

Abstract
Pyrazolopyridine inhibitors with low micromolar potency for CHK1 and good selectivity against CHK2 were previously identified by fragment-based screening. The optimization of the pyrazolopyridines to a series of potent and CHK1-selective isoquinolines demonstrates how fragment-growing and scaffold morphing strategies arising from a structure-based understanding of CHK1 inhibitor binding can be combined to successfully progress fragment-derived hit matter to compounds with activity in vivo. The challenges of improving CHK1 potency and selectivity, addressing synthetic tractability, and achieving novelty in the crowded kinase inhibitor chemical space were tackled by multiple scaffold morphing steps, which progressed through tricyclic pyrimido[2,3-b]azaindoles to N-(pyrazin-2-yl)pyrimidin-4-amines and ultimately to imidazo[4,5-c]pyridines and isoquinolines. A potent and highly selective isoquinoline CHK1 inhibitor (SAR-020106) was identified, which potentiated the efficacies of irinotecan and gemcitabine in SW620 human colon carcinoma xenografts in nude mice.
Introduction
The DNA damage response network ensures the fidelity of DNA replication and controls the repair of damage arising during cellular replication or from exogenous agents such as genotoxic drugs. Checkpoint kinase 1 (CHK1) is a serine/threonine kinase occupying a central position in this complex network of cell regulatory and DNA repair mechanisms.1−3 CHK1 is predominantly activated through phosphorylation on amino acid residues Ser317 and Ser345 by the upstream kinase Ataxia Telangiectasia and Rad3 Related (ATR) in response to single strand breaks in DNA,4,5 and it undergoes autophosphorylation on Ser296.6,7 G1/S, S, or G2/M cell cycle checkpoints are activated in response to genotoxic antitumor drugs to provide an opportunity for repair of damaged DNA or to activate apoptotic pathways.(8) CHK1 is involved in the S-phase checkpoint and stabilizing replication forks, and in the G2/M checkpoint through regulating the stability of the CDC25 phosphatases which control cell cycle progression by regulation of CDK1.9−12 Human cancers frequently have functional defects in the tumor suppressor p53, with consequent loss of G1/S checkpoint control13,14 and greater reliance on S and G2/M checkpoints. Thus, S or G2/M checkpoint inhibitors will selectively sensitize p53 deficient cancer cells to DNA damaging agents.1,8,15,16 CHK1 inhibition by siRNA and several small molecule inhibitors, including the lead compound resulting from the studies reported herein,(17) has confirmed this in preclinical studies.18−23
A range of adenosine 5′-triphosphate (ATP) competitive CHK1 inhibitors have been reported, with varying selectivity for the target enzyme over other kinases.23−26 Inhibitor development has been assisted by structural biology studies of the enzyme, in particular crystallographic analysis of inhibitor binding to the protein.27−30 As well as ATP-competitive inhibitors, allosteric modulators of CHK1 have been identified.(31) Some ATP-competitive CHK1 inhibitors have reached early clinical trials in combination with DNA-damaging agents, although the therapeutic outcomes remain to be determined.2,15,23,32
Results and Discussion
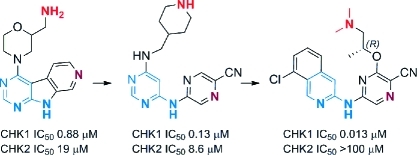
Previously we have shown how a fragment-based screening strategy identified several low molecular weight, ligand efficient templates which were developed into early stage CHK1 inhibitors.33,34 The purine template hit 1 was initially elaborated to the pyrazolopyridine lead 2.(33) Here we describe how structure-based design led, through several scaffold morphing steps, to the evolution of 2 into a potent and selective CHK1 inhibitor (R)-3 (SAR-020106,(17) Figure 1).
Figure 1.

Structures of the template hit 1, early stage lead 2, and (R)-3 (SAR-020106). Features conserved in the elaboration of the template hit and early stage lead into a potent and selective CHK1 inhibitor are highlighted in blue and red.
The crystal structure of 2 bound to CHK1 confirmed that the ligand occupied the ATP pocket and interacted with the hinge region of the kinase. The predicted hydrogen bonds were observed between N1 and N7 of the pyrazolo[3,4-b]pyridine with the backbone amides of Glu85 and Cys87, respectively (Figure 2A). The 2-(aminomethyl)morpholine group was located in the ribose pocket, though no specific polar interactions were observed, while the ethyl ester was directed out onto the solvent exposed surface. The area around the gatekeeper and interior pocket was unexplored, and our initial synthetic effort concentrated on probing this area. CHK1 contains a polar amino acid (Asn59; see Figure 2A) in the interior pocket beyond the gatekeeper residue, which differentiates the enzyme from many other protein kinases where a hydrophobic amino acid is located at the equivalent position.27,29 As a result, the interior pocket of CHK1, defined by Asn59, Glu55, and the Lys38-Asp148 salt bridge, is often observed in crystal structures to contain between 1 and 3 protein bound water molecules (see Figure 2A–E). Ligand interactions with these protein bound water molecules have been shown to be important in conferring both CHK1 potency and selectivity against other kinases.27,29
Figure 2.

Crystal structures of 2 (panel A, PDB 2ym3), 4 (panel B, PDB 2ym4), 6 (panel C, PDB 2ym5), 8 (panel D, PDB 2ym6), 20 (panel E, PDB 2ym7), and (R)-3 (panel F, PDB 2ym8) bound to the CHK1 kinase domain. Red spheres represent water molecules modeled in the refinement. Dashed gray lines represent hydrogen bonds. Panel G: Plan view of the overlay of CHK1 inhibitors 2 (cyan), 8 (gray), 20 (blue), and (R)-3 (gold) relative to the hinge region of CHK1 (cartoon).
Examination of the crystal structure of 2 suggested that a pendant phenyl at the 3-position of the pyrazolo[3,4-b]pyridine could provide a suitable vector toward the interior pocket. Alternatively, fusing a third ring to the core to form a tricycle could also provide a platform for substitution to contact the protein-bound water molecules. Accordingly, sets of analogues exploring these hypotheses were prepared and evaluated. We focused on substitution with functional groups having the potential to form hydrogen bonding interactions with the bound water molecules or the polar residues comprising the Asp148-Lys38 salt bridge in CHK1. Inhibition data for selected compounds tested against CHK1 are described in Table 1. In addition, we investigated the ability of the compounds to inhibit the functionally distinct checkpoint kinase 2 (CHK2).(35) Selectivity for inhibition of CHK1 over CHK2 is desirable, since simultaneous ablation of both CHK1 and CHK2 by RNAi has been shown to be inferior to selective depletion of CHK1 alone in overriding the S-phase checkpoint.(36) The interior pocket of CHK2 contains a hydrophobic leucine residue at the equivalent site to Asn59 in CHK1, providing a less favorable environment for the binding of water molecules or polar ligand functionality. Thus, successful targeting of the interior pocket in CHK1 with polar functionality is predicted to lead to selectivity against CHK2.
Table 1. Inhibition of CHK1 and CHK2 by Bicyclic and Tricyclic Compounds.

| CHK1 Inhibition IC50 (μM)a | CHK2 Inhibition IC50 (μM)a | Selectivity (CHK2/CHK1)b | LE (kcal mol–1 non-H atom–1)c | |
|---|---|---|---|---|
| 2 | 1.0 (0.86, 1.2) | 50 (26, 74) | 50 | 0.38 |
| 4 | 1.5 (1.3, 1.8) | n.d.d | 0.27 | |
| 5 | 3.2 (2.2, 4.2) | 28 (13, 43) | 9 | 0.31 |
| 6 | 0.43 (0.26, 0.60) | 3.4 (±1.16)e | 8 | 0.36 |
| rac-7 | 3.2 (2.4, 3.9) | 12 (11, 13) | 4 | 0.37 |
| (R)-7 | 3.0 (2.9, 3.2) | 13 (11, 16) | 4 | 0.37 |
| (S)-7 | 2.3 (1.55, 3.0) | 3.5 (2.3, 4.6) | 2 | 0.38 |
| 8 | 0.88 (±0.21) e | 19 (16, 23) | 22 | 0.40 |
| 9 | 3.7 (3.2, 4.2) | 10 (9.3, 11) | 3 | 0.33 |
| 10 | 1.7 (1.7, 1.8) | 23 (22, 24) | 14 | 0.47 |
| 11 | 1.7 (1.7, 1.8) | 11 (8.1, 14) | 6 | 0.45 |
| 12 | 0.67 (0.53, 0.80) | 16 (10, 21) | 24 | 0.43 |
| 13 | 0.29 (±0.02)e | 12 (6.8, 16) | 41 | 0.43 |
IC50 determined in a dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA).(33) Mean of two independent determinations; individual values in parentheses. Standard inhibitor staurosporine gave CHK1 IC50 = 2.1 (±1.8) nM and CHK2 IC50 = 27 (±8) nM.
Ratio of IC50 values (CHK2/CHK1).
Ligand efficiency (LE) calculated using LE = [−1.4 log10(IC50 (M))]/(number of heavy atoms).(37)
Not determined.
Mean (±SD) of at least three independent determinations.
Among several analogues prepared,(38) the trisubstituted pyrazolopyridine analogue 4 retained the potency of 2 (Table 1) and was shown by X-ray crystallography to hydrogen bond to a water in the interior pocket through the nitrile group (Figure 2B). However, the crystal structure also indicated that the ligand was unable to simultaneously position all three substituents optimally in the ATP-binding site due to steric congestion. The 2-(aminomethyl)morpholine and ethyl ester were both twisted into potentially undesirable conformations compared to 2, with the ester oriented orthogonal to the bicycle. The ligand attained only equivalent potency to 2 despite the addition of extra interacting functionality, reflected in the reduced ligand efficiency. We attempted to resolve the overcrowding by reverting to a pyrrolo[2,3-d]pyrimidine core(33) and removing the pendant ethoxycarbonyl group. Substitution with the 3-(3-cyanophenyl) group resulted in compound 5, having slightly reduced CHK1 potency relative to 4, while the 3-(3-(hydroxymethyl)phenyl) analogue 6 gave a 3-fold increase in activity. However, the selectivities for CHK1 against CHK2 were reduced for both 5 and 6 compared to 2.
The crystal structure of 6 (Figure 2C) showed that although steric crowding around the core was reduced, the 3-hydroxymethyl substituent had adopted an alternative orientation along a vector into the ribose pocket and away from the interior pocket. The selectivity of compound 2 for CHK1 over CHK2 indicates that, in common with many ATP-competitive inhibitors binding to active kinase conformations,(39) some specificity for inhibition can be obtained through interactions with the selectivity (or solvent-exposed) surface at the entrance to the ATP-binding cleft. The amino acid residues comprising this region vary between kinases and thus alter the properties of the surface. In CHK1 the surface includes contributions from Tyr86, Cys87, and Ser88 (see Figure 2F), while in CHK2 the equivalent residues are leucine, methionine, and glutamate, respectively. In compound 6 the absence of the pendant ethoxycarbonyl group and the failure of the hydroxymethyl group to engage with the water-filled pocket removed the potential contributions to CHK1 selectivity.
One possible approach to restoring selectivity was to replace the morpholine substituent with a less bulky, unbranched alkylamine and reintroduce the 5-substituent to the pyrazolo[3,4-b]pyridine core. Less bulky alkylamine 4-substituents would be anticipated to relieve the crowding seen in 4 and permit both the 3- and 5-substituents to be present in favorable conformations for binding to CHK1. However, the 2-(aminomethyl)morpholine or similar cyclic 4-substituents on the 3-aryl substituted bicyclic cores were important in establishing novelty to provide patentable compounds in an area which has been intensively exemplified as kinase inhibitor scaffolds.40−46 Instead, we maintained the morpholine substituent but elaborated the bicyclic core by the addition of a fused ring.
