

Abstract
Introduction
Methotrexate (MTX) induces macrophage apoptosis in vitro, but there is not much evidence for increased synovial macrophage apoptosis in MTX-treated patients. Macrophage apoptosis is reported, however, during clinical response to anti-tumor necrosis factor-alpha (TNF-α) treatments. This implies that TNF-α promotes macrophage survival and suggests that TNF-α may protect against MTX-induced apoptosis. We, therefore, investigated this proposal and the macrophage signaling pathways underlying it.
Methods
Caspase-3 activity, annexin-V binding/7-aminoactinomycin D (7-AAD) exclusion and cell-cycle analysis were used to measure steps in apoptosis of primary murine macrophages and cells of the RAW264.7 macrophage cell line that had been exposed to clinically-relevant concentrations of MTX and TNF-α.
Results
MTX induces apoptosis in primary murine macrophages at concentrations as low as 100 nM in vitro. TNF-α, which has a context-dependent ability to increase or to suppress apoptosis, efficiently suppresses MTX-induced macrophage apoptosis. This depends on NF-κB signaling, initiated through TNF Receptor Type 1 ligation. Macrophage colony stimulating factor, the primary macrophage survival and differentiation factor, does not activate NF-κB or protect macrophages from MTX-induced apoptosis. A weak NF-κB activator, Receptor Activator of NF-κB Ligand (RANKL) is likewise ineffective. Blocking NF-κB in TNF-α-exposed macrophages allowed pro-apoptotic actions of TNF-α to dominate, even in the absence of MTX. MTX itself does not promote apoptosis through interference with NF-κB signaling.
Conclusions
These findings provide another mechanism by which TNF-α sustains macrophage numbers in inflamed tissue and identify a further point of clinical complementarity between MTX and anti-TNF-α treatments for rheumatoid arthritis.
Introduction
Synovial inflammatory macrophages have a central role in maintaining disease activity in rheumatoid arthritis (RA). Macrophage numbers in tissue are regulated by recruitment, local proliferation, local cell death and emigration to draining lymph nodes [1,2]. Synovial macrophage apoptosis has also been observed in synovium in RA [3,4]. A suppressed rate of apoptosis would contribute to maintaining inflammatory macrophage numbers, and thus clinical activity, in macrophage-dependent conditions. An enhanced rate of synovial macrophage apoptosis is reported in RA patients responding to anti-TNF-α treatments [4] as a delayed, rather than an early phenomenon [5]. Macrophage apoptosis has also been reported in patients with Crohn's disease after anti-TNF-α treatment [6]. These observations suggest that TNF-α directly, or indirectly, sustains macrophage survival in these conditions.
TNF-α activity supports recruitment of macrophages into RA synovium [7], but is not known to enhance the proliferation of macrophages or to prevent the emigration of macrophages through lymphatics [2]. TNF-α, however, may deliver either apoptotic or survival signals, depending on the cell context. TNF-α is a ligand for two related receptors, TNF-R1 (p55) and TNF-R2 (p75). Macrophages and their bone marrow and blood-borne progenitors express both receptors [8]. TNF-α ligation to TNF-R1 leads to assembly of a Death-Inducing Signaling Complex (DISC) and ultimately activation of downstream effector caspases-3/6/7. Where this is the dominant consequence of TNF-R1 ligation, apoptosis follows. In other circumstances, TNF-R1 initiates survival signaling [9] through activation of the NF-κB pathway. TNF-α ligation to TNF-R1 can thus bring apoptosis or survival, depending on the relative activities generated in the DISC-initiated and NF-κB pathways [10]. Macrophage survival is notably dependent on NF-κB signaling. The transcriptionally active RelA/NF-κB1 (p65/p50) complex appears in macrophage nuclei constitutively during late differentiation and is important for continuing survival [10,11]. In vivo, there is enhanced nuclear expression of RelA/NF-κB1 in synovial macrophages in RA [12], consistent with a role for NF-κB activity in maintaining macrophage survival.
The observations of enhanced apoptosis during anti-TNF-α treatment imply that TNF-α predominantly antagonises apoptosis in RA and Crohn's disease [4,6]. On the other hand, methotrexate (MTX), an anti-rheumatic drug that has demonstrable pro-apoptotic effects on monocyte/macrophage cells in vitro, does not appreciably alter the synovial membrane macrophage apoptosis rate in RA patients [3,13]. This led us to question whether the presence of TNF-α in synovium was providing a survival signal to macrophages in MTX-treated patients, thus antagonizing one potential therapeutic function of MTX. That would provide additional explanation for the clinical complementarity of MTX and anti-TNF-α treatments in RA [14]. In the study of Catrina et al., which demonstrated synovial macrophage apoptosis in response to anti-TNF-α therapy, the majority of patients were co-treated with MTX [4].
MTX is a folate analogue with numerous effects on inflammatory cell proliferation and survival, cytokine expression, angiogenesis, cell adhesion and reactive oxygen production [15]. The known direct targets of MTX are dihydrofolate reductase, thymidylate synthase and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase [15]. Consequences of inhibiting these enzymes include reduced availability of purines and pyrimidines for DNA and RNA synthesis and accumulation of AICAR. AICAR, by inhibiting enzymatic deamination of adenosine and adenosine monophosphate, is proposed to increase availability of adenosine extracellularly, for anti-inflammatory effect through adenosine cell surface receptors [16]. These effects are generally believed to be mediated by long-lived polyglutamate forms of MTX, which are generated in many cell types, including normal myeloid precursor cells and myelocytic cancer cells [17].
Materials and methods
Materials
The Recombinant Macrophage-Colony Stimulating Factor (M-CSF) was obtained from R&D Systems (Sydney, NSW, Australia). Annexin V conjugated to phycoerythrin (Annexin V-PE) and 7-amino-actinomycin (7-AAD) were from BD Biosciences (Sydney, NSW, Australia). Parthenolide was purchased from Alexis Biochemicals (Lausen, Switzerland). Rabbit anti-IκBα (C-21) polyclonal antibody, anti-NF-κB p65, anti-NF-κB p50, anti-c-Rel and anti-RelB were obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA. Mouse anti-phospho-IκBα (Ser32/36) (5A5) and horseradish peroxidase-conjugated anti-rabbit were from Amersham Biosciences (Sydney, NSW, Australia). Methotrexate was provided by Mayne Pharma Pty Ltd. (Adelaide, South Australia, Australia). Etanercept came from Wyeth Australia Pty Ltd. (Sydney, NSW, Australia). Recombinant GST-rRANKL was kindly provided by Ming-Hao Zheng and Jiake Xu, Centre for Orthopaedic Research, University of Western Australia [18]. All other reagents were sourced from Sigma-Aldrich (Sydney, NSW, Australia), unless otherwise indicated.
