


Abstract
MicroRNAs (miRNAs) are critical small non-coding RNAs that regulate gene expression by hybridizing to the 3′-untranslated regions (3′-UTR) of target mRNAs, subsequently controlling diverse biological processes at post-transcriptional level. How miRNA genes are regulated receives considerable attention because it directly affects miRNA-mediated gene regulatory networks. Although numerous prediction models were developed for identifying miRNA promoters or transcriptional start sites (TSSs), most of them lack experimental validation and are inadequate to elucidate relationships between miRNA genes and transcription factors (TFs). Here, we integrate three experimental datasets, including cap analysis of gene expression (CAGE) tags, TSS Seq libraries and H3K4me3 chromatin signature derived from high-throughput sequencing analysis of gene initiation, to provide direct evidence of miRNA TSSs, thus establishing an experimental-based resource of human miRNA TSSs, named miRStart. Moreover, a machine-learning-based Support Vector Machine (SVM) model is developed to systematically identify representative TSSs for each miRNA gene. Finally, to demonstrate the effectiveness of the proposed resource, an important human intergenic miRNA, hsa-miR-122, is selected to experimentally validate putative TSS owing to its high expression in a normal liver. In conclusion, this work successfully identified 847 human miRNA TSSs (292 of them are clustered to 70 TSSs of miRNA clusters) based on the utilization of high-throughput sequencing data from TSS-relevant experiments, and establish a valuable resource for biologists in advanced research in miRNA-mediated regulatory networks.
INTRODUCTION
MicroRNAs (miRNAs) are ~22 bp-long, endogenous RNA molecules that act as regulators, leading either mRNA cleavage or translational repression by principally hybridizing to the 3′-untranslated regions (3′UTRs) of their target mRNAs. This negative regulatory mechanism at the post-transcriptional level ensures that miRNAs play prominent roles in controlling diverse biological processes such as carcinogenesis, cellular proliferation and differentiation (1–3).
Recently, an increasing number of miRNA target prediction tools have been developed (4–8). As well as putative miRNA-target interactions, numerous miRNA targets are experimentally validated and collected in TarBase (9), miRecords (10), miR2Disease (11) and miRTarBase (12). According to the latest statistics in miRTarBase, for example, there exist 58 and 43 known target genes of hsa-miR-21 and hsa-miR-122, respectively. It reveals the importance of miRNA functions in contributing to the control of gene expression (Figure 1B). Therefore, transcriptional regulatory networks have been expanded and become rather complex due to the involvement of miRNAs (13).
Figure 1.

The collaboration of miRNAs and TFs makes transcriptional regulatory networks more complex. Shown is (A) a traditional regulatory circuitry that considers only genes and their TFs. (B) A miR-involved regulatory circuitry. (C) The entire regulatory circuitry containing TFs, miRNA genes, miRNAs and their target genes. (D) Identifying TSSs of miRNA genes is the first step to investigate TF-miRNA regulatory relations. (E) Investigation of novel cis- and trans-elements of miRNA genes.
Given the significance of miRNA functions and its role in gene regulation, how miRNA genes are regulated receives considerable attention and directly affects miRNA-mediated gene regulatory networks. Several studies thus elucidated which transcription factors (TFs) can regulate the transcription of miRNA genes (14–16), and which ones should be involved in specific regulatory circuitries (Figure 1C). Moreover, Wang et al. (17) manually identified 243 TF-miRNA regulatory relations by conducting a literature survey and constructing a database, TransmiR. Although such data provide deep insights into the miRNA transcriptional regulation, most of them remain unknown unless a large-scale investigation of novel cis- and trans-elements is undertaken to further determine more TF-miRNA regulatory relations. Hence, precisely locating promoter regions of miRNA genes is of priority concern, in which transcriptional start sites (TSSs) of miRNA genes must be identified first (Figure 1D and E).
Since most miRNA genes are transcribed by RNA polymerase II (18–21), promoter prediction models or genomic annotation based on transcriptional features of RNA polymerase II (class II) gene were used to characterize 5′ boundaries of primary miRNAs (pri-miRNAs) and to identify putative core promoters of miRNA genes (22–24). Additionally, previous studies applied chromatin immunoprecipitation (ChIP) data of RNA polymerase II and histone methylations, which reveal gene promoter signals, for detecting miRNA promoters systematically (25,26). However, all miRNA promoters mentioned above are computationally predicted, without experimental validation to support their reliability. Until now, only few of miRNA promoters predicted by using chromatin signatures have been confirmed by promoter reporter assay (27,28).