Initial compounds prepared from a commercially available pyrimidoindole tricyclic core (7) gave compounds with similar CHK1 potencies and ligand efficiencies to 2. No preference was seen for the enantiomers of the 2-(aminomethyl)morpholine group, (R)-7 and (S)-7. The requirement for the inclusion of a suitable H-bond donor or acceptor capable of interacting with the protein-bound water molecules led to a more involved synthetic effort, resulting in both the nitrile substituted analogue 9 and the introduction of a nitrogen into the fused ring to give the 9H-pyrimido[2,3-b]azaindole 8. While addition of the nitrile group 9 led to a drop in LE, the more compact 8 gave equivalent potency and LE to 2 and restored some of the selectivity over CHK2. X-ray crystallography confirmed that the azaindole nitrogen effectively contacted the network of bound waters in the interior pocket of CHK1, providing a rationale for the recovery of the selectivity in the absence of a substituent directed toward Ser88 and the selectivity surface (Figure 2D).
Importantly, the pyrimido[2,3-b]azaindole core of 8 provided the opportunity to introduce a more varied range of alkylamino substituents to replace the 2-(aminomethyl)morpholine while still generating novel inhibitors.(47) The influence of the alkylamino group was probed with an array of analogues, including 10–13, resulting in the identification of the 4-(aminomethyl)piperidine 13 as a more potent and selective alternative. However, the rigid planar nature of the tricyclic scaffold and synthetic challenges to further elaboration prompted us to consider another scaffold morphing step and to disconnect the middle ring of the tricycle. This was envisaged to introduce a degree of flexibility into the core hinge-binding pharmacophore, potentially allowing the ligand to adopt a better conformation for simultaneous interaction with the hinge region and the water-filled pocket. An efficient synthesis of a small array of compounds allowed this variation to be quickly assessed (Table 2).
Table 2. Inhibition of CHK1 and CHK2 by N-(Pyridin-3-yl)- or N-(Pyrazin-2-yl)pyrimidin-4-amines.


IC50 determined in DELFIA.(33) Mean of two independent determinations; individual values in parentheses. Standard inhibitor staurosporine gave CHK1 IC50 = 2.1 (±1.8) nM and CHK2 IC50 = 27 (±8) nM.
Ratio of IC50 values (CHK2/CHK1).
LE = [−1.4 log10(IC50 (M))]/(number of heavy atoms).(37)
Not determined.
Mean (±SD) of at least three independent determinations.
Single determination.
The potency of simple N-(pyridin-3-yl)pyrimidin-4-amines (see representative example 14) suffered, presumably as the steric clash between the pyrimidine 5-H and pyridine 4′-H would force the core to adopt too extreme a nonplanar conformation. By switching the pyridine to a pyrazine ring, this was overcome, and the first “ring opened” adaptation, N-(pyrazin-2-yl)pyrimidin-4-amine (15), showed some activity, though disappointingly 75-fold less than that of the parent compound, 13. The incorporation of pyrazine substitution into CHK1 inhibitors has been reported previously for a series of pyrazin-2-yl ureas,48,49 where the additional nitrogen atom in the pyrazine ring was shown by crystallographic studies to be important in restraining the urea moiety in the bioactive conformation.(27) Comparisons with these CHK1 inhibitors suggested that appending a nitrile in the 2-position of the pyrazine could be beneficial through achieving additional interactions with the protein in the region of the water-filled pocket. Comparing 15 with 16, the addition of the nitrile increased the CHK1 potency 15-fold. Optimization of the diamine substituent proved fruitful, as 4-(aminomethyl)piperidine gave excellent CHK1 potency while retaining selectivity over CHK2 when incorporated into the N-(pyrazin-2-yl)pyrimidin-4-amine 20.
Crystallographic data explained the increased activity and selectivity of the 2-cyanopyrazines, as there was a clear interaction of the nitrile with the conserved Lys38 and the water network filling the interior pocket (see Figure 2E). Interestingly, a sharp loss of activity was seen when the terminal amine (19–20) was replaced with structurally similar but nonbasic polar groups (21–22). This structure–activity relationship is in contrast with that demonstrated for (6-cyanopyrazin-2-yl)urea CHK1 inhibitors,48,49 where replacement of a pendant amine with nonbasic polar groups was tolerated. Thus, although occupying similar space within the ATP-binding site, as shown by the crystallographic data, and interacting with some similar points on the CHK1 protein, the two series did not have identical structure–activity relationships.
With limited points of substitution that would enable us to probe the solvent-exposed selectivity surface, we sought to exchange the pyrimidine for a pyridine. Consistent with the structure–activity relationships discussed above, the reintroduction of the ester 5-substituent in 23 increased CHK1 affinity by 4-fold (23 vs 20) and CHK1 vs CHK2 selectivity to approximately 300-fold. The 4,5-disubstituted pyridine scaffold presented one opportunity for optimization of the inhibitors, which will be reported separately. We also pursued a further scaffold morphing step, seeking to combine the 4- and 5-substituents into a fused ring to give imidazo[4,5-c]pyridines and isoquinolines. This gave the micromolar inhibitors 24 and 27 (Table 3), with encouraging ligand efficiencies (>0.3 kcal mol–1 heavy atom–1) for the unsubstituted scaffolds.
Table 3. Inhibition of CHK1 and CHK2 by Imidazo[4,5-c]pyridines and Isoquinolines.


IC50 determined in DELFIA.(33) Mean of two independent determinations; individual values in parentheses. Standard inhibitor staurosporine gave CHK1 IC50 = 2.1 (±1.8) nM and CHK2 IC50 = 27 (±8) nM.
LE = [−1.4 log10(IC50 (M))]/(number of heavy atoms).(34)
Not determined.
Mean (±SD) of at least three independent determinations.
Single determination.
Poor solubility gave variability for this analogue in this assay.
We explored the reintroduction of basic amine substituents to mimic the substitution of 20 and 23, originating either from the imidazole nitrogen of the imidazo[4,5-c]pyridines or through tethering via an ether linkage to the pyrazine portion of the scaffolds.(27) As shown by the representative example 25, the analogues with the side chain originating from the imidazo[4,5-c]pyridines had substantially decreased affinity (Table 3). Moving the basic amine substituent to the pyrazine in 26 and 28 gave much improved activity against CHK1 with good selectivity over CHK2. While both imidazo[4,5-c]pyridines (e.g., 26) and isoquinolines (e.g., 28) were potent and selective CHK1 inhibitors, the cellular activities of the imidazo[4,5-c]pyridines were substantially lower than those of the corresponding isoquinolines (see Table 4 and discussion below), and thus, further optimization concentrated on the latter heterocycle.
Table 4. G2 Checkpoint Abrogation and Potentiation of SN38 Cytotoxicity in Human Colon Cancer Cells by Selected Compounds.
| CHK1 IC50 (μM)a | SRB (HT29) GI50 (μM)b | Checkpoint abrogation (HT29) IC50 (μM)c | Potentiation of SN38 cytotoxicity (HT29) P.I.d | ALogPe | TPSAe | |
|---|---|---|---|---|---|---|
| 2 | 1.0 | 64f | 18.5f | –0.22 | 106 | |
| 8 | 0.88 | 62f | 13.5f | –0.44 | 93 | |
| 13 | 0.29 | 17.5f | 3.1f | 1.0 | 84 | |
| 19 | 0.40 | 24f | n.d. | –0.21 | 125 | |
| 20 | 0.12 | 76.5f | n.d. | 0.077 | 111 | |
| 24 | 1.9 | 46 (±39, n = 3) | 23 (±22, n = 3) | 0.19 | 103 | |
| 26 | 0.049 | 125 (120, 130) | 128 (120, 135) | 1.5 | 124 | |
| 27 | 1.2 | 26 (21, 30) | >100, >100 | 1.3 | 2.2 | 74 |
| 28 | 0.017 | 1.2 (1.1, 1.2) | 0.17 (0.16, 0.17) | 3.3 | 2.3 | 96 |
| 29 | 0.74 | 120 (120, 120) | 2.7 (2.0, 3.3) | 2.6 | 2.2 | 74 |
| 30 | 0.022 | 0.53 (0.52, 0.53) | 0.18, 0.008g | 2.9 | 96 | |
| (R)-3 | 0.013 | 0.47 (±0.19, n = 11) | 0.055 (±0.019, n = 5) | 3.1 (±1.6, n = 5) | 3.9 | 87 |
IC50 determined in DELFIA. Mean of at least two independent determinations.
SRB cytotoxicity assay; mean of two independent determinations unless otherwise stated.
G2 checkpoint abrogation assay.(17)
PI; ratio of SRB GI50 for CHK1 inhibitor alone to GI50 for the combination of CHK1 inhibitor and SN38 (see ref (17)).
Single determination.
Poor aqueous solubility gave variability for this analogue in this assay.
Several published urea-based CHK1 inhibitors(49) direct a chlorine substituent toward the selectivity surface. Overlays of our isoquinoline scaffold with these ureas showed that a chlorine in the 8-position could be similarly positioned and have the potential to make a short oxygen–halogen interaction with the carbonyl oxygen of Ser88.(50) To explore this, reoptimization of the basic amine substituent was conducted on isoquinolines with and without 8-chloro substitution (Table 3). In the main, the chlorinated analogues retained CHK1 activity, and this set of compounds led us to the potent and selective compound (rac)-3. The CHK1 enzyme inhibitory activity of (rac)-3 could be attributed, on testing of the enantiomeric pair, to the R-enantiomer, (R)-3.
X-ray crystallography of (R)-3 in the CHK1 enzyme (Figure 2F) showed binding through hydrogen-bonding to Glu85 and Cys87 in the hinge region, the hydrogen-bonding of the nitrile to Lys38, and the dimethylamine nestled in the ribose pocket. The branched methyl on the side chain fitted closely to Val23 while the isoquinoline chloro substituent was close to the specificity surface without projecting too far toward solvent. The distance from the chlorine to the carbonyl oxygen of Ser88 measured in the 2.1 Å resolution crystal structure was 3.44 Å, greater than the sum of the Cl and O van der Waals radii. The potency of (rac)-3 was only approximately twice that of the des-chloro analogue 34, suggesting that an oxygen–halogen interaction was not strongly contributing to the binding energy of (R)-3. However, comparison of 34 and (rac)-3 showed that selectivity against CHK2 was improved from 120-fold to greater than 3000-fold by the addition of the 8-chloro substituent.
Selected compounds were investigated for activity in cellular assays during optimization of CHK1 potency. Cytotoxicity was measured with a sulforhodamine B (SRB) assay(51) using the human colon carcinoma cell line HT29. A cell based enzyme-linked immunosorbent assay (ELISA) was developed to measure G2 checkpoint abrogation by quantifying the level of M-phase phosphoprotein 2 (MPM2) expression (a measure of mitosis) in HT29 cells that passed through an etoposide-induced G2 arrest into mitosis, where they were trapped using nocodazole.(17) The ratio of SRB cytoxicity to cellular ELISA activity gave an activity index (AI), which we found to be a useful indicator of selective inhibition of CHK1 in cells.17,48
As shown in Table 4 with selected examples of the scaffolds described above, the cytotoxity was generally weak, with only 30 and (R)-3 giving GI50 values in the nanomolar range. The potency in the G2 checkpoint abrogation assay in general tracked the CHK1 activity. Isoquinolines 28, 30, and (R)-3, three compounds with CHK1 IC50 <100 nM, abrogated the etoposide-induced G2 checkpoint with IC50 < 200 nM. Conversely, the imidazo[4,5-c]pyridines, e.g. 26, demonstrated good potency in the enzyme assay but showed minimal activity in both the G2 checkpoint abrogation and SRB assays. We attributed this to poor permeability of the imidazo[4,5-c]pyridine core scaffold when paired with the piperidine, consistent with the high polar surface area calculated for 26.