Cell culture
RAW264.7 cells were obtained from the American Type Culture Collection and maintained in Dulbecco's modified Eagle's medium (Life Technologies, Carlsbad, California, USA) with 10% low endotoxin fetal calf serum (CSL, Melbourne, Australia), penicillin and gentamicin (DMEM-FCS). Bone marrow-derived monocytes/macrophages (BMDM) were collected from the femora and tibiae of eight-week old female C57BL or BALB/c mice, as previously described [19], with the approval of the Animal Ethics Committee of The University of Western Australia. Cells were expanded in M-CSF (30 ng/mL)-supplemented DMEM-FCS for seven days at 37°C/5% CO2/95% air. The experiments reported here were conducted with BMDM from C57BL mice, but results were comparable using BALB/c mice.
MTT cytotoxicity assay
Viability was estimated using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazoliumbromide (MTT) assay. BMDM (3 × 104 cells/well) were seeded in 24-well plates in M-CSF-supplemented DMEM-FCS. After 48 hr, cells were treated with TNF-α for 3 hr, followed by MTX exposure for 24 hr. At the end of treatments, 50 μL of MTT (5 mg/mL) was added to each well, and cells were incubated at 37°C for 2 hr. Formazan crystals were then solubilised with 150 μL of 44% dimethyl formamide/20% SDS at room temperature for at least 30 minutes on a rocking platform. A total of 100 μL aliquots were quantitated at 550 nm using a microplate reader (POLARstar OPTIMA, BMG, Germany). Triplicate assays were conducted for all conditions.
Caspase-3 protease activity
RAW264.7 cells and BMDM were cultured in six-well plates at a density of 0.3 × 106 cells/well for 48 hr. After treatments, trypsin-detached RAW264.7 cells and scraped BMDM were lysed and assayed for caspase-3 activity as described previously [20].
Flow cytometry analysis
Apoptotic cells were quantitated by staining with annexin V-PE and 7-AAD, as specified by the manufacturer (BD Biosciences). Flow cytometry was performed on populations of 5000-10,000 cells (Becton Dickinson FACSCalibur, Sydney, NSW, Australia), with fluorescence of annexin V-PE and 7-AAD measured with a 585/42 nm bandpass filter (FL2 channel) and a 670 nm longpass filter (FL3 channel), respectively. Data are expressed as percentages of apoptotic cells, as defined by annexin-V-PE positivity and 7-AAD negativity.
Cell cycle analysis
RAW264.7 cells and BMDM (0.25 × 106/well of a 12-well plate) were collected and washed in cold Dulbecco's Phosphate Buffered Saline (DPBS) before being fixed with 70% ethanol at -20°C. After 24 hr, cells were centrifuged at 500 × g for 10 minutes at 4°C and resuspended in 500 μL of staining buffer (50 μg/mL propidium iodide and 25 μg/mL RNase in 0.1% Triton X-100). After incubating at 37°C for 30 minutes, cells were analysed on a FACSCalibur flow cytometer (Becton Dickinson) using a doublet discrimination protocol.
Western blot analysis
RAW264.7 cells and BMDM (1 × 106 per treatment) were washed with ice-cold DPBS and lysed in RIPA buffer (50 mM Tris, pH 7.5, containing 150 mM NaCl, 1% IGEPAL, 1% sodium deoxycholate, 0.1% SDS and 10 mM EDTA) supplemented with 1X complete protease inhibitors (Roche Applied Science, Sydney, NSW, Australia). A total of 20 μg of total protein was separated by SDS-PAGE and transferred to Hybond-P PVDF membranes (Amersham Biosciences). Membranes were blocked in 1% BSA/5% non-fat milk in TTBS (10 mM Tris, pH 7.6, 150 mM NaCl, 0.1% Tween 20) for one hour at room temperature, before being probed with specific antibodies to phospho-IκBα (1:1000 dilution), IκBα (1:2,000) or β-actin (1:8,000). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse were used at 1:8,000 and 1:20,000 dilutions, respectively. All antibodies were prepared in SignalBoost Immunoreaction Enhancer (Calbiochem, Darmstadt, Germany) and applied for one hour at room temperature. Blots were revealed with enhanced chemiluminescence (ECL) reagents (Amersham Biosciences).
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed on nuclear extracts from RAW264.7 cells (8 × 106 per treatment) that were treated with 10 ng/mL TNF-α for up to one hour, according to previously described protocols [21]. A double-stranded NF-κB consensus oligonucleotide probe 5'-GGGCATGGGAATTTCCAACTC-3' (0.25 pmol) with 5'-G overhangs was filled in with labeled (α-32P)dCTP (Amersham Pharmacia Biotech, Buckinghamshire, England) using the Klenow fragment of E. coli DNA polymerase I (Promega, Madison, Wisconsin, USA). This DNA probe was incubated with 3 μg of nuclear proteins for 10 minutes at room temperature. Where indicated, antibodies (1 μg) to specific NF-κB factors or unlabelled oligonucleotide probes at 100-fold molar excess were also included for supershift EMSA and competition experiments, respectively. Samples were loaded onto a 4% polyacrylamide gel, containing 0.25X Tris-Borate-EDTA buffer, which had been pre-run for two hours in the same buffer. After separation, gels were exposed to Cronex X-ray film, using a single intensifying screen.
Transient transfection of RAW264.7 cells and BMDM with an NF-κB reporter plasmid
RAW264.7 cells (1 × 107) were transiently transfected with 0.5 μg of pNFκB-TA-Luc (Mercury™ Pathway profiling system, BD Biosciences) by the DEAE-dextran procedure, as previously described [21]. Transfected cells were resuspended in DMEM-FCS and distributed into a 24-well culture plate at a density of 3.3 × 105 cells/well. Cells were rested for 48 hr before experimentation. Firefly luciferase expression in transfected RAW264.7 cells was measured using the Promega Luciferase Assay System, according to the manufacturer's instructions. Promoter activity was quantified by measuring light production in a multifunctional microplate reader (POLARstar OPTIMA, BMG, Germany), and presented as relative light units (RLU).
BMDM were transfected with Lipofectamine™ and Plus reagents (Invitrogen) according to manufacturer's instructions. Briefly, for each 24-well, 1 μg pNF-κB-TA-Luc was combined with 0.5 μL Plus reagent and 2 μL Lipofectamine™ in antibiotic-free medium. A total of 1.5 × 105 cells/well was added to the transfection mixture and plated in M-CSF-containing DMEM-FCS. After four to six hours of incubation at 37°C, wells were replaced with fresh growth medium. Luciferase expression was measured at least 48 hr after transfection.
Statistical analysis
Statistical significance (P < 0.05) between groups of experimental data was assessed using paired Student's t-tests.