Obviously, rather than promoter/TSS prediction tools or computational models, experimental datasets derived from high-throughput sequencing analysis of gene initiation reveal how TSS signals are distributed in the genome and provide direct evidence of gene promoters. In this work, we attempt to identify miRNA TSSs by incorporating current datasets, including cap analysis of gene expression (CAGE) tags, TSS Seq libraries and H3K4me3 chromatin signature, to establish an experimental-based resource of miRNA TSSs, named miRStart, with a particular emphasis on the human genome. Moreover, a machine-learning-based support vector machine (SVM) model is developed to select the representative TSSs systematically for each miRNA gene. A user-friendly web resource allows scientists to select miRNA TSSs based on the straightforward display of experimental TSS signals. Besides, this work successfully validates the putative promoter of liver-specific hsa-miR-122 by 5′RACE and luciferase reporter assay, which contains the exhaustive structure and is more authentic than previous one (27). As a novel resource for biologists in advanced research in miRNA-mediated regulatory networks, miRStart integrates abundant data from TSS-relevant experiments, offering reliable human miRNA TSSs to further decipher the miRNA transcription regulation. The resource is currently available at http://mirstart.mbc.nctu.edu.tw/.
MATERIALS AND METHODS
Data collection
Human miRNAs and gene annotation
The genomic coordinates of 940 human pre-miRNAs were obtained from miRBase release 15 (29). According to a previous study, two miRNAs within a distance <50 kb tend to share a common primary transcript (30). Therefore, this work analyzed the 50 kb-long upstream region of each pre-miRNA to identify putative TSSs. Upstream flanking sequences were then downloaded using the BioMart data mining approach provided by Ensembl release 58 (31). Additionally, Homo sapiens genes (GRCh37) with HGNC symbols were also obtained from Ensembl either to define intragenic and intergenic miRNAs or to avoid overlapping an identified TSS with other TSS of an annotated gene. Typically, pre-miRNAs embedded in the same strand of Ensembl genes are defined as ‘intragenic miRNAs’, whereas pre-miRNAs located between Ensembl genes are ‘intergenic miRNAs’.
TSS-relevant datasets derived from high-throughput sequencing
In this work, CAGE tags, TSS Seq tags and H3K4me3 modification were mapped directly to the upstream flanking regions of miRNA precursors for TSS detection (Supplementary Table S1). Totally, 29 million CAGE tags derived from 127 human RNA samples were obtained from FANTOM4 (32). This work also incorporated 75 361 186 and 241 440 055 TSS Seq tags derived from eight human normal tissues (five fetal tissues and three adult tissues) and six human cell lines (DLD1, HEK293, Beas2B, Ramos, MCF7 and TIG) from DBTSS release 7.0, respectively (33). For H3K4me3 modification, high-resolution ChIP-seq data of human CD4+ T cells reported in 2007 (34) were used and downloaded from http://dir.nhlbi.nih.gov/papers/lmi/epigenomes/hgtcell.aspx. Since genomic coordinates of these three experimental datasets are based on NCBI36/hg18, the liftOver program obtained from UCSC Genome browser (35) was applied to convert genomic loci into GRCh37/hg19 (compatible to miRBase release 15).
Supporting evidence of miRNA TSSs
Human expressed sequence tags (ESTs) located in pre-miRNA upstreams and the conservation within those regions provide strong evidences of TSS loci. Here, all human ESTs and conservation among 46 vertebrate species using phastCons method were retrieved from UCSC Genome Browser. They are useful in supporting miRNA TSSs estimated by the proposed SVM model.
SVM-based prediction model
Computational models for miRNA TSS identification are generated by adopting the SVM, which incorporates CAGE tags, TSS Seq tags and H3K4me3 modification as training evidence. Based on the binary classification, SVM maps the input samples into a higher dimensional space using a kernel function and, then, identifies a hyper-plane that discriminates between the two classes with a maximal margin and minimal error. A public SVM library, LibSVM (36), is used to train the predictive model with positive and negative training sets, which are encoded based on different training features. 7286 protein-coding genes with unique TSS were collected from DBTSS as the training sets for establishing a SVM-based TSS prediction model. The total number of CAGE tags, TSS Seq tags and H3K4me3 modification within a 200 bp-long window size from −1100 to +1100 relative to 7286 TSSs was calculated and defined as positive sets, whereas ±10 kb away in relation to 7286 TSSs were defined as negative sets. The comparison between positive and negative sets is illustrated in Supplementary Figure S1B (see Supplementary Data). Then, a matrix with 33 features of 7286 experimentally verified TSS was created (Supplementary Figure S2). This observation reveals how TSS-relevant signals are distributed around exact TSSs and are the inputs for SVM training.