Compounds abrogating the G2 checkpoint at nanomolar concentrations while remaining relatively noncytotoxic (AI > 4) were assessed further to gauge the enhanced efficacy when used in combination with a cytotoxic drug. HT29 colon cancer cells were exposed to a fixed concentration of SN38 (the active metabolite of the DNA topoisomerase I inhibitor irinotecan(52)) that inhibited growth by 50% relative to untreated controls, and they were additionally exposed to increasing concentrations of the CHK1 inhibitor in a 96 h SRB assay. The ability of the CHK1 inhibitor to enhance the cytotoxicity of SN38 was expressed as a potentiation index (PI), which was the ratio of GI50 for the CHK1 inhibitor alone and the GI50 for the CHK1 inhibitor in combination with SN38.(17) Potentiation was observed for 27, 28, 29, and (R)-3, with all except 27 giving a greater than 2-fold potentiation. The two analogues having the most favorable properties overall were 28 and (R)-3, which had GI50 values of 345 nM and 190 nM, respectively, in the presence of SN38. These encouraging cellular assay data led to further profiling of (R)-3.
The inhibitory activity of (R)-3 was assessed at 1 μM concentration against a representative sample of 124 kinases from across the human kinome, using radiometric assay format(56) at [ATP] ∼ Km,ATP for all the enzymes (Figure 3). Only CHK1 and 6 other kinases out of 124 showed >80% inhibition at 1 μM. A further 8 kinases showed some inhibition (40–80%) at 1 μM, but the majority of kinases tested (109/124) showed less than 40% inhibition at 1 μM concentration of (R)-3, indicative of >100-fold selectivity. The kinase inhibitory activities of (R)-3 were also determined in a smaller panel at the higher concentration of 10 μM (see Supporting Information). The compound also displayed good selectivity at this higher concentration, where only CHK1 and 8 other kinases out of 50 tested showed >80% inhibition.
Figure 3.

Selectivity profile of (R)-3 against a panel of 124 kinases measured at 1 μM concentration of the test compound with [ATP] ∼ Km,ATP for each kinase. Kinase dendrogram(58) reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
Metabolic turnover of (R)-3 in mouse and rat liver microsomes was high (80% and 92% metabolized at 30 min, respectively), but the compound was appreciably more stable in human liver microsomes (35% metabolized at 30 min). Good permeability was seen for a monolayer of human intestinal epithelial cells (CaCo-2; A to B Pe 1.43 × 10–6 cm s–1) with no active efflux observed (ratio A → B/B → A 0.25).
(R)-3 was studied in vivo as previously described,(17) including pharmacodynamic and efficacy determinations in SW620 human colon carcinoma xenografts in nude mice.(59) Although having minimal oral bioavailability in mice (F = 5%), distribution of (R)-3 following i.p. dosing (40 mg/kg) was sufficient to inhibit CHK1 in the tumors, as shown by inhibition of the irinotecan-induced CHK1 pS296 autophosphorylation (Figure 4A).(17) At doses giving inhibition of CHK1 activity in vivo, the selective CHK1 inhibitor (R)-3 showed no single agent activity in the SW620 xenograft model, and tumors grew at similar rates to the vehicle-treated controls. When dosed (i.p.) in combination with irinotecan, (R)-3 was observed to potentiate the antitumor activity of the genotoxic drug in the SW620 xenograft model. Tumor growth was delayed when compared to the vehicle-treated control, CHK1 inhibitor alone, or cytotoxic drug alone. Similar data were obtained for the combination of (R)-3 with gemcitabine(57) (Figure 4B and see ref (17)). These experiments demonstrated that the in vitro cellular potency and potentiation of genotoxic cytotoxicity by (R)-3 translated into in vivo biomarker modulation and potentiation of genotoxic drug efficacy expected for a selective CHK1 inhibitor.17,23
Figure 4.

(A) Status of pS296 CHK1 autophosphorylation in SW620 tumor xenografts following treatment with either irinotecan alone or in combination with (R)-(3).(17) Tumor bearing animals were administered either vehicle alone, irinotecan alone (25 or 50 mg/kg ip), or irinotecan combined with a fixed dose of (R)-(3) (40 mg kg–1 ip) 1 h prior to irinotecan administration. Tumors were collected 6 or 24 h following treatment (6 or 24 after administration of the first agent for combinations) and snap frozen. Tumor protein expression was characterized by Western blotting using 50 μg of sample per lane. (B) Antitumor effects of combining (R)-3 with irinotecan in nude mice bearing SW620 xenograft tumors.(17) Symbols: (↑) dose administered; (■) vehicle alone; (shaded up triangles) irinotecan alone (12.5 mg/kg ip); (shaded down triangles) (R)-3 alone (40 mg/kg ip); (⧫) irinotecan and (R)-3 combined ((R)-3 dosed 1 h prior to and 24 h after irinotecan). Values are mean ± SEM; n = 9–11.
Conclusion
The evolution of pyrazolopyridine 2, a micromolar inhibitor of CHK1, to the potent and selective isoquinoline inhibitor (R)-3 demonstrates how fragment-growing and scaffold morphing strategies can be combined to successfully progress fragment-derived hit matter to compounds with appropriate activity in vivo. By incorporating inhibitor selectivity against CHK2 as a goal from the start of the medicinal chemistry, and guided by determinants of CHK1 selectivity apparent from the structural biology of the enzyme, the promising selectivity for CHK1 in the early fragment-derived leads was maintained and improved. Fragment-growing identified two productive vectors for enhancing the potency and selectivity of the initial bicyclic inhibitors: an extension to contact the water-filled pocket of CHK1, best achieved with a tricyclic heteroaromatic framework, and substitution to bind at the specificity surface of the ATP site. The determination of multiple protein–ligand structures during the optimization confirmed the introduction of productive new binding interactions and allowed a rationalization of the poor affinity of sterically crowded bicycles which adopted energetically unfavorable conformations on binding.
Scaffold modification, or morphing, is a common strategy for improvement and diversification of lead molecules derived from screening approaches. In this case, breaking up the tricyclic scaffold to a series of N-(pyrazin-2-yl)pyrimidin-4-amines, followed by optimization of a basic amino-substituted side-chain, maintained the hinge-binding hydrogen bonds and other key interactions for CHK1 potency and selectivity, while addressing challenges of synthetic tractability and crowded intellectual property space encountered with the tricyclic and bicyclic inhibitors, respectively. A further scaffold morphing step was achieved through translocation of the basic amine side-chain, allowing diversification of the hinge-binding group to include a series of 2-amino-isoquinolines. This approach led to the isoquinoline (R)-3 (SAR-020106), a potent inhibitor of CHK1 with excellent selectivity with respect to CHK2 and a broader sample of the kinome, representing one of the most selective CHK1 inhibitors reported to date. Despite the extensive scaffold modification, the isoquinoline (R)-3 (MW = 382) retained the good ligand efficiency of the original template hit, 6-(morpholin-4-yl)purine 1.
Cell-based assays for selective CHK1 inhibition, through measuring the abrogation of an etoposide-induced G2 checkpoint, and for nonselective cytotoxicity were used to confirm that the observed kinase selectivity translated into the expected CHK1-selective pharmacology in cells. The pharmacokinetic properties of the optimized isoquinoline (R)-3 were sufficient to demonstrate inhibition of CHK1 in vivo on i.p. dosing. Pharmacodynamic biomarkers showing the activation of CHK1 by genotoxic drugs were inhibited in human tumor xenografts. At a pharmacodynamically active dose, the inhibitor potentiated the antiproliferative efficacy of the genotoxic agents irinotecan and gemcitabine toward human SW620 colon cancer xenografts in nude mice, and minimal effects of the selective CHK1 inhibitor alone were shown. While optimization of pharmacokinetic properties is desirable to generate compounds suitable for development, the isoquinoline (R)-3 is a useful in vitro and in vivo chemical tool to investigate the effects of selective CHK1 inhibition.
Experimental Section
Synthetic Chemistry
The synthetic procedures for the four scaffolds and the analogues discussed in this paper (3–34) are outlined in Schemes 1–4. The previously described pyrazolopyridine 35(33) was brominated at the 3-position using NBS (Scheme 1). A Suzuki–Miyaura coupling and deprotection of the amine gave the pyrazolopyridine 4. For the pyrrolopyrimidine analogues commercially available 4-chloro-7H-pyrrolo[2,3-d]pyrimidine 37 was first brominated followed by SEM protection of the pyrazole nitrogen to give 38. Displacement of the chlorine substituent with Boc-protected 2-(aminomethyl)morpholine at 100 °C in n-butanol gave an advanced intermediate 39, which was elaborated using Suzuki–Miyaura couplings. Dual deprotection using TBAF and ethylenediamine in DMF followed by HCl in dioxane/MeOH gave 5 and 6.
Scheme 1.

Reagents and conditions: (i) NBS, DMF, rt, 77%; (ii) PdCl2(dppf)·CH2Cl2, 3-cyanophenylboronic acid, Na2CO3, DME/H2O, 140 °C, 2 h, microwave, 48%; (iii) TFA, MeOH, 80 °C, 62%; (iv) NBS, CH2Cl2, rt, 83%; (v) NaH, SEM-Cl, DMF, 0 °C to rt, 93%; (vi) Boc-amine, TEA, n-BuOH, 100 °C, 68%; (vii) Pd(PPh3)4, phenylboronic acid, Na2CO3, DME/H2O, 120 °C, 30 min, microwave, 49–59%; (viii) TBAF, ethane-1,2-diamine, DMF, 60 °C, 7 h and then TFA, MeOH, 80 °C, 21%; (ix) TBAF, ethane-1,2-diamine, DMF, 60 °C, 7 h and then 4 M HCl–dioxane, MeOH, 24 h, rt, 17%.
Scheme 4.

Reagents and conditions: (i) 4-(aminomethyl)piperidine-1-carboxylate, TEA, MeCN, 1.5 h, rt, 93%; (ii) SnCl2·2H2O, EtOH, 70 °C, 2 h, 88%; (iii) (EtO)3CH, Ac2O, 100 °C, 18 h, 40%–100%; (iv) 4-methoxybenzylamine, TEA, MeCN, 1.5 h, rt, 77%; (v) aryl halide (ArCl for 24, 25, 29; ArI for 27), Pd(OAc)2, (±)-BINAP, NaOtBu, DMF/Tol, 150 °C, 30 min, microwave and then MP-TsOH column, 3–41% (11% for 25 including Boc deprotection); (vi) NHMe2 (40% in H2O), 0–20 °C, 2 h 40%; (vii) aq NH3 (28%), 100 °C, o/n, 91%; (viii) NBS, CH2Cl2, 0 °C, 1 h, 42%; (ix) CuI, 18-crown-6, Pd(PPh3)4, KCN, DMF, reflux, 3 h, 82%; (x) NaH (60% in oil), dioxane, aminoalcohol, 100 °C, o/n, 14–32%; (xi) Pd(OAc)2, (±)-BINAP, NatBu, DMF/toluene, microwave, 140–150 °C, 30 min, or Pd2(dba)3, Xantphos, Cs2CO3, toluene, microwave, 140 °C, 45 min (2–45%); (xii) Pd(OAc)2, (±)-BINAP, NatBu, DMF/Tol, microwave, 140–150 °C, 30 min, or Pd2(dba)3, Xantphos, Cs2CO3, toluene, microwave, 140 °C, 45 min followed by TFA, 80 °C, 30 min or TFA, CH2Cl2, rt, 1 h MP-TsOH column (2–26%).