Results
TNF-α protects primary macrophages and a macrophage cell line from MTX-induced apoptosis
M-CSF is the primary growth, differentiation and survival factor for macrophages under physiological conditions. Serum concentrations of M-CSF are elevated to approximately 0.6 ng/ml in patients with RA [22], but concentrations in synovium are unknown. In vitro, a concentration of 30 ng/ml was optimal for sustaining the growth and survival of bone marrow-derived macrophages (BMDM) of C57BL/6 mice (results not shown). In the presence of 30 ng/mL M-CSF, exposure to 10 μM MTX for 24 hr caused an approximately 25% loss of cell numbers in BMDM cultures (Figure 1A). This was related to increased apoptosis, as estimated by caspase-3 activity (Figure 1B), annexin-V binding (early apoptosis; Figure 1B) and increased sub-G0 (apoptotic) fraction on flow cytometric cell cycle analysis (Table 1). The proportion of cells in S-phase also increased with MTX, consistent with its known action on completing DNA synthesis [23] (Table 1).
Figure 1.

TNF-a antagonises MTX-induced apoptosis in macrophages, independent of M-CSF action. A. Exposure of primary BMDM to 10 mM MTX for 24 hr in the presence of 30 ng/mL M-CSF resulted in loss of approximately 25% of cells. This could be entirely prevented by introducing TNF-a three hours before MTX. Cell viability was assessed by the MTT reduction assay. Shown are mean ± SEM of normalised data from three independent experiments. B and D, TNF-a suppresses caspase-3 activation (left axis) and annexin-V binding (right axis) in MTX-exposed primary BMDM (B) and RAW264.7 cells (D). MTX was added three hours after TNF-a treatment, and caspase-3 activity or annexin-V binding was measured after 24 hr for BMDM and 6 hr for RAW264.7 cells. Data are mean ± SEM of at least four independent experiments in each case. C and E, MTX dose-dependently increases apoptosis in BMDM (C) and RAW264.7 cells (E). * = P < 0.05; ** = P < 0.01.
Table 1.
Cell cycle analysis of BMDM treated with TNF-α or its control, followed by MTX exposure for 24 hr or M-CSF withdrawal for 48 hr*.
| Cell cycle distribution (% of cells) | ||||
|---|---|---|---|---|
| sub G0 | G0/G1 | S | G2/M | |
| Control | 3.5 ± 0.5 | 78.6 ± 2.9 | 6.0 ± 1.0 | 10.7 ± 2.7 |
| MTX (10 μM) | 7.5 ± 1.0a | 62.7 ± 2.2a | 16.0 ± 1.2a | 12.0 ± 1.2 |
| TNF-α (10 ng/mL) | 3.6 ± 0.4 | 77.0 ± 2.2 | 7.5 ± 1.7 | 10.7 ± 2.0 |
| TNF-α + MTX | 5.8 ± 0.8b | 65.7 ± 4.8 | 13.8 ± 3.2 | 13.1 ± 2.6 |
| M-CSF (30 ng/mL) | 1.5 ± 0.4 | 74.2 ± 2.6 | 8.4 ± 1.9 | 13.9 ± 2.2 |
| - M-CSF | 15.4 ± 4.1a | 74.7 ± 5.1 | 2.8 ± 0.7a | 5.5 ± 0.9a |
| M-CSF + TNF-α | 1.5 ± 0.2 | 71.8 ± 1.9 | 9.3 ± 1.3 | 15.3 ± 2.7 |
| - M-CSF + TNF-α | 4.4 ± 2.3b | 82.3 ± 3.7b | 1.9 ± 0.4 | 9.7 ± 2.7 |
* Results are mean ± SEM of at least three independent experiments in each case. a P < 0.05 compared to controls. b P < 0.05 compared to MTX alone or no M-CSF.
BMDM, bone marrow-derived macrophages; M-CSF, macrophage colony stimulating factor; MTX, methotrexate; TNF-α, tumor necrosis factor-alpha.
Caspase-3 activation was evident at concentrations as low as 0.1 μM MTX in BMDM, with activity increasing dose-dependently up to 10 μM (Figure 1C). MTX, therefore, induces macrophage apoptosis at levels usually attained in treated human RA patients [24]. This was replicated in the murine RAW264.7 macrophage cell line (Figure 1E). RAW264.7 macrophages do not require exogenous M-CSF to proliferate, having gained growth signaling through introduction of the Abelson leukaemia virus [25]. They have been previously found to model primary macrophage apoptosis response [26]. A concentration of 10 μM MTX was used in subsequent experiments.
TNF-α could also maintain the viability of macrophages exposed to 10 μM MTX. TNF-α, when introduced three hours before MTX, completely prevented the loss of cell numbers in primary BMDM cultures exposed to MTX (Figure 1A). TNF-α also almost completely inhibited MTX-induced caspase-3 activation (Figure 1B) and annexin-V binding (Figure 1B) and significantly suppressed the sub-G0 (apoptotic) fraction on flow cytometric cell cycle analysis of MTX-exposed BMDM (Table 1). TNF-α did not reverse the MTX-induced increase in cells in S-phase. TNF-α, therefore, protected from MTX-induced cell loss by countering apoptosis, not through any action on cell proliferation. TNF-α also reduced the sub-G0 population of M-CSF-deprived BMDM, without restoring S phase progression (Table 1). It is notable that TNF-α was protective even in the presence of M-CSF concentrations that are optimal for apoptosis prevention, suggesting distinct pathways for apoptosis protection. Further increases in M-CSF concentration in culture, up to 120 ng/mL, failed to substitute for the anti-apoptotic action of TNF-α (results not shown). This suggested an action that was independent of M-CSF intracellular signaling and removed the possibility that TNF-α was acting through autocrine induction of M-CSF secretion. At least three hours of pre-exposure to TNF-α was required for optimal protection from MTX-induced apoptosis (results not shown).
TNF-α provided comparable protection from MTX-induced apoptosis in the RAW264.7 macrophage cell line. TNF-α exposure significantly (P < 0.05) suppressed spontaneous apoptosis of RAW264.7 cells as measured by caspase-3 activity and annexin-V binding (Figure 1D). RAW264.7 cells shared the susceptibility of primary macrophages to MTX, demonstrating enhanced caspase-3 activity and annexin-V binding after six hours of exposure (Figure 1D). This increase was markedly suppressed when TNF-α was added three hours before MTX (Figure 1D). Thus, the anti-apoptotic effect of TNF-α is replicated in RAW264.7 cells, allowing use of this cell line for studying the phenomenon.
TNF-α protection from apoptosis is mediated through TNF-R1 (p55), not TNF-R2 (p75) receptor activation
Monocytes/macrophages express both TNF-R1 and TNF-R2 [8]. To determine the receptor subtype(s) that mediated the survival effect of TNF-α, we exploited the species specificity of TNF-α action. Murine TNF-α (mTNF-α) activates both TNF-R1 and TNF-R2 on murine macrophages, while human TNF-α (hTNF-α) activates TNF-R1 only, thereby providing a means to distinguish effects that are mediated through different receptors [27]. mTNF-α and hTNF-α provided comparable protection from MTX-induced caspase-3 activation (Figure 2, P > 0.05), indicating that protection is mediated through TNF-R1, and does not require TNF-R2 signaling.
