

Abstract
Objective
Emerging evidence suggests that protease-activated receptors-1 and 2 (PAR1 and PAR2) can signal together in response to proteases found in the rapidly changing microenvironment of damaged blood vessels. However, it is unknown whether PAR1 and PAR2 promote or mitigate the hyperplastic response to arterial injury. Using cell-penetrating PAR1 pepducins and mice-deficient in PAR1 or PAR2, we set out to determine the respective contributions of the receptors to hyperplasia and phenotypic modulation of smooth muscle cells (SMCs) in response to arterial injury.
Methods and Results
SMCs were strongly activated by PAR1 stimulation as evidenced by increased mitogenesis, mitochondrial activity, and calcium mobilization. The effects of chronic PAR1 stimulation following vascular injury were studied by performing carotid artery ligations in mice treated with PAR1 agonist pepducin, P1pal-13. Histological analysis revealed that PAR1 stimulation caused striking hyperplasia which was ablated in PAR1-/-, and surprisingly in PAR2-/- mice. P1pal-13 treatment yielded an expression pattern consistent with a de-differentiated phenotype in carotid artery SMCs. Detection of PAR1-PAR2 complexes provided an explanation for the hyperplastic effects of the PAR1 agonist requiring the presence of both receptors.
Conclusions
We conclude that PAR2 regulates the PAR1 hyperplastic response to arterial injury leading to stenosis.
Keywords: carotid arteries, neointimal hyperplasia, PAR1, PAR2, smooth muscle cells, pepducins
Smooth muscle cell (SMC) de-differentiation, hyperplasia and neointimal thickening are common responses to iatrogenic injury following percutaneous coronary interventions and can require a secondary intervention to relieve patient discomfort and ischemic symptoms due to restenosis.1 Despite improvements in drug-eluting stent technology, a significant percentage of high risk patients or those with bifurcation lesions develop in-stent restenosis.2, 3 Preventing the formation of stenotic lesions without the emplacement of a permanent wire stent has proven to be challenging, and requires a better understanding of the complex molecular pathways associated with SMC de-differentiation and proliferation in response to vascular injury.
Accumulating evidence indicates that the protease-activated receptors, PAR1, PAR2 and PAR4 play an important role in the various cell types involved in the vascular repair process, including SMCs, endothelium, and inflammatory cells.4 PARs are G-protein coupled receptors activated by proteolytic cleavage of the extracellular domain and exposure of a tethered ligand that binds to the receptor to activate transmembrane signaling.5 Multiple proteases found in the microenvironment of an injured blood vessel wall including thrombin6, plasmin7, and matrix metalloprotease-1 (MMP-1)8, 9 can activate PAR1, whereas PAR2 can be activated by tryptase, matriptase (MT-SP1), FVIIa and FXa, and PAR4 by thrombin.10-12
Over-expression of PAR1 in response to vascular damage or in atherosclerotic lesions has been documented in endothelium and vascular SMCs.13-17 Likewise, PAR2 has been shown to be upregulated in medial and neointimal SMCs, in addition to proliferating adventitial myofibroblasts following damage to the blood vessel.18 Both PAR1 and PAR2 can also mediate endothelial cell proliferation, barrier function and cytokine expression, as well as affect vaso-reactivity and cardiomyocyte hypertrophy.19-23 The shared vascular expression pattern and tight genetic linkage24 of PAR1 and PAR2, in addition to their overlapping functions in promoting cellular migration and proliferation, suggests a potentially critical role for both receptors in vascular remodeling and repair.
Recent studies have suggested that PAR1 and PAR2 may exist in close proximity on the cell surface and can signal together during acute vascular inflammation.20 Although PAR2 is not directly activated by thrombin, the thrombin-generated PAR1 tethered ligand, SFLLRN, serves as a full agonist of PAR2.20, 25 Furthermore, thrombin-cleaved PAR1 has been shown to donate its tethered ligand and transactivate PAR2 in endothelial cells, fibroblasts and HEK cells.20, 26 Genetic and pharmacologic evidence for PAR1-PAR2 association was shown in animal models of sepsis through loss of the protective effects of a PAR1-specific agonist pepducin in PAR2-/- mice.20 These studies revealed that PAR1 and PAR2 can signal as an entwined pair, highlighting a complex, and potentially exploitable, feature of PAR1 and PAR2 complexes. However, it is still not clear whether PAR1 or PAR2 promote or mitigate the hyperplastic response to arterial injury, or affect the differentiation phenotype of vascular SMCs. Here, we sought to identify the roles of PAR1 and PAR2 in SMCs in the process of post-injury arterial repair and stenosis.
We report that chronic activation of PAR1 causes changes in the phenotypic markers of arterial SMCs that reflect a de-differentiated phenotype. Intracellular pepducin-based PAR1 agonists20, 27 stimulated proliferation of vascular SMCs leading to enhanced intimal and medial hyperplasia in a carotid artery injury model which required the presence of both PAR1 and PAR2 in mice. The two receptors were found to form a stable complex providing evidence that PAR1 and PAR2 function in partnership to modulate the smooth muscle cell response to arterial injury.
Experimental Procedures
An expanded Methods section is available in the Supplemental Material, available online at http://atvb.ahajournals.org.
Carotid Artery Ligation Injury
Six-month old C57BL/6 wild-type, PAR1-/-, or PAR2-/- female mice were anesthetized with isoflurane and the left carotid artery was dissected under aseptic conditions. The left carotid artery was then ligated with 6-0 silk sutures and the incision closed with nylon sutures. Buprenorphine analgesia was administered as needed. P1pal-13 (Pal-AVANRSKKSRALF-NH2, s.c. 2.5 mg/kg), P1pal-7 (Pal-KKSRALF-NH2, s.c. 10 mg/kg) pepducins or vehicle (20% DMSO) were administered on a daily basis. Mice were sacrificed at various time points after injury (2 h - 21 days), perfused with 10% formalin at 125 mm Hg. The common carotid arteries were harvested and arterial histopathology and morphometry was analyzed by a pathologist under blinded conditions as described in the Supplemental Material.