(4-(9H-Pyrimido[4,5-b]indol-4-yl)-morpholin-2-yl)methanamines 7 were synthesized by reacting the racemic or single enantiomer(33) Cbz-protected 2-(aminomethyl)morpholine with commercially available 4-chloro-9H-pyrimido[4,5-b]indole (48) and subsequent acid-mediated removal of the Cbz protecting group (Scheme 2). The pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidine and pyrimido[4,5-b]indole-7-carbonitrile tricyclic cores 49 and 50 were constructed from 4-chloro-5-nitropyridine (40) and 4-chloro-3-nitrobenzonitrile (41), respectively (Scheme 2). Nucleophilic aromatic subsitution of the starting material with the anion of ethylcyanoacetate and reduction of the nitro group with zinc in acetic acid allowed an intramolecular cyclization which provided the ethyl 2-amino-1H-indole-3-carboxylate 45 or its 6-aza analogue 44. Condensation with formamide completed the tricyclic cores (46, 47), and chlorination with phosphorus oxychloride installed the reactive chloride functionality of 49 and 50. The diamines were introduced as described for the previous examples to provide 7–13.
Scheme 2.

Reagents and conditions: (i) NaH, NCCH2CO2Et, DMF, 0 °C to rt and then 40 or 41, 2 h, rt, 48%; (ii) Zn/AcOH, 95 °C, 1 h 15 min, 88%; (iii) formamide, ammonium formate, 170 °C, 18 h, 45%; (iv) POCl3, 75 °C, 18 h, 100%; (v) amine, TEA, DMF, 120 °C, 1 h, microwave and then 4 M HCl, dioxane/MeOH, 2.5 h, 3–46%; (vi) amine, TEA, NMP, 140 °C, microwave and then MP-TsOH column, 29%.
The N-(pyridin-2-yl)pyrimidine-4,6-diamines and N-(pyrazin-2-yl)pyrimidine-4,6-diamines were assembled via a palladium-mediated amination of 4,6-dichloropyrimidine (51) with 3-aminopyridine (52), 2-aminopyrazine (53), or 2-amino-5-cyanopyrazine (54) (Scheme 3). The resulting intermediates 55–57 were derivatized by SNAr reactions with amines followed by a deprotection step, where required, to yield 14–22.
Scheme 3.

Reagents and conditions: (i) PdCl2(PPh3)2, LiHMDS, THF, 135 °C, 20 min, 19%–22%; or when X = CH(tBu3P)2Pd(0), NaOtBu, toluene, 80 °C, 2 h, 45%; (ii) amine, NMP, 145 °C, 20 min, (26–41%); (iii) amine, NMP, 145 °C, 20 min, followed by TFA, CH2Cl2, 1 h, rt, or HCl, dioxane, MP-TsOH column (3–63%).
The azabenzimidazoles 60 and 62 were prepared by first stirring 2-bromo-4-chloro-5-nitropyridine (58) at rt with tert-butyl 4-(aminomethyl)piperidine-1-carboxylate or 4-methoxybenzylamine followed by a reduction of the nitro group with tin(II) chloride dihydrate to give the diamine 59 or 61 (Scheme 4). Cyclizations to the azabenzimidazoles were effected by heating with acetic anhydride and triethylorthoformate.
In the course of our investigations into the isoquinoline analogues, several different isoquinoline reagents were coupled using a variety of Buchwald–Hartwig conditions (Scheme 4 and Supporting Information). The lengthy synthesis of 64 has been previously described,(60) while 3-chloroisoquinoline 63 is commercially available and 8-chloroisoquinolin-3-yl trifluoromethanesulfonate 65 was synthesized in large quantities using the method reported by Ventura et al.(61)
Buchwald–Hartwig palladium mediated aminations of 60 and 62 with commercially available 2-amino-5-cyanopyrazine 66 followed by deprotection of Boc or PMB protecting groups using TFA in CH2Cl2 at ambient temperature, or with neat TFA at 80 °C, respectively, gave analogues 24 and 25. Similarly, palladium mediated aminations of 63 and 64 with 66 afforded analogues 27 and 29 directly.
The Buchwald–Hartwig coupling partners for the other isoquinolines and azabenzimidazoles were constructed from 2,6-dichloropyrazine.(60) Substitution of one chlorine using NH3 followed by bromination ortho to the remaining chlorine gave the precursor to the desired 5-amino-3-chloropyrazine-2-carbonitrile 70. Cyanation was achieved by heating with potassium cyanide in DMF with a crown ether and Pd(PPh3)4 to complete the synthesis of 70. SNAr with protected or tertiary aminoalkoxides gave a range of substituted pyrazines 71–76 for Buchwald–Hartwig palladium mediated coupling to the haloheterocycles 62–65. The single enantiomers of 1-(dimethylamino)propan-2-ol, e.g. 68, required for the synthesis of (R)-3 and (S)-3 were prepared by the careful addition of an aqueous solution of dimethylamine to the appropriate, neat, enantiomerically pure propylene oxide, e.g. 67 (Scheme 4). Palladium mediated couplings between isoquinolines 63 or 65 with the relevant substituted pyrazines 73, 74, 75, or 76 afforded products 33, 34, 3, (R)-3, or (S)-3 directly. The optical purities of (R)-3 and (S)-3 were determined to be >95% ee by chiral HPLC analysis (see Supporting Information). Where required, Boc or PMB protecting groups were removed following coupling reactions using TFA in dichloromethane at ambient temperature, or with neat TFA at 80 °C, respectively, to give compounds 26, 28, and 30–32.
General Synthetic Chemistry
Reactions were carried out under N2. Organic solutions were dried over MgSO4 or Na2SO4. Starting materials and solvents were purchased from commercial suppliers and were used without further purification. Microwave reactions were carried out using Biotage Initiator 60 or CEM microwave reactors. Flash silica chromatography was performed using Merck silica gel 60 (0.025–0.04 mm). Ion exchange chromatography was performed using Isolute Flash SCX-II (acidic) or Flash NH2 (basic) resin cartridges. 1H NMR spectra were recorded on a Bruker AMX500 instrument at 500 MHz or Bruker Avance instrument at 400 MHz using internal deuterium locks. Chemical shifts (δ) are reported relative to TMS (δ = 0) and/or referenced to the solvent in which they were measured. Combined HPLC-MS analyses were recorded using either
(1) (LCT) a Waters Alliance 2795 separations module and Waters/Micromass LCT mass detector with electrospray ionization (+ve or −ve ion mode as indicated) with HPLC performed using Supelco DISCOVERY C18, 50 mm × 4.6 mm or 30 mm × 4.6 mm i.d. columns or an Agilent 6210 TOF HPLC-MS with a Phenomenex Gemini 3 μm C18 (3 cm × 4.6 mm i.d.) column [Both were run at a temperature of 22 °C with gradient elution of 10–90% MeOH/0.1% aqueous formic acid at a flow rate of 1 mL/min and a run time of 3.5 or 6 min as indicated. UV detection was at 254 nm, and ionization was by positive or negative ion electrospray. The molecular weight scan range was 50–1000 amu.]
(2) (ZQ) a Micromass ZQ mass spectrometer/Waters Alliance 2795 HT HPLC with a Phenomenex Gemini 5 μm, C18, 30 mm × 4.6 mm i.d. column or a Waters X-Bridge C18, 2.5 μM, 3.0 × 30 mm column. [Both were run at a temperature of 35 °C with a gradient elution of 5–95% [(0.1% ammonia in acetonitrile)/(0.1% ammonia, 5% acetonitrile, and 0.063% ammonium formate in water)] at a flow rate of 2 mL/min and a run time of 4 or 7 min as indicated. UV detection was at 220–400 nm using a Waters 996 photodiode array UV detector, and ionization was by positive or negative ion electrospray. The molecular weight scan range was 80–1000 amu.]
All tested compounds gave >95% purity as determined by these methods. All purified synthetic intermediates gave >95% purity as determined by these methods except where indicated in the text. High-resolution mass spectra were measured using the Agilent TOF system described above.
Ethyl 3-Bromo-4-(2-((tert-butoxycarbonyl)aminomethyl)morpholino)-1H-pyrazolo[3,4-b]pyridine-5-carboxylate (36)
N-Bromosuccinimide (0.044 g, 0.25 mmol) was added in portions at rt to a solution of ethyl 4-(2-((tert-butoxycarbonylamino)methyl)morpholino)-1H-pyrazolo[3,4-b]pyridine-5-carboxylate(33) (35; 0.083 g, 0.20 mmol) in DMF (2 mL). After stirring for 2 h, saturated brine was added and the precipitate was collected by filtration, washed with water, and dried, to yield 36 as a pale yellow powder (0.076 g, 77%). 1H NMR (500 MHz, d4-MeOD) δ 1.40–1.46 (9H + 3H, m), 3.05–3.13 (1H, m), 3.13–3.29 (3H, m), 3.39–3.48 (1H, m), 3.55–3.62 (1H, m), 3.95–4.00 (2H, m), 4.02–4.07 (1H, m), 4.44 (2H, q, J = 7.5 Hz), 8.53 (1H, s); LC-MS (LCT, 6 min) Rt 4.83 min; m/z (ESI) 484, 486 [MH+].
Ethyl 4-(2-(Aminomethyl)morpholino)-3-(3-cyanophenyl)-1H-pyrazolo[3,4-b]pyridine-5-carboxylate (4)
A mixture of 36 (14 mg, 0.029 mmol), PdCl2(dppf)·CH2Cl2 (18 mg, 10 mol %), 3-cyanophenylboronic acid (9 mg, 0.06 mmol), and Na2CO3 (8 mg, 0.075 mmol) in DME (2 mL) and water (0.5 mL) was heated to 140 °C in a microwave reactor for 2 h. The mixture was partitioned between brine (10 mL) and EtOAc (2 × 8 mL). The combined organic layers were washed with brine (10 mL) and water (10 mL), dried, filtered, and concentrated. Preparative TLC, eluting with 1:2 hexane/EtOAc, gave ethyl 4-(2-((tert-butoxycarbonylamino)methyl)morpholino)-3-(3-cyanophenyl)-1H-pyrazolo[3,4-b]pyridine-5-carboxylate as a light yellow oil (7 mg, 48%). 1H NMR (500 MHz, d4-MeOD) δ 1.44 (3H, t, J = 7.5 Hz), 1.46 (9H, s), 2.86–3.05 (4H, m), 3.06–3.26 (4H, m), 3.57–3.65 (1H, m), 4.45 (2H, q, J = 7.5 Hz), 7.75 (1H, dd, J = 7.5, 7.5 Hz), 7.87 (1H, d, J = 7.5 Hz), 8.01 (1H, d, J = 7.5 Hz), 8.08 (1H, s), 8.55 (1H, s); LC-MS (LCT, 6 min) Rt 4.74 min; m/z (ESI) 507 [MH+]. Ethyl 4-(2-((tert-butoxycarbonylamino)methyl)morpholino)-3-(3-cyanophenyl)-1H-pyrazolo[3,4-b]pyridine-5-carboxylate (2 mg, 0.004 mmol) was dissolved in MeOH (2 mL), and TFA (1 mL) was added. After being refluxed at 80 °C for 16 h, the solvents were evaporated and the residue was purified on SCX-II acidic resin (2 g), eluting with MeOH and then 2 M NH3/MeOH. The basic fractions were combined and concentrated. The crude oil was purified by preparative TLC, eluting with EtOAc, to give 4 as a yellow oil (1 mg, 62%). 1H NMR (500 MHz, d4-MeOD) δ 1.43 (3H, t, J = 7.5 Hz), 2.46–2.51 (2H, m), 2.85–2.91 (1H, m), 2.97–3.05 (2H, m), 3.08–3.25 (3H, m), 3.62–3.67 (1H, m), 4.44 (2H, q, J = 7.5 Hz), 7.75 (1H, dd, J = 7.5, 7.5 Hz), 7.90 (1H, d, J = 7.5 Hz), 8.00 (1H, d, J = 7.5 Hz), 8.08 (1H, s), 8.56 (1H, s); LC-MS (LCT, 6 min) Rt 2.56 min; m/z (ESI) 407 [MH+]. HRMS (ESI) m/z calcd for C21H23N6O3 (M + H) 407.1826, found 407.1831.