Figure 2.

Human (h) and murine (m) TNF-a confer comparable protection against apoptosis in RAW264.7 cells exposed to 10 mM MTX. Caspase-3 activity was measured after six hours with MTX, without TNF-a, or pre-treated with 10 ng/mL mTNF-a or hTNF-a for three hours. Shown are mean ± SEM of three independent experiments. * = P < 0.05.
NF-κB is required for the survival effect of TNF-α in macrophages
TNF-R1 ligation activates several pro-survival pathways via its association with TRAF2 (TNFR-Associated Factor 2), RIP (Receptor-Interacting Protein) c-Src and Jak2. These include the NF-κB pathway, Phosphatidylinositol-3-Kinase/Protein Kinase B (PI3K/AKT) and Mitogen-Activated Protein Kinase pathways [28,29]. Constitutive NF-κB activity is critical to macrophage survival [26]. Therefore, we next examined the kinetics of NF-κB activation in TNF-α-stimulated macrophages and investigated whether NF-κB activity was required for the anti-apoptotic effect of TNF-α.
Proteosomal degradation of the NF-κB inhibitory protein, IκBα, is an early indicator of activation in the canonical NF-κB pathway [30]. TNF-α (10 ng/mL) addition in RAW264.7 cells induced the degradation of IκBα within 15 minutes (Figure 3A, P < 0.05). This corresponded to the appearance of DNA-binding NF-κB in the nucleus, with activity peaking at 30 minutes before returning to baseline levels by one hour (Figure 3B). Supershift analyses with specific antibodies to RelA, NF-κB1, c-Rel and Rel-B identified RelA (p65) and NF-κB1 (p50) as the main components of the NF-κB complex induced by TNF-α (Figure 3B). A light c-Rel supershifted band was also detected (Figure 3B). IκBα levels recovered by one hour (Figure 3A). The IKBA gene is also NF-κB-responsive, so early recovery of IκBα levels indicates functional NF-κB signaling [30]. Lastly, TNF-α stimulation of RAW264.7 cells transiently expressing a NF-κB-responsive reporter construct showed that NF-κB transcriptional activity was enhanced in a time-dependent manner (Figure 3C). These results confirm that TNF-α is a rapid activator of NF-κB function in macrophages.
Figure 3.
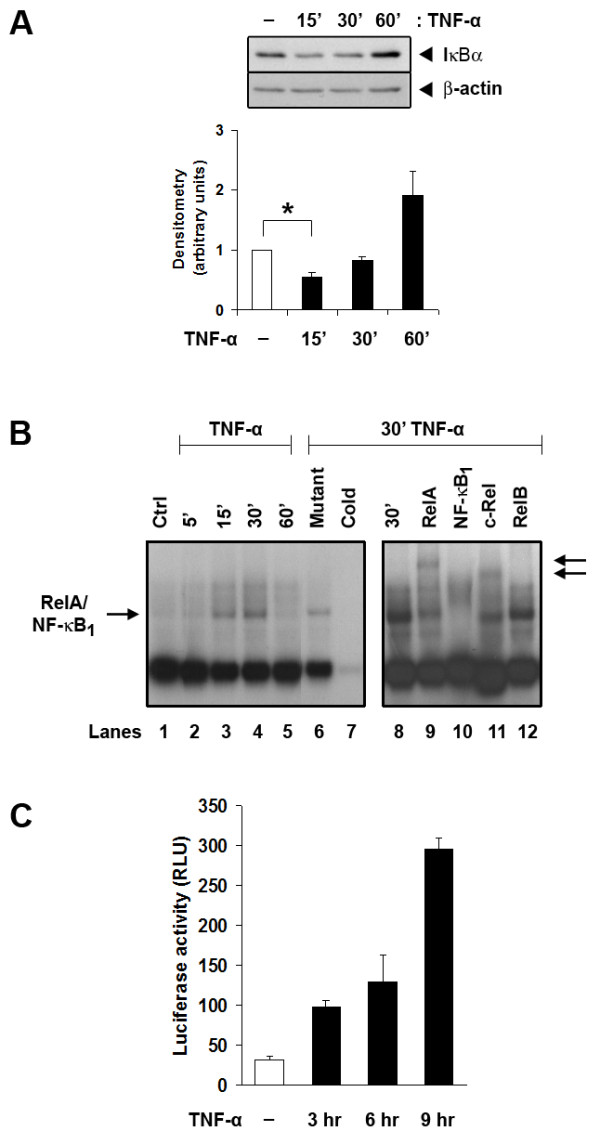
TNF-a activates NF-kB in RAW264.7 cells. A. TNF-a promotes degradation of IkBa and resynthesis over 60 minutes. IκBα and β-actin protein levels were assessed by Western blot analysis and quantified by densitometry (lower panel). Densitometry data show IκBα levels that were corrected by β-actin and represent normalised data from four independent experiments. * = P < 0.05. B. EMSA for NF-kB proteins in nuclear extracts of RAW264.7 cells over six hours of TNF-a exposure. The arrow on the left indicates the position of the RelA/NF-kB1 heterodimer. Competition for binding by an unlabeled specific oligonucleotide (lane 7) but not by an irrelevant sequence at 100-fold molar excess (lane 6) confirmed specificity of DNA binding. Supershift analyses with specific antibodies to RelA, NF-kB1 (p50), c-Rel and RelB (lanes 9, 10, 11 and 12, respectively, of the right panel) confirmed the identities of RelA and NF-kB1 in the complex with a lighter supershift c-Rel band. Arrows on the right indicate the locations of supershifted bands. The figure is representative of two independent experiments. C. TNF-a (10 ng/ml) induces NF-kB activity, as estimated by luciferase activity in RAW264.7 cells that had been transiently transfected with the pNF-kB-TA-Luc reporter. Results are representative of three independent experiments.
To investigate whether NF-κB activity is essential for TNF-α activity against MTX-induced apoptosis, the sesquiterpene lactone parthenolide (PAR) and BAY11-7085 (BAY) were used to specifically prevent NF-κB signaling [19,31]. PAR and BAY pre-treatment for 30 minutes abolished both constitutive and TNF-α-stimulated NF-κB transcriptional activity (Figure 4A). They also completely prevented TNF-α from rescuing RAW264.7 cells (Figure 4B,C) and primary macrophages (Figure 4D-F) from MTX-induced apoptosis and caspase-3 activation. These findings indicate that NF-κB activation is required for TNF-α protection against apoptosis induced by MTX. Notably, TNF-α became pro-apoptotic, rather than anti-apoptotic when NF-κB signaling was blocked, when estimated by either annexin-V binding (P < 0.05) or caspase-3 activity (P < 0.05).
Figure 4.