Coimmuno-precipitation of PAR1 and PAR2
These assays have been described previously; full details are given in the Supplemental Material.
Statistical Analyses
Statistical analyses were performed using Graphpad Prism (version 5.0a or Microsoft Excel, Version 11.6). Data are represented as mean ± se. For two group comparisons of parametric data, a two-tailed distribution unequal variance Student’s t test was performed. For multiple-group comparisons, 2-way ANOVA tests were performed followed by Bonferroni posttest analysis. Statistical significance was defined as * p < 0.05 or ** p < 0.005.
Results
Proliferative Responses of PAR Agonists in Vascular Smooth Muscle Cells
To assess the relative contributions of PAR1, PAR2 and PAR4 in proliferation of arterial SMCs we began by assessing PAR surface expression and signaling in primary smooth muscle cells derived from mouse aorta (MOVAS). PAR1, PAR2, and PAR4 were all expressed on the MOVAS cell surface, as determined by flow cytometry (Figure 1A). Robust calcium signals were obtained from the PAR1 agonist thrombin and the PAR1 tethered ligand peptide SFLLRN, which fully activates both PAR1 and PAR2 (Figure 1B). The PAR2-selective ligand, SLIGRL, gave a weaker calcium signal than the PAR1-selective agonist TFLLRN. The well-characterized cell-penetrating PAR1 i3-loop pepducin, P1pal-13, which activates PAR1, PAR1-PAR2 complexes, but not PAR2 alone,20, 27 was a highly potent stimulator of SMC calcium flux. A calcium response was not observed with PAR4-specific agonist, AYPGKF, despite its apparent expression in the mouse SMCs. Likewise, P1pal-13 did not stimulate aggregation of mouse platelets which express PAR4 (but not PAR1 or PAR2) and did not have an additional effect on aggregation induced by the PAR4 agonist AYPGKF (Supplemental Figure I). These data indicate that MOVAS generate stronger calcium signals through PAR1 as compared to PAR2, and do not respond to PAR4 agonist.
Figure 1. PAR1 agonists stimulate calcium mobilization and proliferation of smooth muscle cells (SMCs).
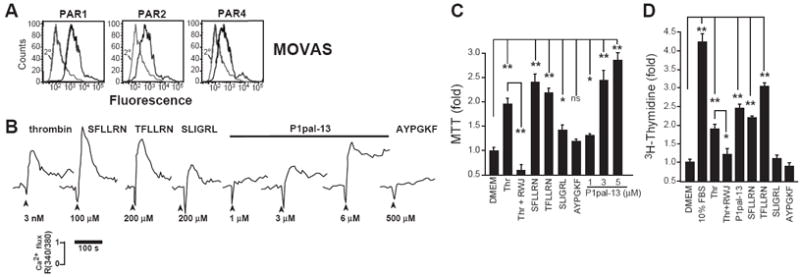
(A) Mouse vascular aorta SMCs (MOVAS) were analyzed for surface expression of PAR1, PAR2, and PAR4 by flow cytometry. (B) Profile of PAR agonist activity in mobilizing calcium in MOVAS, (C) in mitochondrial activity as assessed by MTT (0.3 nM thrombin, 3 μM RWJ-56110, 100 μM SFLLRN, TFLLRN, or SLIGRL, 200 μM AYPGKF, or P1pal-13 as indicated for 4 d), (D) and in mitogenesis assays as assessed by 3[H]-Thymidine (P1pal-13 was used at 3 μM, other agonists and inhibitors were used at the same concentrations in C, for 2 d).*, P <0.05 and **, P <0.005.
Next, we compared the ability of PAR1 versus PAR2 agonists to stimulate mitochondrial activity (MTT) and DNA synthesis (3H-thymidine incorporation) in MOVAS. Thrombin significantly increased mitochondrial activity and DNA synthesis in the SMCs by 2-fold, which was suppressed by the PAR1 small-molecule inhibitor, RWJ-56110 (Figure 1C-D). PAR1 agonists SFLLRN and TFLLRN gave significant increases in both mitochondrial activity and DNA synthesis. In contrast, PAR2 peptide agonist, SLIGRL, gave a slight increase in mitochondrial activity but had no effect on DNA synthesis (Figure 1C-D). PAR4 peptide agonist, AYPGKF, had no effect on mitochondrial activity or mitogenesis. The PAR1 P1pal-13 pepducin conferred a robust dose-dependent increase in mitochondrial activity and significantly increased mitogenesis. These data indicate that stimulation of PAR1 triggers mitogenesis in SMCs whereas activation of PAR2 alone does not.
PAR1 i3-loop Pepducin Agonist P1pal-13 Induces Medial and Intimal Hyperplasia in Injured Carotid Arteries in Wild-Type but not PAR1-/- or PAR2-/- mice
To examine in vivo functions of PAR1 and PAR2 in vascular remodeling and restenosis following injury, we performed carotid artery ligation injuries in C57BL/6 mice. To document the stages of injury and subsequent repair and vascular remodeling processes, we harvested carotid arteries from wild-type mice at multiple time-points throughout the 21-day post-injury period (Supplemental Figure II). Ligation of the common carotid artery resulted in vessel occlusion leading to acute edema and cytoplasmic swelling of the medial SMCs by the 2 h time point. The edematous phase persisted for 2 d and was mostly resolved by day 4. Acute inflammatory cell infiltration was observed in both intima and adventitia in the early stages following vascular injury, with the peak at day 7. This was followed by a medial and intimal proliferative stage characterized by SMC hyperplasia and peri-arterial inflammation. Ki67 staining, a marker for cellular proliferation, revealed pronounced adventitial proliferation at days 4-7, medial proliferation at days 7-14, and intimal proliferation at day 14 (Supplemental Figure II).