5-Bromo-4-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (38)
A suspension of 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (37; 0.49 g, 3.2 mmol) and N-bromosuccinimide (0.68 g, 3.8 mmol) in dry CH2Cl2 (20 mL) was stirred at rt for 2.5 h. The suspension was diluted with MeOH and evaporated onto silica. The crude product was purified using flash column chromatography, eluting with 2:1 hexanes/EtOAc, to yield 5-bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidine as an off-white solid (0.613 g, 2.64 mmol, 83%). 1H NMR (500 MHz, d4-MeOD) δ 7.64 (1H, s), 8.56 (1H, s); LC-MS (LCT, 6 min) Rt 3.34 min; m/z (ESI) 236, 234, 232 [MH+]. To a solution of 5-bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1.03 g, 4.43 mmol) in DMF (12 mL) at 0 °C was added NaH (0.21 g, 60% suspension in oil, 5.05 mmol). After the suspension was stirred at this temperature for 15 min, SEM-Cl (0.89 g, 5.34 mmol) in DMF (2 mL) was added. The resulting suspension was then stirred at 0 °C for 20 min and at rt for 1 h. Water (30 mL) was added; the precipitate was collected by filtration, washed with water (2 × 30 mL), and dried in vacuo. Compound 38 was obtained as light pink crystals (1.5 g, 93%). 1H NMR (500 MHz, d6-DMSO) δ 0.00 (9H, s), 0.90 (2H, t, J = 7.5 Hz), 3.60 (2H, t, J = 7.5 Hz), 5.70 (2H, s), 7.25 (1H, s), 8.70 (1H, s); LC-MS (LCT, 6 min) Rt 4.34 min; m/z (ESI) 366, 364, 362 [MH+].
tert-Butyl (4-(5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)morpholin-2-yl)methylcarbamate (39)
A solution of tert-butyl morpholin-2-ylmethylcarbamate (0.075 g, 0.35 mmol), 38 (0.12 g, 0.33 mmol), and Et3N (0.13 mL, 0.75 mmol) in n-BuOH (1.5 mL) was heated at 100 °C in a microwave reactor for 1 h. The solution was concentrated onto silica gel. The crude product was purified using flash column chromatography, eluting with 2:1 hexanes/EtOAc, to yield 39 as a viscous oil (0.122 g, 0.225 mmol, 68%). 1H NMR (500 MHz, CDCl3) δ −0.03 (9H, s), 0.93 (2H, t, J = 7 Hz), 1.27 (9H, s), 2.95 (1H, dd, J = 7, 7 Hz), 3.21–3.23 (2H, m), 3.40–3.45 (1H, m), 3.56 (2H, t, J = 7 Hz), 3.85–3.90 (2H, m), 4.02–4.16 (2H, m), 4.90–4.95 (1H, m), 5.58 (2H, s), 7.28 (1H, s), 8.43 (1H, s); LC-MS (LCT, 6 min) Rt 4.30 min; m/z (ESI) 544, 542 [MH+].
3-(4-(2-(Aminomethyl)morpholino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)benzonitrile (5)
A mixture of 39 (44 mg, 0.08 mmol), Pd(PPh3)4 (5 mg, 4 mol %), 3-cyanophenylboronic acid (24.5 mg, 0.22 mmol), and sodium carbonate (30 mg, 0.17 mmol) in DME (1.5 mL) and water (0.5 mL) was heated to 120 °C in a microwave reactor for 30 min. The mixture was partitioned between brine (10 mL) and EtOAc (2 × 8 mL). The combined organic layers were washed with brine (10 mL) and water (10 mL), dried (Na2SO4), filtered, and concentrated. Preparative TLC, eluting with EtOAc/n-hexane/1:1 (Rf = 0.27), gave tert-butyl (4-(5-bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)morpholin-2-yl)methylcarbamate as a yellow oil (27 mg, 59%). 1H NMR (500 MHz, CDCl3) δ 0.00 to −0.04 (9H, s), 0.95 (2H, t, J = 8.0 Hz), 1.45 (9H, s), 2.65–2.72 (1H, m), 2.82–2.98 (2H, m), 3.09–3.19 (1H, m), 3.35–3.50 (2H, m), 3.61 (2H, t, J = 8.0 Hz), 3.62–3.78 (2H, m), 4.58 (1H, m), 4.78 (1H, broad s), 5.67 (2H, s), 7.30 (1H, s), 7.51–7.53 (2H, m), 7.78 (1H, d, J = 7.5 Hz), 7.84 (1H, s), 8.55 (1H, s); LC-MS (LCT, 6 min) Rt 5.57 min; m/z (ESI) 565 [MH+]. A mixture of tert-butyl (4-((5-(3-cyanophenyl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)morpholin-2-yl)methylcarbamate (37 mg, 0.066 mmol), 1.0 M TBAF/THF (0.55 mL, 0.55 mmol), and ethane-1,2-diamine (20 μL) in DMF (1.5 mL) was stirred at 60 °C under N2 for 16 h. It was diluted with brine (8 mL) and extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL) and water (10 mL), dried (Na2SO4), filtered, and concentrated. The crude oil (12 mg) was purified by preparative TLC, yielding 8 mg (LC-MS Rt 4.51 min; m/z (ESI) 435 [MH+]). It was then dissolved in a mixture of MeOH (3 mL) and TFA (2 mL). The solution was stirred at 80 °C for 12 h. The solvents were evaporated, and the residue was purified on SCX-II acidic resin (1 g) eluting with MeOH and then 2 M NH3–MeOH. The basic fractions were combined and evaporated to give 5 as a yellow oil (4.5 mg, 21%). 1H NMR (500 MHz, d4-MeOD) δ 2.52–2.60 (1H, m), 2.63–2.70 (1H, m), 2.85–2.95 (2H, m), 3.41–3.51 (2H, m), 3.55–3.60 (1H, m), 3.74 (2H, d, J = 12 Hz), 7.48 (1H, s), 7.79–7.55 (2H, m), 7.88 (J = 7.7 Hz, 1H, d), 7.93 (1H, s), 8.39 (1H, s); LC-MS (LCT, 6 min) Rt 2.12 min; m/z (ESI) 335 [MH+]. HRMS (ESI) m/z calcd for C18H19N6O (M + H) 335.1620, found 335.1618.
C-[4-(9H-Pyrimido[4,5-b]indol-4-yl)-morpholin-2-yl]methylamine (rac-7)
A mixture of 4-chloro-9H-pyrimido[4,5-b]indole (47; 0.045 g, 0.221 mmol), morpholin-2-ylmethylcarbamic acid tert-butyl ester (0.055 g, 0.25 mmol), and Et3N (0.10 mL, 0.75 mmol) in DMF (0.70 mL) was heated to 120 °C in a microwave reactor for 1 h. The cooled solution was partitioned between water (20 mL) and EtOAc (20 mL). The organic extract was dried (Na2SO4), filtered, and concentrated. Flash column chromatography, eluting with EtOAc, gave [4-(9H-pyrimido[4,5-b]indol-4-yl)morpholin-2-ylmethyl]carbamic acid tert-butyl ester as a yellow solid (0.021 g, 0.0548 mmol, 25%). LC-MS (LCT, 6 min) Rt 5.57 min; m/z (ESI) 384 [MH+]. A solution of [4-(9H-pyrimido[4,5-b]indol-4-yl)morpholin-2-ylmethyl]carbamic acid tert-butyl ester (0.021 g, 0.548 mmol) and 4 M HCl–dioxane (1 mL) in MeOH (5 mL) was stirred at rt for 2.5 h. The solution was evaporated to dryness and purified by ion exchange on SCX-II acidic resin (2 g), eluting with MeOH and then 2 M NH3–MeOH. The basic fractions were combined. Preparative silica TLC, eluting with 1% NH3–9% MeOH–90% CH2Cl2, gave rac-7 (0.007 g, 0.025 mmol, 46%) as a beige powder. 1H NMR (500 MHz, d4-MeOD) δ 2.75–2.80 (2H, m), 3.00–3.06 (1H, m), 3.35–3.40 (1H, m), 3.75–3.85 (1H, m), 3.90 (1H, dd, J = 10, 10 Hz), 4.10–4.13 (1H, m), 4.19 (1H, d, J = 13 Hz), 4.25 (1H, d, J = 13 Hz), 7.35 (1H, dd, J = 8.8 Hz), 7.47 (1H, dd, J = 8.8 Hz), 7.57 (1H, d, J = 8 Hz), 7.79 (1H, d, J = 8 Hz), 8.46 (1H, s); LC-MS (LCT, 6 min) Rt 1.98 min; m/z (ESI) 284 [MH+].
Ethyl 2-Cyano-2-(3-nitropyridin-4-yl)acetate (42)
Ethylcyanoacetate (3.75 g, 38 mmol) in DMF (3 mL) was added dropwise at 0 °C to a suspension of NaH (1.5 g, 60% in mineral oil, 38 mmol) in DMF (9 mL). The reaction mixture was stirred at rt for 30 min and then cooled to 0 °C, and 3-nitro-4-chloropyridine (41; 2.63 g, 19 mmol, 1 equiv) in DMF (3 mL) was added slowly. Two molar HCl (20 mL) and EtOAc (20 mL) were added after 2 h stirring at rt, and the organic layer was washed with water and brine, dried, and concentrated to give a red-orange oil. The crude oil was triturated with EtOAc, and the resultant solid was collected by filtration to give 42 (2.88 g, 48%). 1H NMR (d6-DMSO) δ 1.18 (3H, t, J = 7.0 Hz), 4.06 (2H, q, J = 7.0 Hz), 7.58 (1H, br s), 7.88 (1H, dd, J = 1.0, 7.0 Hz), 8.70 (1H, s), 13.2 (1H, br s); LC-MS (ZQ, 4 min) Rt 1.13 min; m/z (ES–) 190 [M – EtO], 234 [M – H].
2-Amino-1H-pyrrolo[2,3-c]pyridine-3-carboxylic Acid Ethyl Ester (44)
A solution of 42 (2.20 g, 9.35 mmol) in AcOH (20 mL) was heated to 80 °C under nitrogen. Zinc dust (6.12 g, 94 mmol) in 500 mg portions was added, and then the reaction mixture was heated at 95 °C for 1 h 15 min. Upon cooling, the insoluble material was filtered off and washed with fresh AcOH. The filtrate was concentrated, and the residue was treated with saturated NaHCO3 to give a light brown solid. This was filtered, washed with water, and dried to give 44 as a light brown solid (1.69 g, 8.24 mmol, 88% yield). 1H NMR (d6-DMSO) δ 1.32 (3H, t, J = 7.0 Hz), 4.23 (2H, q, J = 7.0 Hz), 7.0 (2H, br s), 7.41 (1H, d, J = 4.5 Hz), 8.0 (1H, br), 8.31 (1H, br), 11.0 (1H, s, br). LC-MS (TOF, 4 min), Rt = 1.70 min; m/z (ES+) 206.