TNF-a must activate NF-kB signaling to protect macrophages from MTX-induced apoptosis. A. PAR and BAY (10 μM) prevent TNF-a-induced NF-kB activity in RAW264.7 cells, as measured by activity of the pNF-kB-TA-Luc reporter. Inhibitors were introduced 30 minutes before TNF-a and luciferase activity was measured nine hours later. Results are mean ± SEM of at least four independent experiments. B-F. PAR and BAY (10 mM or 7.5 mM) prevent TNF-a from rescuing MTX-treated RAW264.7 cells (B and C) and BMDM (D-F) from apoptosis. Cells were exposed to PAR (B, D, and E) or BAY (C and F) for 30 minutes before TNF-a stimulation for three hours, after which MTX was introduced. Annexin-V binding and/or caspase-3 activity was measured 6 hours later in RAW264.7 cells and 24 hours later in BMDM. Results are mean ± SEM of at least three independent experiments in each case. Normalised data are shown for BAY experiments. * = P < 0.05; ** = P < 0.01.
The involvement of PI3K/AKT, ERK, JNK and p38 MAP kinases was also explored using their specific inhibitors, but they were found to be dispensable for TNF-α-induced survival of macrophages, as assessed by annexin-V staining and caspase-3 activity (results not shown).
Autocrine TNF-α signaling is not required for basal survival of M-CSF-maintained macrophages
Macrophages are also sources of TNF-α, raising the possibility that constitutive NF-κB activity (and thus survival) depended on autocrine TNF-α stimulation. We, therefore, treated BMDM cultures with etanercept, a chimeric protein comprising Fc domains of human IgG1 and TNF-α-binding domains of the Type 2 TNF receptor, which neutralises both murine and human TNF-α [32]. An irrelevant human myeloma-derived IgG1 served as a control. Etanercept, however, did not increase caspase-3 activation, indicating that autocrine TNF-α stimulation was not important for survival of macrophages cultured with optimal concentrations of M-CSF (results not shown).
RANKL, a weak activator of NF-κB in macrophages, does not protect from MTX-induced apoptosis
The observation that TNF-α protected macrophages from MTX-induced apoptosis led us to question whether other cytokines present in rheumatoid synovium or erosions may also protect through NF-κB induction. Receptor Activator of NF-κB Ligand (RANKL) circulates at elevated concentration in RA and is demonstrable in rheumatoid synovium and erosions [33]. It is required for osteoclast formation from monocyte/macrophage precursors. Unlike TNF-α, however, RANKL did not suppress caspase-3 activation in MTX-exposed primary macrophage cultures at concentrations up to 400 ng/mL (Figure 5A). A dose of 200 ng/mL of RANKL is sufficient to elicit classical osteoclastogenesis in our hands [34], so was used for other experiments. Comparing effects on NF-κB activation, we found that RANKL (six hours) stimulated an approximately three-fold increase in NF-κB-luciferase reporter expression in transfected BMDM, well short of the approximately 22-fold increase with TNF-α (Figure 5B). Western blot analyses also indicated weaker phosphorylation and degradation of IκBα after RANKL treatment, compared to TNF-α (Figure 5C). Therefore, RANKL does not protect differentiated primary macrophages from MTX, possibly due to insufficient activation of NF-κB.
Figure 5.
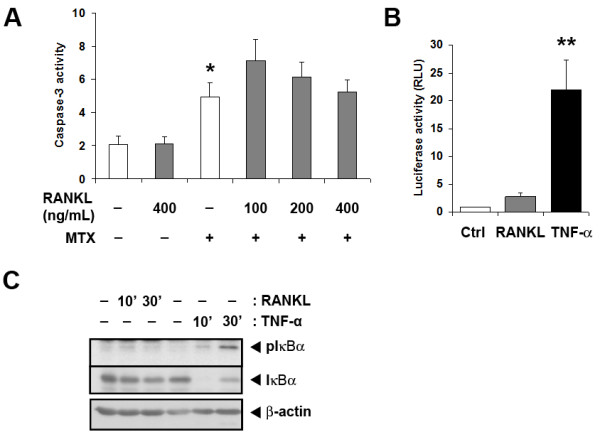
RANKL, a weak NF-κB activator, does not protect primary BMDM from MTX-induced apoptosis. A. RANKL (100 to 400 ng/mL) was introduced to BMDM 6 hours before MTX (10 mM) treatment. Caspase-3 activity was measured 24 hours later. Results are mean ± SEM from at least six independent experiments. B. and C. Compared to RANKL (200 ng/mL), TNF-a (10 ng/mL) treatment causes greater activation of NF-kB in BMDM. Cells transiently expressing the pNF-kB-TA-Luc reporter were stimulated with RANKL or TNF-a for six hours (B). Shown are mean ± SEM of normalised data from three independent experiments. C. Cells were M-CSF-starved overnight before stimulation with RANKL or TNF-a for 10 minutes and 30 minutes. Whole-cell lysates were subjected to western blot analysis for phosphorylated IkBa, total IkBa and b-actin. Results are representative of three independent experiments. * = P < 0.05; ** = P < 0.01.
MTX itself does not induce apoptosis through NF-κB suppression
The observation that NF-κB induction countered the apoptotic action of MTX led us to test whether MTX itself acted through inhibiting NF-κB. This has been previously observed in Jurkat T-cells and in the poorly differentiated myelo-monocytic U937 cell line [35]. However, MTX, at concentrations sufficient to induce apoptosis, failed to suppress either basal or TNF-α-induced NF-κB activity in RAW264.7 cells transiently transfected with a specific NF-κB reporter construct (pNF-κB-TA-Luc) (Figure 6A). In BMDM, it also failed to prevent the TNF-α-stimulated phosphorylation of IκBα, an essential step in IκBα degradation and RelA/NF-κB1 complex release (Figure 6B). MTX, therefore, does not exert its apoptotic actions through NF-κB suppression.
Figure 6.
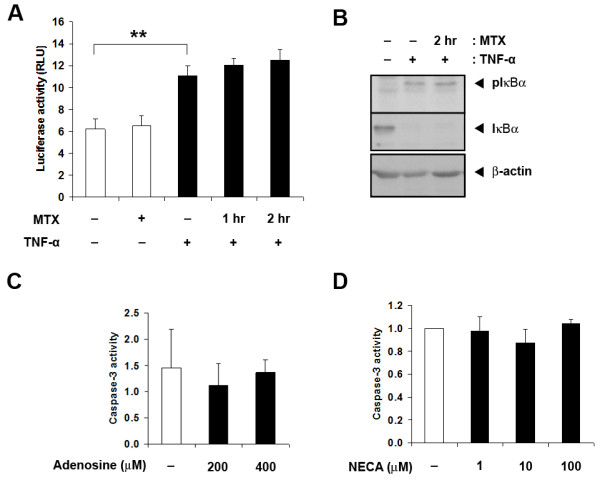
MTX does not inhibit TNF-a-induced NF-kB activity. A. Reporter-expressing RAW264.7 cells were exposed to MTX for one hour or two hour before TNF-a stimulation for a further three hour. Shown are mean ± SEM of four independent experiments. B. BMDM were pre-treated with MTX for two hour before TNF-a stimulation for 10 minutes. Whole-cell lysates were probed with antibodies for phosphorylated IkBa, total IkBa and b-actin. C. Adenosine and D. NECA do not increase basal macrophage apoptosis. BMDM were exposed to increasing concentrations of adenosine or NECA for 24 hour before caspase-3 measurements. Shown are mean ± SEM of results from three independent experiments in each case. ** = P < 0.01.