We then determined whether PAR1-/- or PAR2-/- mice had altered proliferative responses in the media following carotid artery ligation injury as compared to wild-type C57BL/6 mice. Surprisingly, neither PAR1- nor PAR2-deficiency had a significant effect on medial growth as compared to wild-type (WT) mice (Figure 2). As neither PAR1- nor PAR2-deficiency alone had a signficant effect on medial area, we tested whether the presence of PAR1 was compensating for the loss of PAR2 using the PAR1 pepducin antagonist P1pal-7. Treatment of injured WT mice with P1pal-7 for 21 days had no effect (Supplemental Figure III), consistent with the observed lack of an effect of PAR1-deficiency relative to WT mice on the media. As expected, the PAR1 inhibitor P1pal-7 also had no effect in the PAR1-deficient mice. Plpal-7 treatment of PAR2-deficient mice caused a slight increase in medial area in the PAR2-deficient mice, thus ruling out a compensation mechanism by PAR1 since medial proliferation was not inhibited by PAR1 blockade. Highly similar to the effects in the media, P1pal-7 had no effect on intimal area in injured wild-type, PAR1-/-, and a slight non-significant increase in the PAR2-/- intima (data not shown).
Figure 2. The PAR1 pepducin agonist, P1pal-13, causes a significant increase in medial hyperplasia that is lost in PAR1- and PAR2-deficient mice.
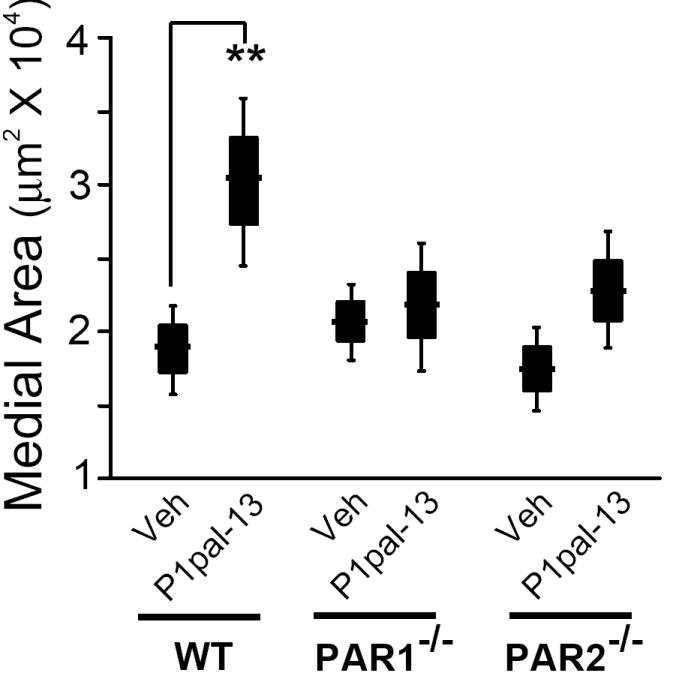
C57BL/6 wild-type (WT, n=12-17), PAR1-/- (n=7-10), and PAR2-/- (n=9-19) mice underwent ligation injury of the left common carotid artery and were treated daily for 21 d with P1pal-13 (2.5 mg/kg) or vehicle (20% DMSO). The mean of each treatment group is indicated as a horizontal line within a black box that depicts ± se and the error bars depict ± 2 se (to represent the 95% confidence interval) for each treatment cohort calculated from cross sections of the arteries as described in the Supplemental Material. **, P <0.005.
We then treated the injured mice for 21-days with P1pal-13, which activates both PAR1 and PAR1-PAR2 complexes. As shown in Figure 2, the P1pal-13 treated wild-type mice had a significant 60% increase in medial area compared to vehicle-treated animals, suggesting that strong PAR1 and/or PAR1-PAR2 stimulation causes a hyperproliferative response. P1pal-13 had no effect on medial area on the uninjured contralateral carotid arteries in any of the mice (data not shown). In contrast, the medial SMC proliferative response to the PAR1 agonist pepducin was completely lost in the PAR1-/- mice, demonstrating the requirement for its cognate receptor. Likewise, the medial hyperplasia in response to P1pal-13 was lost in the PAR2-/- mice. This indicates that the presence of PAR2 was also required for the proliferative response to the PAR1 pepducin agonist, and suggests that PAR2 may either directly or indirectly interact with PAR1 in the vascular SMCs.
We then quantified the effects of P1pal-13 on intimal morphology of the injured carotids in wild-type, PAR1- and PAR2-deficient mice. The PAR1 pepducin agonist P1pal-13 caused a striking 13-fold increase in mean neointimal area leading to nearly complete lumenal stenosis as compared to vehicle-treated wild-type mice (Figure 3A-B). In contrast, genetic deficiency of PAR1 or PAR2 did not attenuate the intimal growth relative to wild-type in the vehicle cohorts, and in fact caused a mean increase in intimal area following arterial injury (Figure 3A-B). This would indicate that without a strong PAR agonist present such as P1pal-13, PAR1 and PAR2 may provide negative feedback to suppress the post-injury hyperplastic response in this carotid artery injury model. As expected, the P1pal-13 PAR1 agonist had no effect on intimal hyperplasia in the PAR1-/- mice as compared to vehicle treatment (Figure 3A-B). Intimal hyperplasia in response to P1pal-13 was also completely lost in the PAR2-/- mice as compared to wild-type mice (Figure 3A-B), demonstrating that the hyperplastic response triggered by the PAR1 agonist requires the presence of both PAR1 and PAR2.
