google Scholar]
- Yu W.; Kim Chiaw P.; Bear C. E. Probing Conformational Rescue Induced by a Chemical Corrector of F508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Mutant. J. Biol. Chem. 2011, 286, 24714–24725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Given a half-life for [3H] of 4 500 days or 3.888 × 108 seconds, 185 GBq/g is equivalent to 3.10 × 10–3 mol [3H] per mol H2O.
- If pure D2O (which has 2 mol [2H] per mol water) provides 80% deuteration, then tritiation should proceed to a level of 0.80 ÷ 2 × 3.10 × 10–3 = 1.24 × 10–3 mol [3H] per mol 1, or 0.124% incorporation. This amounts to a level of 1.33 × 1012 Bq/mol.
- Chang X. B.; Cui L.; Hou Y. X.; Jensen T. J.; Aleksandrov A. A.; Mengos A.; Riordan J. R. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Mol. Cell 1999, 4, 137–142. [DOI] [PubMed] [Google Scholar]
- Du K.; Sharma M.; Lukacs G. L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nature Struct. Mol. Biol. 2005, 12, 17–25. [DOI] [PubMed] [Google Scholar]
- Reddy N. J.; Sharma T. C. Reactions of 3-Bromoflavanones with Hydrazine Hydrate. Indian J. Chem. 1985, 24B, 715–718. [Google Scholar]