After the establishment of SVM-based model for miRNA TSS prediction, the model performance was evaluated by 5-fold cross-validation. Next, the SVM model scanned up to 50-kb upstream regions for each pre-miRNA with a 2200 bp-long window and a 100 bp-long step to identify a 200 bp-long region containing high-confidence TSS. The putative region containing the most possible TSS of a miRNA is selected as a priority if:
The region is classified into ‘positive’ by the SVM model.
The positive region does not overlap with exons of protein-coding genes.
ESTs and conservation are supported around the region.
The positive region is nearest to the 5′ end of pre-miRNA.
Finally, the tag density in representative regions is calculated using the following density function:
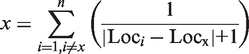 |
where x denotes the density of each locus within a representative region possibly contained miRNA TSSs; Loci represents the location of site i; and Locx denotes the location of sitex. The total number of sites detected in the representative region is denoted as n. We recommend a putative miRNA TSS if the locus has the highest density of CAGE tags and TSS Seq tags. Since polycistronic miRNAs tend to be transcribed from a common transcription unit (30), it is logical to provide putative TSSs of miRNA clusters rather than the TSS of each miRNA. For this reason, human miRNAs with identical putative TSSs in miRStart were defined as a miRNA cluster. Besides, as suggested in the previous study, miRNAs within a distance <50 kb were assigned to a new cluster or the existed clusters. Such miRNAs were excluded if they are reported to be embedded in different host genes and not all of them are intragenic or intergenic.
Cell lines and RNA interference with shRNA
The human HCC cell lines, HuH-7, Hep3B and human embryonic kidney HEK293T cells were cultured as described previously (37). HuH7 cells were plated and infected with lentiviruses expressing shDGCR8 in the presence of 8 μg/ml protamine sulfate for 24 h, which was followed by puromycin (2 μg/ml; 48 h) selection. The shRNA target sequences for DGCR8 were 5′GCTCGATGAGTTAGAAGATTT3′ (TRCN0000159003). The shLuc (TRCN0000072243, shLuc) targeting the luciferase gene was used as a control for RNA interference. Gene expression and the knockdown efficiency of DGCR8 were examined using RT–PCR and standard gel electrophoresis. Expression of pri-miR-122 and mature miR-122 was detected by RT–PCR and low stringency northern blotting, respectively (38). [γ-32P]-labeled 5′-ACAAACACCATTGTCACACTCCA-3′ was used in detecting miR-122 by northern blotting. U6 snRNA was used as an internal control. The primer sequences are listed in Supplementary Table S2.
RNA ligase mediated rapid amplification of cDNA ends
PolyA+ RNA was purified from HuH7 that were infected with lentiviruses expressing shDGCR8 using OligotexR mRNA Kit (Qiagen). RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) was performed using FirstChoice RLM-RACE Kit (Ambion) and 250 ng of polyA+ RNA, following the manufacturer's instructions. The gene-specific primer for the 5′-RACE was reverse primer R01863_R' (5′-AGGGACCTAGAACAGAAATCG-3′). For the 3′-RACE, three forward gene-specific primers were used: 122-D1 (5′-CAATGGTGTTTGTGTCTAAACT-3′), 122-D2 (5′-CTACCGTGTGCCTGAC-3′), and 122-D3 (5′-CTCCTGGCACCATCTAC-3′).
Plasmid constructs
Luciferase reporter constructs containing several upstream regions of pri-miR-122 (nucleotides −1 to −182, −1 to −391, −1 to −1358, −375 to −1358 and −1329 to −2221) were subcloned in pGL3-basic vector (Promega) and designated as pGL3-miR-122-U, −U1, −U12, −U2 and −U3, respectively. Mutations of two putative TATA boxes were generated using a QuickChange Site-Directed Mutagenesis Kit (Stratagene). The TATA boxes were mutated at −23 to −28 (mTATA1) and −81 to −87 (mTATA2). The RT–PCR primers used in mutagenesis are listed in Supplementary Table S2.