Figure 3. The PAR1 pepducin agonist causes significant neointimal hyperplasia in the injured carotid arteries of wild-type but not PAR1- or PAR2-deficient mice.
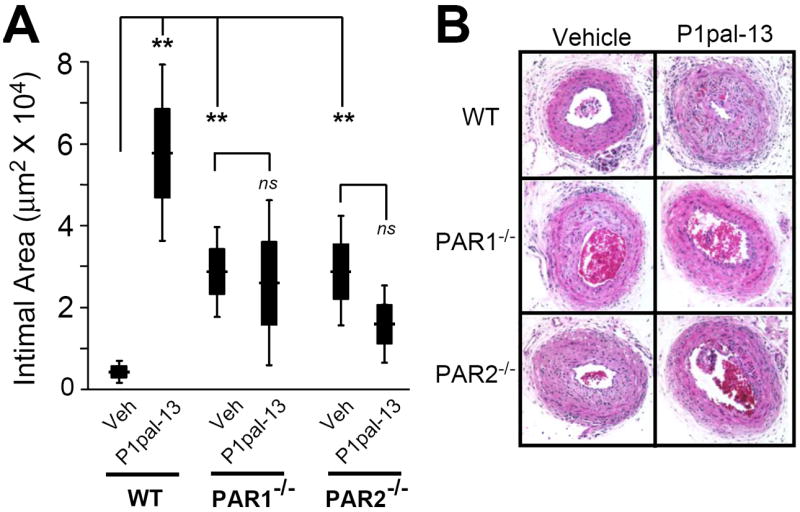
C57BL/6 wild-type (WT, n=12-17), PAR1-/- (n=7-10), and PAR2-/- (n=9-19) underwent ligation injury of the left common carotid artery and were treated as described in Figure 2. (A) The mean intimal areas (black boxes represent ± se and error bars represent ± 2se) for each treatment cohort were calculated from cross sections of the arteries as described in the Supplemental Material. **, P <0.005. (B) Representative H&E cross-sections of carotids from different treatment groups and genotypes.
To validate the requirement for both PAR1 and PAR2 to mediate the observed P1pal-13-dependent SMC proliferative response in the injured arteries, primary SMCs were isolated from carotid arteries of wild-type, PAR1-/- and PAR2-/- mice. Immunofluorescent staining of markers of differentiated SMCs including α-smooth muscle cell actin (α-SMA), and SM-22 calcium-binding cytoskeletal protein that regulates actin-myosin contraction confirmed that the isolated cells were SMCs (Figure 4A). The specific loss of PAR1 and PAR2 protein expression was also confirmed in the SMCs isolated from the carotids of the respective knockout mice (Figure 4A). PAR1 agonist pepducin stimulated ex vivo mitogenesis of the primary carotid arterial SMCs from wild-type mice (Figure 4B). However, SMCs from PAR1-/- and PAR2-/- mice had a complete loss of the proliferative response to P1pal-13 treatment, confirming that the presence of both PAR1 and PAR2 are necessary to support the mitogenic signal from the PAR1 agonist pepducin (Figure 4B).
Figure 4. Effect of genetic loss of PAR1 or PAR2 on proliferation.
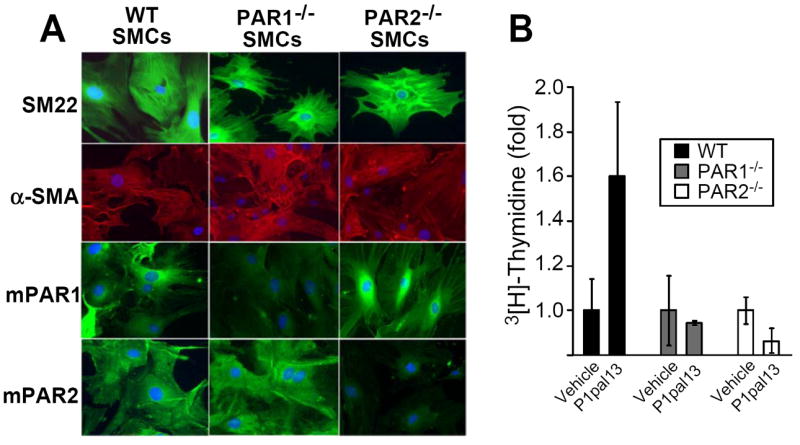
(A) Primary SMCs were isolated from the carotid arteries of wild-type (WT), PAR1-/- and PAR2-/- mice. SMCs were plated on culture slides and stained with DAPI (blue), or antibodies against SM-22 (top row, green/FITC), α-SMA (second row, red/TRITC), mouse PAR1 (third row, green/FITC), or mouse PAR2 (bottom row, green/FITC). (B) Primary SMCs were subjected to daily treatment of vehicle (0.2% DMSO) or P1pal-13 (3 μM) for 3 d and mitogenesis was measured by incorporation of 3[H]-Thymidine.
PAR2 Forms a Stable Complex with PAR1
Previous studies also showed that PAR1 and PAR2 can interact pharmacologically with each other on the surface of cells whereby the proteolytically-cleaved tethered ligand of PAR1 can access and activate PAR2 in the context of endothelial barrier protection and coagulation/fibrinolytic responses.20, 26, 29 Moreover, fluorescence resonance energy transfer (FRET) analysis20 showed that PAR1 and PAR2 reside in close molecular contact (≤100 Å) both in cytoplasmic vesicles and on the cell surface. To provide direct evidence that PAR1 and PAR2 can form stable heterodimers or hetero-oligomers, we performed co-immunoprecipitation experiments using T7-tagged PAR1 and PAR2-myc. We found that PAR2-myc strongly associated with T7-PAR1 when the receptors were co-expressed, demonstrating a physical interaction or complex when immunoprecipitations were performed with either T7-Ab beads (Figure 5) or Myc-Ab beads (Supplemental Figure IVA). Conversely, HA-tagged CXCR4, a receptor that is not expected to bind PAR1 or PAR2, was used as a negative control and did not detectably immunoprecipitate with T7-PAR1 (Supplemental Figure IVB). These data provide evidence that PAR1 and PAR2 can specifically associate within a complex, possibly as a heterodimer or hetero-oligomer.