Anti-inflammatory actions of MTX on macrophages have been attributed to enhanced extracellular adenosine generation. However, neither adenosine itself (Figure 6C), or the pan-adenosine receptor agonist NECA (Figure 6D), affected caspase-3 activity. Thus, adenosine accumulation alone is not a sufficient explanation for MTX-induced macrophage apoptosis.
Discussion
TNF-α protects macrophages from apoptosis induced by MTX. This offers an explanation for the clinical observation that anti-TNF-α treatments cause apoptosis of monocyte/macrophage lineage cells in peripheral blood and synovium of patients with RA [4], in a study where most participants were also taking MTX. The survival signal from TNF-α is transduced through TNF-R1 and the canonical NF-κB pathway. M-CSF, which directs macrophage differentiation, growth and survival, but does not activate NF-κB signaling, cannot substitute for TNF-α. This is notable, because M-CSF is present in peripheral blood and synovial fluid in RA [22], and might have been expected to render anti-apoptotic effects of TNF-α redundant. M-CSF signals for growth and survival primarily through the PI3K/AKT pathway.
TNF-α can activate both pro-apoptotic and anti-apoptotic pathways, with outcomes that differ between cell types and conditions. The importance of NF-κB signaling in survival is highlighted by the outcomes of PAR and BAY exposure, which converted TNF-α from a dominantly pro-survival signal to an apoptotic signal. NF-κB-regulated proteins that have been linked to inhibition of apoptotic signaling include TRAF-1, TRAF-2, cIAP-1, cIAP-2, XIAP, FLIP, A20, GADD45β, the antioxidant Mn-SOD, and anti-apoptotic members of the Bcl-2 family, such as A1 and Bcl-xL [36].
The canonical NF-κB pathway is activated in macrophages of rheumatoid synovium [12], consistent with a role in maintaining cell survival in untreated patients [10,11]. Other products of the inflamed synovium, including IL-1, RANKL, GM-CSF, VEGF, PDGF and leukotrienes [37] can promote NF-κB signaling in responsive cells and may contribute, with TNF-α, to NF-κB activation in macrophages of rheumatoid synovium. However, the experiments with RANKL indicated that not all NF-κB activators protect from apoptosis. RANKL failed to protect primary murine macrophages from MTX-induced apoptosis, even at concentrations that promote osteoclastogenesis in our hands [34] and which exceed the concentrations reported in rheumatoid synovial fluid [38]. The difference may arise because RANKL activates NF-κB less intensely, or because of differences in other pathways recruited by TNF-R1 and RANK. Finding that RANKL is not a survival factor for macrophages mirrors findings with osteoclast precursors, where RANKL promotes activation and differentiation, but not survival [39]. The mature osteoclast, however, is protected from apoptosis by RANKL with mixed reports of NF-κB involvement [40]. The finding that RANKL-induced NF-κB activation is insufficient to protect primary macrophages from apoptosis is also in accord with the conclusion from clinical studies, that RANKL is not an important driver of synovial inflammation in RA, even though it is critical for erosion formation [41].
MTX has been reported to suppress NF-κB activity in a different cell context [35]. However, NF-κB suppression could not be demonstrated in MTX-exposed macrophages, so cannot be the mechanism of MTX-induced macrophage apoptosis. MTX anti-inflammatory response has also been linked with the extracellular accumulation of adenosine. Adenosine is reported to promote apoptosis of colon cancer cells in vitro [42], but we could find no evidence that adenosine alone causes apoptosis in macrophages. The experiments could not disprove a permissive role for adenosine. The experiments do, however, prove that MTX and TNF-α do not just act conversely on NF-κB signaling. MTX has many potentially apoptotic actions [3,13,15] and NF-κB has many anti-apoptotic actions [36]. It, therefore, seems more likely that the fate of TNF-α and MTX-exposed macrophages is decided by the late effectors of apoptosis signaling, particularly at mitochondrial steps, that integrate and balance multiple converging inputs [43]. This requires better definition.
Macrophage NF-κB signaling is an attractive therapeutic target in RA because of roles in inflammation, differentiation and survival. However, NF-κB pathways are employed in diverse tissues, with broad biological consequences. Anti-TNF-α treatments suppress NF-κB activity in circumstances where it is promoted by TNF-α, such as in the synovial macrophage. Currently available NF-κB-suppressing small molecules, however, lack such cellular specificity. Glucocorticoids suppress macrophage NF-κB activity, but with poor specificity [21]. The proteosomal inhibitor, bortezomib, suppresses NF-κB signaling by preventing IκBα degradation and is reported to reduce synovial macrophage inflammatory activity in experimental murine collagen arthritis [44] but, again, is not specifically targeted to the macrophage. Thalidomide likewise suppresses clinical activity of systemic onset juvenile RA [45], and leads to monocyte apoptosis in vitro through actions on AKT-1 [46] and NF-κB signaling [47], but with the attendant threat of teratogenesis and other serious off-target effects. Developing macrophage-specific NF-κB suppressants may also have to contend with internal redundancy, with RelA and cRel able to serve similar functions for apoptosis suppression in haematopoietic cells [48].
Conclusions
We were able to confirm that the potent NF-κB activator, TNF-α, counters MTX-induced apoptosis in primary murine macrophages. A less potent activator, RANKL, could not. Neither could the primary macrophage differentiation and survival factor, M-CSF, which we found did not activate NF-κB signaling. However, MTX did not suppress either constitutive or induced NF-κB signaling, ruling out a direct apoptotic action through NF-κB inhibition. These findings provide an additional explanation for the clinical complementarity between MTX and anti-TNF-α treatments for rheumatoid arthritis.
Abbreviations
7-AAD: 7-aminoactinomycin D; AICAR: 5-aminoimidazole-4-carboxamide ribonucleotide; BAY: BAY11-7085; BMDM: bone marrow-derived macrophages; DISC: death-inducing signaling complex; DMEM: Dulbecco's Modified Eagle Medium; ERK: extracellular signal-regulated kinase; FCS: foetal calf serum; FLIP FLICE: inhibitory protein; GADD45β: growth arrest and DNA damage inducible protein; GM-CSF: Granulocyte macrophage-colony stimulating factor; IκB: inhibitor of NF-κB; IAP: inhibitors of apoptosis; IL-1, interleukin-1; JNK: jun N-terminal kinase; MAPK: mitogens-activated protein kinase; M-CSF: macrophage colony stimulating factor; Mn-SOD: manganese superoxide dismutase; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MTX: methotrexate; NF-κB: nuclear factor-kappa-light-chain-enhancer of activated B cells; PAR: parthenolide; PDGF: platelet-derived growth factor; PI3K/AKT: phosphatidylinositol-3-kinase/protein kinase B; RANKL: receptor activator of NF-κB; RIP: receptor-interacting protein; TNF-α: tumor necrosis factor-alpha; TRAF TNF: receptor associated factor; VEGF: vascular endothelial growth factor; XIAP: X-linked inhibitor of apoptosis protein.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SL performed the experiments and co-wrote the manuscript. DJ conceived of the study and co-wrote the manuscript. All authors were involved in the design of the study and interpretation of results.