9H-Pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidin-4-ol (46)
A mixture of 44 (1.80 g, 8.8 mmol) and ammonium formate (0.62 g, 9.8 mmol) in formamide (10 mL) was heated at 170 °C for 18 h. One molar HCl was added to the cooled reaction mixture, and the resulting suspension was filtered to remove insolubles. The filtrate was then adjusted to pH 7 with saturated sodium bicarbonate solution. The resulting precipitate was collected by filtration and dried to a constant weight in a vacuum oven to give 46 (0.74 g, 45%). 1H NMR (d6-DMSO) δ 7.89 (1H, d, J = 4.0), 8.26 (1H, s), 8.37 (1H, d, J = 4.0), 8.83 (1H, s), 12.5 (s, br); LC-MS (ZQ, 4 min) Rt = 0.75 min; m/z (ES–) 185 [M – H].
4-Chloro-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidine (49)
A mixture of 46 (264 mg, 1.4 mmol) and Et3N (750 μL) in POCl3 (5 mL) was heated at 75 °C for 18 h. Toluene was added to the cooled reaction mixture, and the solvents were then removed in vacuo to give 49 as a brown oil (290 mg, 100%) used directly in the subsequent reaction. LC-MS (ZQ, 4 min) Rt 1.25 min; m/z (ES+) 205/207.
4-(2-(Aminomethyl)morpholino)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidine (8)
A mixture of 49 (20 mg, 0.098 mmol), morpholin-2-ylmethylcarbamic acid tert-butyl ester (32 mg, 0.147 mmol), and Et3N (68 mL, 0.489 mmol) in NMP (1 mL) was heated in a microwave reactor at 140 °C for 15 min. The reaction mixture was diluted with MeOH and applied to a MP-TsOH cartridge which had been preconditioned with MeOH. The cartridge was flushed with MeOH and left for 20 min. The product was eluted using 2 M NH3 in MeOH, affording a brown oil, which was purified using preparative HPLC to give 4-(2-(aminomethyl)morpholino)-9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyrimidine (11 mg, 40%). 1H NMR (d4-MeOD) δ 3.10–3.04 (1H, m), 3.25–3.19 (2H, m), 3.58–3.51 (1H, m), 3.83 (1 H, td, J = 11.6, 2.5), 4.00–3.94 (1H, m), 4.14 (1H, dd, J = 11.6, 2.0), 4.32 (1H, d, J = 13.4), 4.46 (1H, d, J = 12.9), 7.78 (1H, d, J = 5.6), 8.43 (1H, d, J = 5.6), 8.46 (2H, br s), 8.54 (1H, s), 8.84 (1H, s); LC-MS (ZQ, 7 min) Rt 1.78 min; m/z (ES+)285 [M + H]. HRMS (ESI) m/z calcd for C14H17N6O (M + H) 285.1458, found 285.1457.
5-(6-Chloropyrimidin-4-ylamino)pyrazine-2-carbonitrile (57)
A mixture of 4,6-dichloropyrimidine (51; 1.0 g, 6.7 mmol), 2-amino-5-cyanopyrazine (54; 806 mg, 6.7 mmol), and bis(triphenylphosphine)palladium(II) chloride (94 mg, 0.134 mmol) in dry THF (24 mL) was degassed under a stream of nitrogen gas over 10 min with stirring. 1 M LiHMDS in THF (7.38 mL, 7.4 mmol) was added, and the mixture was heated at 135 °C for 20 min in a microwave reactor. The reaction mixture was adsorbed onto silica and purified by flash chromatography, eluting with 30% EtOAc in hexane, to give 57 (300 mg, 19%). LC-MS (ZQ, 4 min) Rt = 1.64; min m/z (ES–) 231 [M – H].
5-(6-(Piperidin-4-ylmethylamino)pyrimidin-4-ylamino)pyrazine-2-carbonitrile (20)
A mixture of 57 (124 mg, 0.533 mmol), 4-(aminomethyl)-1-N-Boc-piperidine (228 mg, 1.066 mmol), and Et3N (150 μL, 1.07 mmol) in 1-methyl-2-pyrrolidinone (1 mL) was heated at 145 °C for 15 min in a microwave reactor. The mixture was concentrated in vacuo, and the residue was purified by preparative HPLC. The purified solid was dissolved in CH2Cl2 (2 mL) and trifluoroacetic acid (3 mL) and was stirred for 1 h at rt before being applied to a MP-TsOH cartridge. After washing with MeOH, the pure product was eluted using 7 M NH3 to give 20 (10 mg, 6.2%). 1H NMR (d6-DMSO, 400 MHz): δ 1.30 (3H, m), 1.85 (4H, m), 2.71 (2H, m), 3.18 (2H, m), 3.26 (1H, br s), 7.14 (1H, br s), 8.21 (1H, s), 8.62 (s, 1H), 8.79 (1H, br s); LCMS (LCT, 4 min) Rt = 0.78 min m/z (ES+) 311. HRMS (ESI) m/z calcd for C15H19N8 (M + H) 311.1742, found 311.1727.
N4-(4-Methoxybenzyl)-6-bromopyridine-3,4-diamine (61)
A solution of 4-methoxybenzylamine (0.756 g, 5.51 mmol) in acetonitrile (2 mL) was added to a mixture of 2-bromo-4-chloro-5-nitropyridine (53) (1.19 g, 5.01 mmol) and Et3N (0.768 mL, 5.51 mmol) in acetonitrile (8 mL). After stirring for 1.5 h, the solution was diluted with EtOAc (100 mL), and the resulting solution was then washed successively with water and brine before being concentrated in vacuo to a light brown oil which solidified on standing to give N-(4-methoxybenzyl)-2-bromo-5-nitropyridin-4-amine (1.31 g, 3.87 mmol, 77%). 1H NMR (d6-DMSO, 400 MHz) δ 3.70 (3H, s), 4.60 (2H, d, J = 6 Hz), 6.95 (2H, d, J = 9 Hz), 7.10 (1H, s), 7.35 (2H, d, J = 9 Hz), 8.80 (s, 1H), 9.00 (br t, 1H, J = 6 Hz). LC-MS (ZQ, 4 min) Rt = 2.13 min; m/z (ESI–) 336, 338 [M – H]. Tin(II) chloride dihydrate (4.37 g, 19.4 mmol) was added portionwise to N-(4-methoxybenzyl)-2-bromo-5-nitropyridin-4-amine (1.31 g, 3.87 mmol) in absolute EtOH (10 mL) at rt. The mixture was heated at 70 °C for 2 h before being concentrated in vacuo. The residue was suspended in a mixture of EtOAc and saturated sodium bicarbonate solution and filtered. The insoluble solids were washed with EtOAc. The aqueous phase was re-extracted with EtOAc, and the combined organic layers were washed with brine, dried (Na2SO4), and concentrated to give 61 as a brown solid (1.05 g, 3.41 mmol, 88%). 1H NMR (d6-DMSO, 400 MHz) δ 3.75 (3H, s), 4.30 (2H, d, J = 5.5 Hz), 4.85 (2H, br s), 6.35 (1H, br t, 5.5 Hz), 6.45 (1H, s), 6.90 (2H, d, J = 8.5 Hz), 7.30 (2H, d, J = 8.5 Hz), 7.40 (1H, s); LC-MS (ZQ, 4 min) Rt = 1.77 min; m/z (ESI–) 306, 308; (ESI+) 308, 310 [MH+].
1-(4-Methoxybenzyl)-6-bromo-1H-imidazo[4,5-c]pyridine (62)
Acetic anhydride (1.28 mL, 13.6 mmol) was added to a solution of 61 (1.05 g, 3.41 mmol) in triethylorthoformate (13 mL). The mixture was heated at 100 °C for 18 h and then concentrated to give 62 as a brown oil (1.18 g, quantitative). 1H NMR (d6-DMSO, 400 MHz) δ 3.70 (3H, s), 5.45 (2H, s), 6.90 (2H, d, J = 9.0 Hz), 7.35 (2H, d, J = 9 Hz), 8.00 (1H, s), 8.60 (1H, s), 8.75 (1H, s). LC-MS (ZQ, 7 min) Rt = 2.27 min; m/z (ESI+) 318, 320 (MH+).
5-(1H-Imidazo[4,5-c]pyridin-6-ylamino)pyrazine-2-carbonitrile (24)
Palladium(II) acetate (3.5 mg, 16 μmol) was added to (±)-2,2″-bis(diphenylphosphino)-1,1″-binaphthalene (59 mg, 94 μmol) in DMF/toluene (1:2), and the resulting mixture was degassed under a stream of nitrogen gas for 10 min. 2-Amino-5-cyanopyrazine (66; 19 mg, 0.16 mmol), sodium tert-butoxide (45 mg, 0.47 mmol), and 62 (50 mg, 0.16 mmol) were added, and the mixture was degassed for a further 5 min before heating at 150 °C for 30 min in a microwave reactor. The reaction mixture was partitioned between water and CH2Cl2. The aqueous phase was extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and concentrated. The residue was dissolved in MeOH, passed through a PS-Thiol column, and concentrated. The product was purified using preparative HPLC to give 5-(1-(4-methoxybenzyl)-1H-imidazo[4,5-c]pyridin-6-ylamino)pyrazine-2-carbonitrile (22.4 mg, 40%). 1H NMR (d6-DMSO, 400 MHz) δ 3.72 (3H, s), 5.41 (2H, s), 6.96 (2H, d, J = 8.8 Hz), 7.34 (2H, d, J = 8.8 Hz), 8.17 (1H, s), 8.49 (1H, s), 8.73–8.76 (2H, m), 8.77 (1H, d, J = 1.3 Hz), 10.84 (1H, br s). LC-MS (ZQ, 7 min) Rt = 2.32 min; m/z (ESI+) 358 (MH+), (ESI–) 356 (M – H). 5-(1-(4-Methoxybenzyl)-1H-imidazo[4,5-c]pyridin-6-ylamino)pyrazine-2-carbonitrile (10.6 mg, 30 μmol) was treated with TFA at 80 °C over 30 min. Isolation by SPE on a MP-TsOH cartridge, eluting with 2M NH3 in MeOH, followed by concentration, gave 24 as a white solid (7.02 mg, 100%). 1H NMR (d6-DMSO, 400 MHz) δ 8.26 (1H, s), 8.35 (1H, s), 8.75–8.77 (3H, m), 10.85 (1H, br s), 12.79 (1H, br s); LC-MS (ZQ, 7 min) Rt = 1.36 min; m/z (ESI+) 238 (MH+), (ESI–) 236 (M – H). HRMS (ESI) m/z calcd for C11H8N7 (M + H) 238.0836, found 238.0845.
(R)-1-(Dimethylamino)propan-2-ol (68)
Dimethylamine 40% in water (11.39 mL, 90 mmol) was slowly added to (R)-propylene oxide (5.25 mL, 74.9 mmol) which had been cooled in an ice bath. This solution was stirred at rt for 2 h before being extracted with CH2Cl2 (4 × 5 mL). The combined organic layers were dried over Na2SO4, and the pure (R)-1-(dimethylamino)propan-2-ol (5.12 g, 49.6 mmol, 40.0% yield) was isolated as a clear oil using distillation under reduced pressure (50 mbar). 1H NMR (CDCl3, 500 MHz) δ 1.12 (3H, d, J = 6.0 Hz), 2.16–2.12 (1H, m), 2.25–2.21 (1H, m), 2.27 (6H, s), 3.40 (1H, br s), 3.82–3.76 (1H, m).