Figure 5. Co-immunoprecipitation of PAR1 with PAR2.
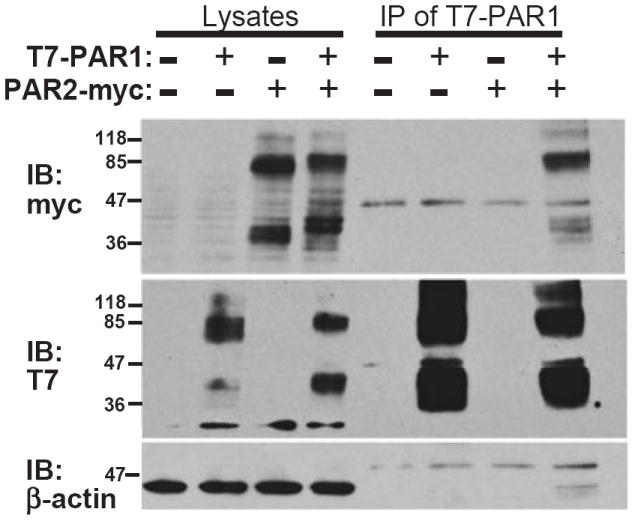
(top) T7-PAR1 was transiently co-expressed in COS7 cells with PAR2-myc or pcDEF3 vector alone (-) as indicated. T7 agarose beads were used to immunoprecipitate bound PAR2-myc from cell lysates as shown by the myc immunoblot. (middle) Immunoblot of T7 confirmed the presence of T7-PAR1, (bottom) and β-actin staining confirmed equal loading of the lysates.
Treatment with a PAR1 Agonist Causes De-differentiation of Carotid Artery SMCs
The observation that the P1pal-13 PAR1 agonist caused significant neointimal hyperplasia following arterial injury suggests that the neointimal SMCs may have originated from previously differentiated, contractile SMCs that underwent de-differentiation to a ‘synthetic’ phenotype to allow migration from the media and increased proliferation.30 To distinguish between the contractile and synthetic state of the SMCs isolated from the carotid arteries of vehicle versus P1pal-13 treated mice, real-time quantitative PCR was performed with SMC differentiation and de-differentiation markers (Supplemental Table I). SMCs harvested after 14 days from P1pal-13 and vehicle-treated injured and uninjured arteries were assessed and normalized to GAPDH expression (rER or relative expression ratio). We found that injury of the carotid arteries in vehicle and P1pal-13 treated animals caused a significant decrease in mRNA expression of contractile SMC differentiation markers α-SMA, SM-22, and SM-MHC (Figure 6A). Quite interestingly, treatment with PAR1 agonist pepducin also caused a suppression in the mRNA of the SMC differentiation genes in uninjured carotid arteries (Figure 6A). This suggests that PAR1 stimulation even in the absence of injury can suppress the differentiation markers within quiescent SMCs. However, there were no differences in intimal or medial area, or obvious changes in histology or morphology in the uninjured arteries (data not shown). Therefore, the P1pal-13-dependent decrease in the SMC contractile marker mRNAs did not appear to have an effect on the architecture of the healthy blood vessels.
Figure 6. The PAR1 agonist pepducin causes de-differentiation of SMCs isolated from the carotid arteries of mice.
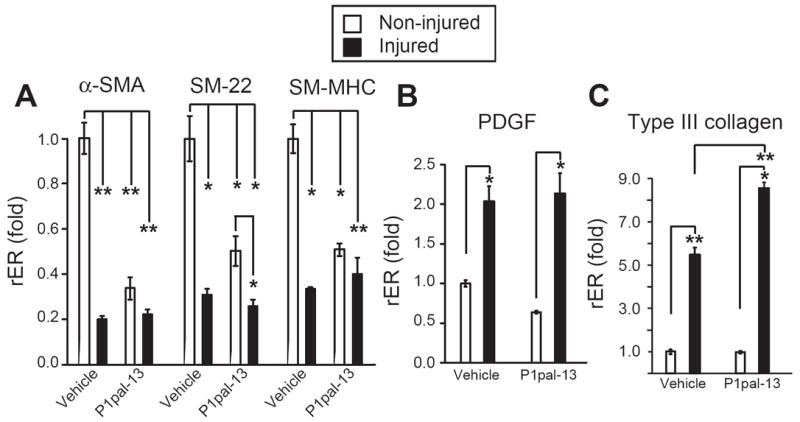
(A) RNA was extracted from uninjured and injured (ligated) carotid arteries of C57BL/6 wild-type mice treated with P1pal-13 or vehicle for 14 days. Real-time PCR was performed with primers for markers of SMC differentiation, (B,C) or markers of SMC de-differentiation. Mean relative gene expression was normalized to GAPDH expression (rER, relative expression ratio) and fold change was determined by comparing the relative expression ratios (rER) of each group to uninjured vehicle treated arteries (n=3), *, P <0.05 and **, P <0.005.
Arterial injury also increased the expression of de-differentiation factors PDGF-B and Type III collagen in SMCs harvested after 14 days from the carotids of either vehicle or P1pal-13-treated animals (Figure 6B-C). P1pal-13 treatment caused a significantly greater increase in expression of Type III collagen mRNA in injured blood vessels as compared to the vehicle cohort (Figure 6C). However, P1pal-13 treatment did not cause an increase in PDGF-B and Type III collagen mRNA levels in the uninjured arteries. Together, these data indicate that strong stimulation of PAR1 can cause phenotypic changes in arterial SMCs consistent with conversion from a differentiated to a de-differentiated state which could contribute to the observed increases in neointimal hyperplasia in response to arterial injury in mice.