Contributor Information
Susan ZY Lo, Email: szylo40@gmail.com.
James H Steer, Email: james.steer@uwa.edu.au.
David A Joyce, Email: david.joyce@uwa.edu.au.
Acknowledgements
The authors acknowledge the facilities, scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation and Analysis, The University of Western Australia, a facility funded by The University, State and Commonwealth Governments. We thank Dr. Kathy Hill-Miller for technical support with flow cytometry.
Supported by National Health and Medical Research Council (Australia) Project Grant No. 572638 and an Australian Commonwealth Postgraduate Scholarship awarded to Susan ZY Lo.
References
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Bitsch A, Stadelmann C, Siebert H, Bruck W. Macrophages are eliminated from the injured peripheral nerve via local apoptosis and circulation to regional lymph nodes and the spleen. J Neurosci. 2001;21:3401–3408. doi: 10.1523/JNEUROSCI.21-10-03401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, Weedon H, Papangelis V, Walker J, Roberts-Thomson PJ, Ahern MJ. Apoptosis in the rheumatoid arthritis synovial membrane: modulation by disease-modifying anti-rheumatic drug treatment. Rheumatology (Oxford) 2010;49:862–875. doi: 10.1093/rheumatology/kep467. [DOI] [PubMed] [Google Scholar]
- Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y, Klareskog L, Ulfgren AK. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. 2005;52:61–72. doi: 10.1002/art.20764. [DOI] [PubMed] [Google Scholar]
- Smeets TJ, Kraan MC, van Loon ME, Tak PP. Tumor necrosis factor alpha blockade reduces the synovial cell infiltrate early after initiation of treatment, but apparently not by induction of apoptosis in synovial tissue. Arthritis Rheum. 2003;48:2155–2162. doi: 10.1002/art.11098. [DOI] [PubMed] [Google Scholar]
- Lugering A, Schmidt M, Lugering N, Pauels HG, Domschke W, Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn's disease by using a caspase-dependent pathway. Gastroenterology. 2001;121:1145–1157. doi: 10.1053/gast.2001.28702. [DOI] [PubMed] [Google Scholar]
- Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, Feldmann M, Maini RN. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Gehr G, Gentz R, Brockhaus M, Loetscher H, Lesslauer W. Both tumor necrosis factor receptor types mediate proliferative signals in human mononuclear cell activation. J Immunol. 1992;149:911–917. [PubMed] [Google Scholar]
- Yuan J. Transducing signals of life and death. Curr Opin Cell Biol. 1997;9:247–251. doi: 10.1016/S0955-0674(97)80069-5. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Pennington KN, Taylor JA, Bren GD, Paya CV. IkappaB kinase-dependent chronic activation of NF-kappaB is necessary for p21(WAF1/Cip1) inhibition of differentiation-induced apoptosis of monocytes. Mol Cell Biol. 2001;21:1930–1941. doi: 10.1128/MCB.21.6.1930-1941.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel ML, McMorrow LB, Gravallese EM. Nuclear factor-kappa B in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum. 1995;38:1762–1770. doi: 10.1002/art.1780381209. [DOI] [PubMed] [Google Scholar]
- Herman S, Zurgil N, Deutsch M. Low dose methotrexate induces apoptosis with reactive oxygen species involvement in T lymphocytic cell lines to a greater extent than in monocytic lines. Inflamm Res. 2005;54:273–280. doi: 10.1007/s00011-005-1355-8. [DOI] [PubMed] [Google Scholar]
- Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, Teoh LA, Fischkoff SA, Chartash EK. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48:35–45. doi: 10.1002/art.10697. [DOI] [PubMed] [Google Scholar]
- Wessels JA, Huizinga TW, Guchelaar HJ. Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 2008;47:249–255. doi: 10.1093/rheumatology/kem279. [DOI] [PubMed] [Google Scholar]
- Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–39. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Curt GA, Fine RL, Griffin JD, Chabner BA. Formation of methotrexate polyglutamates in purified myeloid precursor cells from normal human bone marrow. J Clin Invest. 1985;75:1008–1014. doi: 10.1172/JCI111761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Tan JW, Huang L, Gao XH, Laird R, Liu D, Wysocki S, Zheng MH. Cloning, sequencing, and functional characterization of the rat homologue of receptor activator of NF-kappaB ligand. J Bone Miner Res. 2000;15:2178–2186. doi: 10.1359/jbmr.2000.15.11.2178. [DOI] [PubMed] [Google Scholar]
- Yip KH, Zheng MH, Feng HT, Steer JH, Joyce DA, Xu J. Sesquiterpene lactone parthenolide blocks lipopolysaccharide-induced osteolysis through the suppression of NF-kappaB activity. J Bone Miner Res. 2004;19:1905–1916. doi: 10.1359/JBMR.040919. [DOI] [PubMed] [Google Scholar]
- Yip KH, Zheng MH, Steer JH, Giardina TM, Han R, Lo SZ, Bakker AJ, Cassady AI, Joyce DA, Xu J. Thapsigargin modulates osteoclastogenesis through the regulation of RANKL-induced signaling pathways and reactive oxygen species production. J Bone Miner Res. 2005;20:1462–1471. doi: 10.1359/JBMR.050324. [DOI] [PubMed] [Google Scholar]
- Steer JH, Kroeger KM, Abraham LJ, Joyce DA. Glucocorticoids suppress tumor necrosis factor-alpha expression by human monocytic THP-1 cells by suppressing transactivation through adjacent NF-kappa B and c-Jun-activating transcription factor-2 binding sites in the promoter. J Biol Chem. 2000;275:18432–18440. doi: 10.1074/jbc.M906304199. [DOI] [PubMed] [Google Scholar]
- Rioja I, Hughes FJ, Sharp CH, Warnock LC, Montgomery DS, Akil M, Wilson AG, Binks MH, Dickson MC. Potential novel biomarkers of disease activity in rheumatoid arthritis patients: CXCL13, CCL23, transforming growth factor alpha, tumor necrosis factor receptor superfamily member 9, and macrophage colony-stimulating factor. Arthritis Rheum. 2008;58:2257–2267. doi: 10.1002/art.23667. [DOI] [PubMed] [Google Scholar]