5-Amino-3-chloropyrazine-2-carbonitrile (70)
2,6-Dichloropyrazine (69; 2.89 g, 19.4 mmol) was stirred in aqueous NH3 (28%, 10 mL) and heated to 100 °C overnight in a sealed tube. The reaction mixture was cooled, and the resultant precipitate was filtered. Trituration with water and then ether gave 6-chloropyrazin-2-amine as a white solid (2.28 g, 17.6 mmol, 91%). 1H NMR (d6-DMSO, 400 MHz) δ 6.9 (2H, br s), 7.70 (1H, d, J = 0.4 Hz), 7.80 (1H, d, J = 0.4 Hz); LC-MS (ZQ, 7 min) Rt = 1.05 min; m/z (ESI+) 130, 132 (MH+). 6-Chloropyrazin-2-amine (2.50 g, 19.3 mmol) was stirred in CH2Cl2 (60 mL) and cooled to 0 °C. N-Bromosuccinimide (2.92 g, 16.4 mmol) was added slowly, and the reaction mixture was stirred at 0 °C for 60 min. The reaction mixture was filtered through Celite and concentrated to give a brown oil. Purification by flash chromatography, eluting with 0–25% EtOAc–hexanes, gave 5-bromo-6-chloropyrazin-2-amine as a yellow solid (1.69 g, 8.16 mmol, 42%). 1H NMR (d6-DMSO, 400 MHz) δ 7.1 (2H, br s), 7.65 (1H, s); LC-MS (ZQ, 4 min) Rt = 1.46 min; m/z (ESI–) 205 (M – H). A mixture of 5-bromo-6-chloropyrazin-2-amine (1.00 g, 4.8 mmol), copper(I) iodide (914 mg, 4.8 mmol), 18-crown-6 (95 mg, 0.36 mmol), and tetrakis(triphenylphosphine)palladium (0) (83 mg, 0.072 mmol) was suspended in dry DMF (20 mL), and a stream of nitrogen was passed through it for 5 min. Potassium cyanide (312 mg, 4.8 mmol) was added, and the mixture was stirred at rt for 30 min and then refluxed at 200 °C for 3 h. The mixture was cooled and diluted with EtOAc and absorbed onto silica gel (10 g). DMF was removed by evaporation. The product was purified by flash chromatography, eluting with 1:1 EtOAc–hexanes, to yield 70 as a yellow solid (607 mg, 3.93 mmol, 82%). 1H NMR (d6-DMSO, 400 MHz) δ 7.87 (1H, s), 8.1 (2H, br s); LC-MS (ZQ, 4 min) Rt = 1.20 min; m/z (ESI–) 153 (M – H).
(R)-5-Amino-3-(1-(dimethylamino)propan-2-yloxy)pyrazine-2-carbonitrile (74)
(R)-1-(Dimethylamino)propan-2-ol (68; 0.667 g, 6.47 mmol) was added dropwise to a suspension of NaH 60% (0.388 g, 9.71 mmol) in dioxane (16.18 mL) and stirred for 30 min. 5-Amino-3-chloropyrazine-2-carbonitrile (70; 1.000 g, 6.47 mmol) was added in one portion and heated at 90 °C for 14 h. After cooling water (200 mL) was added and the solution was extracted with diethyl ether (4 × 100 mL) and dried over MgSO4, the volatiles were removed under vacuum. Column chromatography using a gradient of MeOH in CH2Cl2 (+ 1% NH3) gave 74 (0.558 g, 2.52 mmol, 39.0% yield) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 1.35 (3H, d, J = 6.5 Hz), 2.37 (6H, s), 2.52 (1H, dd, J = 4.0, 13.5 Hz), 2.76 (1H, dd, J = 7.5, 13.5 Hz), 5.31 (2H, br s), 5.42–5.35 (1H, m), 7.54 (1H, s). LC-MS (TOF, 3.5 min) Rt = 0.80 min; m/z (ESI) 222 (M + H).
(R)-5-(8-Chloroisoquinolin-3-ylamino)-3-(1-(dimethylamino)propan-2-yloxy)pyrazine-2-carbonitrile ((R)-3)
To four microwave vials was added Xantphos (66.8 mg, 0.116 mmol), Pd2(dba)3 (52.9 mg, 0.058 mmol), 8-chloroisoquinolin-3-yl trifluoromethanesulfonate (180 mg, 0.578 mmol), 74 (128 mg, 0.578 mmol), and Cs2CO3 (376 mg, 1.155 mmol) followed by dry toluene (4 mL). The vials were sealed, and dry nitrogen was bubbled through the stirred solution for 8 min. The reactions were irradiated at 130 °C for 45 min in a Biotage microwave. Upon cooling, the mixtures were combined, diluted with MeOH, and purified using an acidic ion exchange column which was washed with MeOH before the basic components eluted with 2 M NH3 in MeOH followed by CH2Cl2/2 M NH3 in MeOH. The residue was purified by column chromatography eluting with a gradient of MeOH in CH2Cl2 (+1% NH3) to give (R)-3 (301 mg, 0.786 mmol, 45% yield) as a light yellow powder. 1H NMR (500 MHz, d6-DMSO) δ 1.46 (3H, d, J = 6.5 Hz), 2.21 (6H, s), 2.54 (2H, dd, J = 5, 13 Hz), 2.66 (1H, dd, J = 7, 13 Hz), 5.54–5.46 (1H, m), 7.66 (1H, d, J = 7.5 Hz), 7.72 (1H, dd, J = 7.5, 8.0 Hz), 7.84 (1H, d, J = 8.0 Hz), 8.26 (1H, s), 8.48 (1H, s), 9.40 (1H, s), 11.16 (1H, s). LC-MS (TOF, 3.5 min) Rt = 2.58 min; m/z (ESI) 383 (M + H). HRMS (ESI) m/z calcd for C19H20ClN6O (M + H) 383.1382, found 383.1366.
Acknowledgments
We thank Dr. A. Mirza, M. Richards, and Dr. M. Liu for assistance in the spectroscopic characterization of test compounds and Dr. M. Lamers for the preparation of CHK1 protein. This work was supported by Cancer Research UK [CUK] grant numbers C309/A2187, C309/A8274, and C309/A8365 and by The Institute of Cancer Research. We acknowledge NHS funding to the NIHR Biomedical Research Centre.
Glossary
Abbreviations Used
- ATP
adenosine 5′-triphosphate
- ATR
ataxia telangiectasia and rad3 related
- CHK1
checkpoint kinase 1
- CHK2
checkpoint kinase 2
- DELFIA
dissociation-enhanced lanthanide fluorescent immunoassay
- ELISA
enzyme-linked immunosorbent assay
- LE
ligand efficiency
- MPM2
M-phase phosphoprotein 2
- SRB
sulforhodamine B
Supporting Information Available
Experimental details for the preparation and characterization of compounds (rac)-3, (S)-3, 6, (R)-7, (S)-7, 9–19, 21, 22, and 25–34; experimental details for the determination of the enantiopurity of (R)-3 and (S)-3; experimental details for CHK1 and CHK2 enzyme inhibition assays; summary of crystal structure determinations of CHK1 in complex with 2, 4, 6, 8, 20, and (R)-3; and kinase inhibitory profiles of (R)-3 at 1 μM and 10 μM test concentrations. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally to this work.
Accession Codes
PDB ID Codes: 2ym3, 2ym4, 2ym5, 2ym6, 2ym7, 2ym8.
Supplementary Material
References
- Collins I.; Garrett M. D. Targeting the cell division cycle in cancer: CDK and cell cycle checkpoint kinase inhibitors. Curr. Opin. Pharmacol. 2005, 5, 366–373. [DOI] [PubMed] [Google Scholar]
- Dai Y.; Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010, 16, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan M. B.; Bartek J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Kumagai A.; Wang S. X.; Dunphy W. G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000, 14, 2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke C. A.; Clarke P. R. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem. J. 2005, 388, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits V. A. J. Spreading the signal. Dissociation of Chk1 from chromatin. Cell Cycle 2006, 5, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Zhou B. B.; Elledge S. J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [DOI] [PubMed] [Google Scholar]
- Feijoo C.; Hall-Jackson C.; Wu R.; Jenkins D.; Leitch J.; Gilbert D. M.; Smythe C. Activation of mammalian Chk1 during DNA replication arrest: A role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 2001, 154, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C. Y.; Graves P. R.; Thoma R. S.; Wu Z.; Shaw A. S.; Piwnica-Worms H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997, 277, 1501–1505. [DOI] [PubMed] [Google Scholar]
- Sanchez Y.; Wong C.; Thoma R. S.; Richman R.; Wu Z.; Piwnica-Worms H.; Elledge S. J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through cdc25. Science 1997, 277, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Chen Z.; Gunasekera A. H.; Sowin T. J.; Rosenberg S. H.; Fesik S.; Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 2003, 278, 21767–21773. [DOI] [PubMed] [Google Scholar]
- Hollstein M.; Sidransky D.; Vogelstein B.; Harris C. C. p53 mutations in human cancers. Science 1991, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Oren M. Regulation of the p53 tumor suppressor protein. J. Biol. Chem. 1999, 274, 36031–36034. [DOI] [PubMed] [Google Scholar]
- Bucher N.; Britten C. D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol. Cancer Ther. 2004, 3, 513–519. [PubMed] [Google Scholar]
- Walton M. I.; Eve P. D.; Hayes A.; Valenti M.; De Haven Brandon A.; Box G.; Boxall K. J.; Aherne G. W.; Eccles S. A.; Raynaud F. I.; Williams D. H.; Reader J. C.; Collins I.; Garrett M. D. The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106. Mol. Cancer Ther. 2010, 9, 89–100. [DOI] [PubMed] [Google Scholar]
- Blasina A.; Hallin J.; Chen E.; Arango M. E.; Kraynov E.; Register J.; Grant S.; Ninkovic S.; Chen P.; Nichols T.; O’Connor P.; Anderes K. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 2008, 7, 2394–2404. [DOI] [PubMed] [Google Scholar]
- Ganzinelli M.; Carrassa L.; Crippa F.; Tavecchio M.; Broggini M.; Damia G. Checkpoint kinase 1 down-regulation by an inducible small interfering RNA expression system sensitized in vivo tumors to treatment with 5-fluorouracil. Clin. Cancer Res. 2008, 14, 5131–5141. [DOI] [PubMed] [Google Scholar]
- Tse A. N.; Rendahl K. G.; Sheikh T.; Cheema H.; Aardalen K.; Embry M.; Ma S.; Moler E. J.; Ni Z. J.; Lopes de Menezes D. E.; Hibner B.; Gesner T. G.; Schwartz G. K. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin. Cancer Res. 2007, 13, 591–602. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Xue J.; Sowin T. J.; Zhang H. Differential roles of checkpoint kinase 1, checkpoint kinase 2, and mitogen-activated protein kinase-activated protein kinase 2 in mediating DNA damage-induced cell cycle arrest: implications for cancer therapy. Mol. Cancer Ther. 2006, 5, 1935–1943. [DOI] [PubMed] [Google Scholar]
- Zabludoff S. D.; Deng C.; Grondine M. R.; Sheehy A. M.; Ashwell S.; Caleb B. L.; Green S.; Haye H. R.; Horn C. L.; Janetka J. W.; Liu D.; Mouchet E.; Ready S.; Rosenthal J. L.; Queva C.; Schwartz G. K.; Taylor K. J.; Tse A. N.; Walker G. E.; White A. M. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 2008, 7, 2955–2966. [DOI] [PubMed] [Google Scholar]