Discussion
In this study, we demonstrated that both PAR1 and PAR2 regulate the hyperplastic response following carotid artery injury in mice. Chronic PAR1 stimulation triggered SMC de-differentiation and proliferation to promote medial and neointimal growth that led to highly stenotic arterial lesions. However, for PAR1 agonist-driven hyperplasia to occur, PAR2 must also be present, consistent with an interaction or linkage between the two protease-activated receptors. In agreement with previous reports of PAR1-PAR2 transactivation,20, 26, 29 we detected stable PAR1 and PAR2 complexes, thereby providing evidence that the two receptors may exist as a heterodimer or hetero-oligomer. The possibility remains that PAR2 may also act as a scaffold for another unknown component which regulates the PAR1-hyperplastic response.
Unexpectedly, we found that PAR1-/- and PAR2-/- mice have no change in medial area and have increased intimal hyperplasia in response to carotid artery ligation injury as compared to wild-type mice. Cheung et al.,16 found no effect in medial area and no statistically significant effects in intimal area in carotid arteries of PAR1-/- mice as compared to wild-type using a flexible wire injury model. However, they also found a significant 2-fold increase in cell density in the intima of the PAR1-/- mice as compared to wild-type.16 Possible explanations for this increase in intimal area or cell number in the PAR1-deficient mice could be due to a loss of negative feedback signals from PAR1, over-compensation from PAR2 in the absence of a PAR1 partner, and/or changes in PAR1-dependent control of extracellular matrix deposition and remodeling. An additional explanation is that both PAR1 and PAR2 are well known to regulate barrier function and vascular permeability.20 Therefore, the complete absence of either PAR1 or PAR2 might cause enhanced vascular permeability and increased exposure to proteases and other inflammatory/proliferative mediators following injury that might offset the potential anti-proliferative effects of the knockouts.
Unlike the case in mice, studies in rats using pharmacologic inhibitors of PAR114, 31 or blocking antibodies32 have shown inhibition of neointimal hyperplasia following injury induced by balloon angioplasty. As we observed with PAR1-deficient mice or treatment of WT mice with the PAR1 inhibitor P1pal-7, neither PAR1 inhibitors or blocking antibodies had a suppressive effect on medial area in the balloon-injured rat carotid arteries.31, 32 Differences in the type of injuries, e.g. ligation or wire injury in mice versus balloon angioplasty in rat, or rodent species may also contribute to these different outcomes. In this regard, mPAR2 and rPAR2 share 89% identity with each other and are 81% and 82% identical with hPAR2, respectively. The PAR2 exodomains have considerable divergence in the first 30-32 aa, but otherwise are highly identical in the transmembrane and intracellular domains including the the 3rd intracellular (i3) loop. The PAR2 cleavage sites are identical in mouse, rat and human (SKGR-S). mPAR1 and rPAR1 share 91% identity with each other and are 78% and 79% identical with hPAR1, respectively. Interestingly, hPAR1 has a LDPR-S thrombin cleavage site, mPAR1 has a VNPR-S cleavage site, whereas rPAR1 has a PNPR-S cleavage site which is typical of PAR4 receptors which use a signature PXPR motif with a thrombin-optimized second proline at the P4 position.33 Therefore, it is possible that rat PAR1 is cleaved at a higher rate by thrombin as compared to mouse PAR1 which could contribute to the larger amount of restenosis injury and consequently the greater inhibition observed by thrombin receptor antagonists in rat as compared to mouse. Aside from other divergences in the N-terminal extracellular domains, the three PAR1 sequences are otherwise highly similar in the transmembrane domains and intracellular loops with complete identity in the i3 loop which is targeted by the PAR1 pepducins used in this study.
Cooperative signaling between PAR1 and PAR2 in vivo has been previously shown in a model of acute systemic inflammation, where the protective effect of PAR1 agonist activity required the presence of PAR2 in endothelial barrier function and survival in mice.20 Confocal fluorescence resonance energy transfer studies detected PAR1 and PAR2 receptors in close molecular proximity in cytoplasmic vesicles and on the plasma membrane in endothelial cells.20 PAR1 was found to couple to G12/13-rho pathways whereas PAR2 coupled to Gi-rac pathways. PAR2 appeared to dominate over PAR1 in endothelial cells and transactivation of PAR2 by PAR1 stimulated barrier protective Gi-rac signaling.20 Additionally, cooperative signaling between PAR1 and PAR2 has recently been observed on tumor cells which regulates the coagulation/fibrinolytic responses.29 Interestingly, thrombin but not Xa activated the PAR1/PAR2 response in breast cancer cells suggesting that the receptor complexes may reside in different membrane micro-domains.29 These studies are all consistent with the notion that PAR1/PAR2 complexes may be a mechanism to confer diverse signaling options to cells.