- Bruce-Gregorios JH, Soucy D, Chen MG, Benson N. Effect of methotrexate on cell cycle and DNA synthesis of astrocytes in primary culture: flow cytometric studies. J Neuropathol Exp Neurol. 1991;50:63–72. doi: 10.1097/00005072-199101000-00005. [DOI] [PubMed] [Google Scholar]
- Hamilton RA, Kremer JM. Why intramuscular methotrexate may be more efficacious than oral dosing in patients with rheumatoid arthritis. Br J Rheumatol. 1997;36:86–90. doi: 10.1093/rheumatology/36.1.86. [DOI] [PubMed] [Google Scholar]
- Raschke WC, Baird S, Ralph P, Nakoinz I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 1978;15:261–267. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- Pagliari LJ, Perlman H, Liu H, Pope RM. Macrophages Require Constitutive NF-kappa B Activation To Maintain A1 Expression and Mitochondrial Homeostasis. Mol Cell Biol. 2000;20:8855–8865. doi: 10.1128/MCB.20.23.8855-8865.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiers W. Tumor necrosis factor. Characterization at the molecular, cellular and in vivo level. FEBS Lett. 1991;285:199–212. doi: 10.1016/0014-5793(91)80803-B. [DOI] [PubMed] [Google Scholar]
- Natoli G, Costanzo A, Guido F, Moretti F, Bernardo A, Burgio VL, Agresti C, Levrero M. Nuclear factor kB-independent cytoprotective pathways originating at tumor necrosis factor receptor-associated factor 2. J Biol Chem. 1998;273:31262–31272. doi: 10.1074/jbc.273.47.31262. [DOI] [PubMed] [Google Scholar]
- Pincheira R, Castro AF, Ozes ON, Idumalla PS, Donner DB. Type 1 TNF receptor forms a complex with and uses Jak2 and c-Src to selectively engage signaling pathways that regulate transcription factor activity. J Immunol. 2008;181:1288–1298. doi: 10.4049/jimmunol.181.2.1288. [DOI] [PubMed] [Google Scholar]
- Place RF, Haspeslagh D, Giardina C. Induced stabilization of IkappaBalpha can facilitate its re-synthesis and prevent sequential degradation. J Cell Physiol. 2003;195:470–478. doi: 10.1002/jcp.10262. [DOI] [PubMed] [Google Scholar]
- Nasu K, Nishida M, Ueda T, Yuge A, Takai N, Narahara H. Application of the nuclear factor-kappaB inhibitor BAY 11-7085 for the treatment of endometriosis: an in vitro study. Am J Physiol Endocrinol Metab. 2007;293:E16–23. doi: 10.1152/ajpendo.00135.2006. [DOI] [PubMed] [Google Scholar]
- Egberts JH, Cloosters V, Noack A, Schniewind B, Thon L, Klose S, Kettler B, von Forstner C, Kneitz C, Tepel J, Adam D, Wajant H, Kalthoff H, Trauzold A. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008;68:1443–1450. doi: 10.1158/0008-5472.CAN-07-5704. [DOI] [PubMed] [Google Scholar]
- Pettit AR, Walsh NC, Manning C, Goldring SR, Gravallese EM. RANKL protein is expressed at the pannus-bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatology (Oxford) 2006;45:1068–1076. doi: 10.1093/rheumatology/kel045. [DOI] [PubMed] [Google Scholar]
- Ang ES, Pavlos NJ, Chai LY, Qi M, Cheng TS, Steer JH, Joyce DA, Zheng MH, Xu J. Caffeic acid phenethyl ester, an active component of honeybee propolis attenuates osteoclastogenesis and bone resorption via the suppression of RANKL-induced NF-kappaB and NFAT activity. J Cell Physiol. 2009;221:642–649. doi: 10.1002/jcp.21898. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Aggarwal BB. Methotrexate suppresses NF-kappaB activation through inhibition of IkappaBalpha phosphorylation and degradation. J Immunol. 2001;167:2911–2920. doi: 10.4049/jimmunol.167.5.2911. [DOI] [PubMed] [Google Scholar]
- Nakshatri H, Goulet RJ Jr. NF-kappaB and breast cancer. Curr Probl Cancer. 2002;26:282–309. doi: 10.1067/mcn.2002.129977. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- Kotake S, Udagawa N, Hakoda M, Mogi M, Yano K, Tsuda E, Takahashi K, Furuya T, Ishiyama S, Kim KJ, Saito S, Nishikawa T, Takahashi N, Togari A, Tomatsu T, Suda T, Kamatani N. Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. 2001;44:1003–1012. doi: 10.1002/1529-0131(200105)44:5<1003::AID-ANR179>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Ikeda F, Matsubara T, Tsurukai T, Hata K, Nishimura R, Yoneda T. JNK/c-Jun signaling mediates an anti-apoptotic effect of RANKL in osteoclasts. J Bone Miner Res. 2008;23:907–914. doi: 10.1359/jbmr.080211. [DOI] [PubMed] [Google Scholar]
- Schett G, David JP. Denosumab--a novel strategy to prevent structural joint damage in patients with RA? Nat Clin Pract Rheumatol. 2008;4:634–635. doi: 10.1038/ncprheum0927. [DOI] [PubMed] [Google Scholar]
- Yasuda Y, Saito M, Yamamura T, Yaguchi T, Nishizaki T. Extracellular adenosine induces apoptosis in Caco-2 human colonic cancer cells by activating caspase-9/-3 via A(2a) adenosine receptors. J Gastroenterol. 2009;44:56–65. doi: 10.1007/s00535-008-2273-7. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Lee SW, Kim JH, Park YB, Lee SK. Bortezomib attenuates murine collagen-induced arthritis. Ann Rheum Dis. 2009;68:1761–1767. doi: 10.1136/ard.2008.097709. [DOI] [PubMed] [Google Scholar]
- Lehman TJ, Schechter SJ, Sundel RP, Oliveira SK, Huttenlocher A, Onel KB. Thalidomide for severe systemic onset juvenile rheumatoid arthritis: A multicenter study. J Pediatr. 2004;145:856–857. doi: 10.1016/j.jpeds.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Gockel HR, Lugering A, Heidemann J, Schmidt M, Domschke W, Kucharzik T, Lugering N. Thalidomide induces apoptosis in human monocytes by using a cytochrome c-dependent pathway. J Immunol. 2004;172:5103–5109. doi: 10.4049/jimmunol.172.8.5103. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Lamothe B, Aggarwal BB. Thalidomide suppresses NF-kappa B activation induced by TNF and H2O2, but not that activated by ceramide, lipopolysaccharides, or phorbol ester. J Immunol. 2002;168:2644–2651. doi: 10.4049/jimmunol.168.6.2644. [DOI] [PubMed] [Google Scholar]
- Grossmann M, Metcalf D, Merryfull J, Beg A, Baltimore D, Gerondakis S. The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc Natl Acad Sci USA. 1999;96:11848–11853. doi: 10.1073/pnas.96.21.11848. [DOI] [PMC free article] [PubMed] [Google Scholar]