- Garrett M. D.; Collins I. Anticancer therapy with checkpoint inhibitors: What, where and when?. Trends Pharmacol. Sci. 2011, 32, 308–316. [DOI] [PubMed] [Google Scholar]
- Janetka J. W.; Ashwell S. Checkpoint kinase inhibitors: A review of the patent literature. Expert Opin. Ther. Pat. 2009, 19, 165–197. [DOI] [PubMed] [Google Scholar]
- Janetka J. W.; Ashwell S.; Zabludoff S.; Lyne P. Inhibitors of checkpoint kinases: From discovery to the clinic. Curr. Opin. Drug Discovery Dev. 2007, 10, 473–486. [PubMed] [Google Scholar]
- Prudhomme M. Novel checkpoint 1 inhibitors. Recent Pat. Anti-Cancer Drug Discovery 2006, 55–68. [DOI] [PubMed] [Google Scholar]
- Tao Z.-F.; Wang L.; Stewart K. D.; Chen Z.; Gu W.; Bui M.-H.; Merta P.; Zhang H.; Kovar P.; Johnson E.; Park C.; Judge R.; Rosenberg S.; Sowin T.; Lin N.-H. Structure-based design, synthesis, and biological evaluation of potent and selective macrocyclic checkpoint kinase 1 inhibitors. J. Med. Chem. 2007, 50, 1514–1527. [DOI] [PubMed] [Google Scholar]
- Chen P.; Luo C.; Deng Y.; Ryan K.; Register J.; Margosiak S.; Tempczyk-Russell A.; Nguyen B.; Myers P.; Lundgren K.; Kan C. C.; O’Connor P. M. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell 2000, 100, 681–692. [DOI] [PubMed] [Google Scholar]
- Foloppe N.; Fisher L. M.; Francis G.; Howes R.; Kierstan P.; Potter A. Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification. Bioorg. Med. Chem. 2006, 14, 1792–1804. [DOI] [PubMed] [Google Scholar]
- Foloppe N.; Fisher L. M.; Howes R.; Kierstan P.; Potter A.; Robertson A. G.; Surgenor A. E. Structure-based design of novel Chk1 inhibitors: Insights into hydrogen bonding and protein-ligand affinity. J. Med. Chem. 2005, 48, 4332–4345. [DOI] [PubMed] [Google Scholar]
- Converso A.; Hartingh T.; Garbaccio R. M.; Tasber E.; Rickert K.; Fraley M. E.; Yan Y.; Kreatsoulas C.; Stirdivant S.; Drakas B.; Walsh E. S.; Hamilton K.; Buser C. A.; Mao X.; Abrams M. T.; Beck S. C.; Tao W.; Lobell R.; Sepp-Lorenzino L.; Zugay-Murphy J.; Sardana V.; Munshi S. K.; Jezequel-Sur S. M.; Zuck P. D.; Hartman G. D. Development of thioquinazolinones, allosteric Chk1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1240–1244. [DOI] [PubMed] [Google Scholar]
- Ma C. X.; Janetka J. W.; Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends. Mol. Med. 2011, 17, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews T. P.; Klair S.; Burns S.; Boxall K.; Cherry M.; Fisher M.; Westwood I. M.; Walton M. I.; McHardy T.; Cheung K.-M. J.; Van Montfort R.; Williams D.; Aherne G. W.; Garrett M. D.; Reader J.; Collins I. Identification of inhibitors of checkpoint kinase 1 through template screening. J. Med. Chem. 2009, 52, 4810–4819. [DOI] [PubMed] [Google Scholar]
- Matthews T. P.; McHardy T.; Klair S.; Boxall K.; Fisher M.; Cherry M.; Allen C. E.; Addison G. J.; Ellard J.; Aherne G. W.; Westwood I. M.; Montfort R. v.; Garrett M. D.; Reader J. C.; Collins I. Design and evaluation of 3,6-di(hetero)aryl imidazo[1,2-a]pyrazines as inhibitors of checkpoint and other kinases. Bioorg. Med. Chem. Lett. 2010, 20, 4045–4049. [DOI] [PubMed] [Google Scholar]
- Antoni L.; Sodha N.; Collins I.; Garrett M. D. CHK2 kinase: cancer susceptibility and cancer therapy—two sides of the same coin?. Nat. Rev. Cancer 2007, 7, 925–936. [DOI] [PubMed] [Google Scholar]
- Guzi T. J.; Paruch K.; Dwyer M. P.; Labroli M.; Shanahan F.; Davis N.; Taricani L.; Wiswell D.; Seghezzi W.; Penaflor E.; Bhagwat B.; Wang W.; Gu D.; Hsieh Y.; Lee S.; Liu M.; Parry D. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011, 10, 591–602. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficency: A useful tool for metric lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Collins I.; Reader J. C.; Cheung K. M.; Matthews T. P.; Proisy N.; Klair S. S.. Morpholino-bicyclo-heteroaryl compounds as CHK1 kinase inhibitors and their preparation, pharmaceutical compositions and use in the treatment of cancer. Patent Appl. WO2008075007, 2008; Chem Abstr. 2008, 149, 104737
- Cherry M.; Williams D. H. Recent kinase and kinase inhibitor X-ray structures: Mechanisms of inhibition and selectivity insights. Curr. Med. Chem. 2004, 11, 663–673. [DOI] [PubMed] [Google Scholar]
- Anand N. K.; Blazey C. M.; Bowles O. J.; Bussenius J.; Canne Bannen L.; Chan D. S.-M.; Chen B.; Co E. W.; Costanzo S.; Defina S. C.; Dubenko L.; Franzini M.; Huang P.; Jammalamadaka V.; Khoury R. G.; Kim M. H.; Klein R. R.; Le D. T.; Mac M. B.; Nuss J. M.; Parks J. J.; Rice K. D.; Tsang T. H.; Tsuhako A. L.; Wang Y.; Xu W.. Preparation and structure activity of pyrazolo-pyrimidine derivatives as antitumor agents and kinase modulators. Patent WO 2005117909, 2005; Chem Abstr. 2006, 144, 104737
- Bold G.; Frei J.; Lang M.; Traxler P.; Furet P.. Preparation of pyrazolo[3,4-d]pyrimidines as antitumor agents. Patent WO 9814449, 1998; Chem Abstr. 1998, 128, 282843.
- Caldwell J. J.; Davies T. G.; Donald A.; McHardy T.; Rowlands M. G.; Aherne G. W.; Hunter L. K.; Taylor K.; Ruddle R.; Raynaud F. I.; Verdonk M.; Workman P.; Garrett M. D.; Collins I. Identification of 4-(4-aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as selective inhibitors of protein kinase B through fragment elaboration. J. Med. Chem. 2008, 51, 2147–2157. [DOI] [PubMed] [Google Scholar]
- Hoehn H.; Denzel T.. 1H-Pyrazolo[3,4-b]pyridines. Patent DE 2301268, 1973; Chem Abstr. 1973, 79, 115578.
- McHardy T.; Caldwell J. J.; Cheung K. M.; Hunter L. J.; Taylor K.; Rowlands M.; Ruddle R.; Henley A.; de Haven Brandon A.; Valenti M.; Davies T. G.; Fazal L.; Seavers L.; Raynaud F. I.; Eccles S. A.; Aherne G. W.; Garrett M. D.; Collins I. Discovery of 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides as selective, orally active inhibitors of protein kinase B (Akt). J. Med. Chem. 2010, 53, 2239–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler P.; Bold G.; Frei J.; Lang M.; Lydon N.; Mett H.; Buchdunger E.; Meyer T.; Mueller M.; Furet P. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(Phenylamino)pyrazolo[3,4-d]pyrimidines. J. Med. Chem. 1997, 40, 3601–3616. [DOI] [PubMed] [Google Scholar]
- Traxler P.; Furet P.; Bold G.; Frei J.; Lang M.. Preparation of 4-amino-1H-pyrazolo[3,4-d]pyrimidines as EGF-receptor-specific protein tyrosine kinase inhibitors. Patent WO 9631510, 1996; Chem Abstr. 1997, 126, 8129.
- Collins I.; Reader J. C.; Klair S.; Scanlon J.; Addison G.; Cherry M.. Preparation of 9H-pyrimido[4,5-b]indole, 9H-pyrido[4′,3′:4,5]pyrrolo[2,3-d]pyridine and 9H-1,3,6,9-tetraazafluorene derivatives as CHK1 kinase inhibitors. Patent WO 2009004329, 2009; Chem Abstr. 2009, 150, 98355.
- Wang G. T.; Li G.; Mantei R. A.; Chen Z.; Kovar P.; Gu W.; Xiao Z.; Zhang H.; Sham H. L.; Sowin T.; Rosenberg S. H.; Lin N.-H. 1-(5-Chloro-2-alkoxyphenyl)-3-(5-cyano-pyrazin-2-yl)ureas as potent and selective inhibitors of Chk1 kinase: Synthesis, preliminary SAR, and biological activities. J. Med. Chem. 2005, 48, 3118–3121. [DOI] [PubMed] [Google Scholar]
- Li G.; Hasvold L. A.; Tao Z.-F.; Wang G. T.; Gwaltney S. L. II; Patel J.; Kovar P.; Credo R. B.; Chen Z.; Zhang H.; Park C.; Sham H. L.; Sowin T.; Rosenberg S. H.; Lin N.-H. Synthesis and biological evaluation of 1-(2,4,5-trisubstituted phenyl)-3-(5-cyanopyrazin-2-yl)ureas as potent Chk1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2293–2298. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Hays F. A.; Westhof E.; Ho P. S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Kunimoto T.; Nitta K.; Tanaka T.; Uehara N.; Baba H.; Takeuchi M.; Yokokura T.; Sawada S.; Miyasaka T.; Mutai M. Antitumor activity of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothecin, a novel water-soluble derivative of camptothecin, against murine tumors. Cancer Res. 1987, 47, 5944–5947. [PubMed] [Google Scholar]
- Ghose A. K.; Viswanadhan V. N.; Wendoloski J. J. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: an analysis of ALOGP and CLOGP methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [DOI] [PubMed] [Google Scholar]
- PipelinePilot, version 7.0; Accelrys: San Diego, CA, USA, http://accelrys.com.
- The National Centre for Protein Kinase Profiling, MRC Protein Phosphorylation Unit, University of Dundee, U.K. (http://www.kinase-screen.mrc.ac.uk)
- Matthews D. J.; Yakes F. M.; Chen J.; Tadano M.; Bornheim L.; Clary D. O.; Tai A.; Wagner J. M.; Miller N.; Kim Y. D.; Robertson S.; Murray L.; Karnitz L. M. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 2007, 6, 104–110. [DOI] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- All procedures followed U.K. Home Office and local ethical guidelines and were performed according to U.K. National Cancer Research Institute guidelines, see:Workman P.; Aboagye E. O.; Balkwill F.; Balmain A.; Bruder G.; Chaplin D. J.; Double J. A.; Everitt J.; Farningham D. A.; Glennie M. J.; Kelland L. R.; Robinson V.; Stratford I. J.; Tozer G. M.; Watson S.; Wedge S. R.; Eccles S. A. Committee of the National Cancer Research Institute. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins I.; Reader J. C.; Williams D. H.; Klair S. S.; Scanlon J. E.; Piton N.; Cherry M.. Bicyclylaryl-aryl-amine compounds, their preparation, and their use as CHK1 kinase inhibitors for treating proliferative diseases. Patent WO 2009103966, 2009; Chem Abstr. 2009, 151, 289187.
- Ventura B.; Barigelletti F.; Durola F.; Flamigni L.; Sauvage J.-P.; Wenger O. S. Fe(II), Ru(II) and Re(I) complexes of endotopic, sterically non-hindering, U-shaped 8,8′-disubstituted-3,3′-biisoquinoline ligands: syntheses and spectroscopic properties. Dalton Trans. 2008, 491–498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.