Promoter reporter assay
Cells (5 × 104/well) were seeded in 24-well plate and co-transfected with 0.5 μg of pGL3-basic or pGL3-basic-promoter constructs and 0.05 μg of pRL-TK (Promega) using jetPEI reagent (Polyplus-Transfection). After 48 h, the luciferase activity was measured using the Dual-Luciferase Reporter Assay System kit (Promega). pGL3-NRP1 promoter construct (37) was used as a positive control for the promoter reporter assay.
Statistical analysis
All data are expressed as mean ± SD and compared between groups using the Student's t-test. A P<0.05 was considered to be statistically significant.
RESULTS
CAGE tags, TSS Seq tags and H3K4me3 enriched loci reveal TSSs of RNA polymerase II genes
As ~20-nt sequences are derived from the 5′ terminal of cDNAs, CAGE tags can be massively generated using a biotinylated cap-trapper with specific linkers to ensure that the sequences after 5′ cap of cDNAs are reserved (39). Based on this attribute, CAGE tags are extensively adopted to identify the TSSs of genes with 5′ cap transcripts, i.e. RNA polymerase II (class II) genes (40). Similar to CAGE tags, TSS Seq tags initially denominated by DBTSS are also the 5′-end sequences of human and mice cDNAs based on use of the TSS Seq method (33). More than 300 million TSS Seq tags were generated by integrating the oligo-capping method and Solexa sequencing technology, offering an abundant resource to detect class II TSSs. Besides, histone methylation significantly influences gene expression. H3K4me3, which represents histone H3 as trimethylated at its lysine 4 residue, is enriched around TSS and positively correlated with gene expression, regardless of whether or not the genes are transcribed productively. As a massive parallel signature sequencing technique, ChIP-seq performs well in chromatin modifications and provides high-resolution profiling of histone methylations in the human genome (34).
To evaluate the feasibility of using these three experimental-based datasets to identify miRNA TSS, the occurrence distributions of CAGE tags, TSS Seq tags, and H3K4me4 modification around experimentally verified TSSs of RNA polymerase II genes were examined (Figure 2). After obtaining 7286 annotated genes with Entrez Gene ID and unique TSS from DBTSS, genes with multiple TSSs were omitted to avoid the overlapping tags between adjacent TSSs (Supplementary Figure S3). Next, the averages of CAGE tags, TSS Seq tags and H3K4me4-enriched loci from −2500 to +2500 (window size = 200 bp) relative to each TSS were mapped and analyzed. Figure 2A depicts the tag occurrence distributions of three sets of experimental evidence, and the peaks of CAGE and TSS Seq tags are positively correlated with the locations of the experimentally verified TSSs as well as H3K4me3-enriched loci. It implies that CAGE tags, TSS Seq tags and H3K4me3-enriched loci can be considered as effective supporting evidences for revealing the TSSs of RNA polymerase II genes, including TSSs of miRNAs (Figure 2B).
Figure 2.

CAGE tags, TSS Seq tags and H3K4me3 modification aggregate around 7286 experimentally verified TSSs and can be used to identify miRNA TSSs. (A) Tag occurrence distribution of the three sets of experimental evidence near TSSs. (B) Examples of three identified miRNA TSSs in miRStart. Notably, the three miRNA TSSs are experimentally verified, revealing that miRNA TSSs can be determined based on the tag occurrence distributions of CAGE tags, TSS Seq tags and H3K4me3 modification.
TSS candidates of intragenic and intergenic human miRNAs
To identify miRNA TSSs in the human genome, three sets of experimental evidence including CAGE tags, TSS Seq tags and H3K4me4 modification were mapped to the 50 kb upstream region of each miRNA precursor to observe their occurrence distribution. According to the evaluation process mentioned above, genomic loci that aggregated by three sets of experimental evidence with apparent peaks reveals a highly probable regions for miRNA TSSs. Additionally, expressed sequence tags (ESTs) and evolutionarily conserved genomic regions around putative miRNA TSSs also provide strong evidence to increase the reliability of corresponding miRNA TSSs.