The protective effect of PAR2 activation has also been observed in reperfusion injury in rat hearts by decreasing neutrophil accumulation, improving ventricular function and coronary flow, while reducing the ischemic risk zone and markers of myocardial necrosis.34, 35 These results led to the hypothesis that early PAR2 activation leads to endogenous protection. However, a recent study has suggested that PAR2 also has detrimental effects based on the observation that PAR2-/- mice had significantly smaller infarct size, less impairment of the heart, and reduced inflammatory cytokines in heart lysates after injury36. This suggested that PAR2 might have a dual role, with a protective effect on endothelial cells and a damaging effect on cardiomyocytes and infiltrating leukocytes36. Moreover, models of acute inflammation,37 neurogenic pain,38 and arthritis39 have all shown damaging effects of PAR2 activation. These divergent outcomes could be due to temporal or cell-type specific signaling differences between PAR1/PAR2 heterodimers versus PAR2 homodimers.28, 29 The observation that PAR1 and PAR2 have been linked to overlapping G protein signaling pathways as well as distinct subsets of Gα proteins suggests that the net signal may be a product of whether the receptor homodimer or heterodimer is being activated.40
We found that ligation injury caused highly significant decreases in the expression of differentiation markers α-SMA, SM-22, and SM-MHC and increases in the de-differentiation markers PDGF and Type III collagen in vivo. De-differentiated SMCs have been shown to exhibit a high rate of production of extracellular matrix components in vitro, such as Type III collagen,30, 41 a critical component of the arterial wall during development.42 This could help explain the occlusive stenosis observed in the injured mice treated with the PAR1 agonist pepducin which had further increases in type III collagen expression in the SMCs.30 Our observation that chronic PAR1 stimulation caused significant decreases in the expression of contractile markers of SMCs from uninjured carotid arteries indicates that PAR1 may also impact early phenotypic changes in the medial SMCs that take place prior to intimal migration and proliferation. As SMC hyperplasia is a common response to iatrogenic injury following percutaneous coronary interventions and can require repeat and urgent revascularization procedures to prevent ischemic morbidity and mortality, it is important to develop new pharmacologic approaches such as targeting the PAR1-PAR2 system in SMCs to suppress either the early events of de-differentiation and/or the late events of proliferation to prevent in-stent restenosis.1-3
Supplementary Material
Acknowledgments
We thank Mark Aronovitz for his expertise in developing the arterial ligation injury models.
Sources of Funding This work was funded in part by grants from the National Institutes of Health R01 HL-57905 and R01 HL-64701 (to A.K.).
Footnotes
Disclosures None.
References
- 1.Orford JL, Selwyn AP, Ganz P, Popma JJ, Rogers C. The comparative pathobiology of atherosclerosis and restenosis. Am J Cardiol. 2000;86:6H–11H. doi: 10.1016/s0002-9149(00)01094-8. [DOI] [PubMed] [Google Scholar]
- 2.Douglas JS., Jr Pharmacologic approaches to restenosis prevention. Am J Cardiol. 2007;100:10K–16K. doi: 10.1016/j.amjcard.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Sukhija R, Mehta JL, Sachdeva R. Present status of coronary bifurcation stenting. Clin Cardiol. 2008;31:63–66. doi: 10.1002/clc.20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114:1070–1077. doi: 10.1161/CIRCULATIONAHA.105.574830. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien PJ, Molino M, Kahn M, Brass LF. Protease activated receptors: theme and variations. Oncogene. 2001;20:1570–1581. doi: 10.1038/sj.onc.1204194. [DOI] [PubMed] [Google Scholar]
- 6.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 7.Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;38:4572–4585. doi: 10.1021/bi9824792. [DOI] [PubMed] [Google Scholar]
- 8.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 9.Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O’Callaghan K, Covic L, Kuliopulos A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 11.Seitz I, Hess S, Schulz H, Eckl R, Busch G, Montens HP, Brandl R, Seidl S, Schomig A, Ott I. Membrane-type serine protease-1/matriptase induces interleukin-6 and -8 in endothelial cells by activation of protease-activated receptor-2: potential implications in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:769–775. doi: 10.1161/01.ATV.0000258862.61067.14. [DOI] [PubMed] [Google Scholar]
- 12.Camerer E, Huang W, Coughlin SR. Tissue Factor- and Factor X-dependent Activation of Protease-activated Receptor 2 by Factor VIIa. Proc Natl Acad Sci (USA) 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelken NA, Soifer SJ, O’Keefe J, Vu TK, Charo IF, Coughlin SR. Thrombin receptor expression in normal and atherosclerotic human arteries. J Clin Invest. 1992;90:1614–1621. doi: 10.1172/JCI116031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chieng-Yane P, Bocquet A, Letienne R, Bourbon T, Sablayrolles S, Perez M, Hatem SN, Lompre AM, Le Grand B, David-Dufilho M. Protease-activated receptor-1 antagonist F 16618 reduces arterial restenosis by down-regulation of tumor necrosis factor alpha and matrix metalloproteinase 7 expression, migration, and proliferation of vascular smooth muscle cells. J Pharmacol Exp Ther. 2010;336:643–651. doi: 10.1124/jpet.110.175182. [DOI] [PubMed] [Google Scholar]
- 15.Marutsuka K, Hatakeyama K, Sato Y, Yamashita A, Sumiyoshi A, Asada Y. Protease-activated receptor 2 (PAR2) mediates vascular smooth muscle cell migration induced by tissue factor/factor VIIa complex. Thromb Res. 2002;107:271–276. doi: 10.1016/s0049-3848(02)00345-6. [DOI] [PubMed] [Google Scholar]
- 16.Cheung WM, D’Andrea MR, Andrade-Gordon P, Damiano BP. Altered vascular injury responses in mice deficient in protease-activated receptor-1. Arterioscler Thromb Vasc Biol. 1999;19:3014–3024. doi: 10.1161/01.atv.19.12.3014. [DOI] [PubMed] [Google Scholar]
- 17.Wilcox JN, Rodriguez J, Subramanian R, Ollerenshaw J, Zhong C, Hayzer DJ, Horaist C, Hanson SR, Lumsden A, Salam TA, et al. Characterization of thrombin receptor expression during vascular lesion formation. Circ Res. 1994;75:1029–1038. doi: 10.1161/01.res.75.6.1029. [DOI] [PubMed] [Google Scholar]
- 18.Damiano BP, D’Andrea MR, de Garavilla L, Cheung WM, Andrade-Gordon P. Increased expression of protease activated receptor-2 (PAR-2) in balloon-injured rat carotid artery. Thromb Haemost. 1999;81:808–814. [PubMed] [Google Scholar]
- 19.Mirza H, Yatsula V, Bahou WF. The Proteinase Activated Receptor-2 (PAR-2) Mediates Mitogenic Responses in Human Vascular Endothelial Cells. J Clin Inv. 1996;97:1705–1714. doi: 10.1172/JCI118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, Kuliopulos A. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tennant GM, Wadsworth RM, Kennedy S. PAR-2 mediates increased inflammatory cell adhesion and neointima formation following vascular injury in the mouse. Atherosclerosis. 2008;198:57–64. doi: 10.1016/j.atherosclerosis.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 22.Major CD, Santulli RJ, Derian CK, Andrade-Gordon P. Extracellular mediators in atherosclerosis and thrombosis: lessons from thrombin receptor knockout mice. Arterioscler Thromb Vasc Biol. 2003;23:931–939. doi: 10.1161/01.ATV.0000070100.47907.26. [DOI] [PubMed] [Google Scholar]
- 23.Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM, Andrade-Gordon P, Kotzsch M, Spring D, Luther T, Abe J, Pohlman TH, Verrier ED, Blaxall BC, Mackman N. Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Circulation. 2007;116:2298–2306. doi: 10.1161/CIRCULATIONAHA.107.692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt VA, Nierman WC, Feldblyum TV, Maglott DR, Bahou WF. The human thrombin receptor and proteinase activated receptor-2 genes are tightly linked on chromosome 5q13. Br J Haematol. 1997;97:523–529. doi: 10.1046/j.1365-2141.1997.922907.x. [DOI] [PubMed] [Google Scholar]
- 25.Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, Sundelin J, Scarborough RM. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- 26.O’Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275:13502–13509. doi: 10.1074/jbc.275.18.13502. [DOI] [PubMed] [Google Scholar]
- 27.Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci (USA) 2002;99:643–648. doi: 10.1073/pnas.022460899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sevigny LM, Zhang P, Bohm A, Lazarides K, Perides G, Covic L, Kuliopulos A. Interdicting protease-activated receptor-2-driven inflammation with cell-penetrating pepducins. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1017091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McEachron TA, Pawlinski R, Richards KL, Church FC, Mackman N. Protease-activated receptors mediate crosstalk between coagulation and fibrinolysis. Blood. 2010;116:5037–5044. doi: 10.1182/blood-2010-06-293126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 31.Andrade-Gordon P, Derian CK, Maryanoff BE, Zhang H-C, Addo MF, Cheung W-M, Damiano BP, D’Andrea MR, Darrow AL, DeGaravilla L, Eckardt AJ, Giardino EC, Haertlein BJ, McComsey DF. Administration of a Potent Antagonist of Protease-Activated Receptor-1 (PAR-1) Attenuates Vascular Restenosis Following Balloon Angioplasty in Rats. J Pharm Exp Therap. 2001;298:34–42. [PubMed] [Google Scholar]
- 32.Takada M, Tanaka H, Yamada T, Ito O, Kogushi M, Yanagimachi M, Kawamura T, Musha T, Yoshida F, Ito M, Kobayashi H, Yoshitake S, Saito I. Antibody to thrombin receptor inhibits neointimal smooth muscle cell accumulation without causing inhibition of platelet aggregation or altering hemostatic parameters after angioplasty in rat. Circ Res. 1998;82:980–987. doi: 10.1161/01.res.82.9.980. [DOI] [PubMed] [Google Scholar]
- 33.Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic recognition motif for thrombin recognition and cleavage. Biochem J. 2003;376:733–740. doi: 10.1042/BJ20030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Napoli C, Cicala C, Wallace JL, de Nigris F, Santagada V, Caliendo G, Franconi F, Ignarro LJ, Cirino G. Protease-activated receptor-2 modulates myocardial ischemia-reperfusion injury in the rat heart. Proc Natl Acad Sci U S A. 2000;97:3678–3683. doi: 10.1073/pnas.97.7.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Napoli C, De Nigris F, Cicala C, Wallace JL, Caliendo G, Condorelli M, Santagada V, Cirino G. Protease-activated receptor-2 activation improves efficiency of experimental ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2002;282:H2004–2010. doi: 10.1152/ajpheart.00909.2001. [DOI] [PubMed] [Google Scholar]
- 36.Antoniak S, Rojas M, Spring D, Bullard T, Verrier E, Blaxall B, Mackman N, Pawlinski R. Protease-Activated Receptor 2 Deficiency Reduces Cardiac Ischemia/Reperfusion Injury. Arterioscler Thromb Vasc Biol. 2010 doi: 10.1161/ATVBAHA.110.213280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vergnolle N, Hollenberg MD, Sharkey KA, Wallace JL. Characterization of the inflammatory response to proteinase-activated receptor-2 (PAR2)-activating peptides in the rat paw. Br J Pharmacol. 1999;127:1083–1090. doi: 10.1038/sj.bjp.0702634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vergnolle N, Bunnett NW, Sharkey KA, Brussee V, Compton SJ, Grady EF, Cirino G, Gerard N, Basbaum AI, Andrade-Gordon P, Hollenberg MD, Wallace JL. Proteinase-activated receptor-2 and hyperalgesia: A novel pain pathway. Nat Med. 2001;7:821–826. doi: 10.1038/89945. [DOI] [PubMed] [Google Scholar]
- 39.Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, Smith AJ, Hunter GD, McLean JS, McGarry F, Ramage R, Jiang L, Kanke T, Kawagoe J. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCoy KL, Traynelis SF, Hepler JR. PAR1 and PAR2 couple to overlapping and distinct sets of G proteins and linked signaling pathways to differentially regulate cell physiology. Mol Pharmacol. 2010;77:1005–1015. doi: 10.1124/mol.109.062018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45(Suppl A):A25–32. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci U S A. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.