Among the 940 human miRNAs in miRBase release 15 (29), 483 (51%) are classified as intragenic and 457 (49%) are classified as intergenic in miRStart. As is generally assumed, intragenic miRNAs, whose precursors are located within introns, exons or UTRs of protein-coding transcripts, share common promoters with their host genes and are expressed simultaneously (30,41,42). However, for intergenic miRNAs, their primary transcripts are transcribed from individual, non-protein-coding genes and have their own promoters (18). The human miRNA let-7a-1 provides a typical example of how to use the above-mentioned experimental evidence to define intergenic miRNA TSSs (Supplementary Figure S4). In total, 1083 CAGE tags and 208 TSS Seq tags are within the 50 kb upstream region of let-7a-1 precursor (Genomic coordinates Chr9: 96938239-96938318 [+]). The aggregation of CAGE tags, TSS Seq tags and H3K4me3 modification is apparently around the 9000–10 000 upstream region of precursor, implying that TSS candidates of let-7a-1 may be located between 96928239 and 96929239. It is noticed that CAGE tags are strikingly assembled at 96928529 that denotes the putative TSS of let-7a-1. As anticipated, an EST BG326593 at 96928570 nearby putative TSS provides supporting evidence that the determined TSS is reliable. The upstream region immediately adjacent to putative TSS is quite conserved between 44 vertebrate species, implying that this motif may have promoter activity. Furthermore, two miRNAs, let-7f-1 and let-7d, close to let-7a-1 (distance less than 3000 bps) have identical TSS coordinates in miRStart. This observation suggests that the three miRNAs should be clustered and may be transcribed as a single primary transcript. In sum, TSSs of either intragenic or intergenic miRNAs in human are defined properly in miRStart and can be further analyzed to elucidate TF-miRNA regulatory relations.
Systematically identifying human miRNA TSSs by the SVM model
SVM, a machine-learning method, has been adopted to solve pattern identification problems with an obvious correlation with the underlying statistical learning theory (43). SVM focuses on mapping input vectors to a higher dimensional space in which a maximal separating hyperplane is defined. Two parallel hyperplanes are constructed on each side of the hyperplane that separates the data into two groups. The separating hyperplane maximizes the distance between the two parallel hyperplanes. Moreover, SVM can solve a classification problem when the number of training data is extremely small (44). Therefore, to identify 940 human miRNA TSSs efficiently, a SVM model was developed to systematically select the representative TSSs for each miRNA gene. The model performance was evaluated by a 5-fold cross-validation test, indicating the following: sensitivity of 90.36%, specificity of 90.05%, accuracy of 90.21% and precision of 90.08%. The randomization test was carried out to avoid the occurrence of overfitting as well (Supplementary Table S3). After scanning the 50 kb upstream region of miRNA precursors with SVM model and then executing the filtering process, miRStart provides 10 TSS candidates for each intergenic miRNA gene. As for intragenic miRNA genes, although miRStart officially uses their host gene starts as TSSs, putative TSSs identified by SVM model are still provided because several investigations have demonstrated that intragenic miRNA genes may have their own promoters (26,45). Figure 3 depicts the system flow of miRStart.
Figure 3.

System flow of miRStart.
In total, miRStart identified 90% (847 out of 940) putative TSSs of human miRNAs, among them are 365 putative TSSs of intergenic miRNAs. miRStart also clustered 292 human miRNAs with 70 putative TSSs of transcription units. Users can access the suggested TSSs of individual miRNAs or miRNA clusters (by switching to the ‘cluster list’ view). Table 1 lists 30 TSSs of intergenic miRNA genes identified by SVM model (Supplementary Table S4 for the entire list). Notably, the distances between intergenic miRNA TSSs and their precursors significantly fluctuate from less than 100 bps to 50 kb. A comparison was made of the distance between intergenic pre-miRNA and its TSS with the 5′UTR length of protein-coding gene by calculating the distance between 7286 experimentally verified TSSs and their CDS starts. Figure 4 indicates that in contrast with intergenic miRNAs, the 5′UTR lengths of 7286 protein-coding genes are nearly within 50 bps to 100 bps, results of which correspond to a previous study (46).
Table 1.
Partial list of intergenic miRNA TSSs identified by SVM model in miRStart
| miRNA/miRNA cluster | Genomic coordinates | Putative TSS | Distance from precursor | Supporting evidences |
||||
|---|---|---|---|---|---|---|---|---|
| No. of CAGE tags | No. of TSS tags | H3K4me3 | ESTs | Conservation | ||||
| hsa-mir-9-3 | Chr15: 89911248 [+] | 89905739 | 5509 | 56 | 11 | + | + | |
| hsa-mir-223 | ChrX: 65238712 [+] | 65235302 | 3410 | 92 | 10 | + | + | |
| hsa-mir-183~96~182 | Chr7: 129414854 [−] | 129420061 | 5207 | 27 | + | + | ||
| hsa-mir-3132 | Chr2: 220413869 [−] | 220462760 | 48891 | 2 | + | + | ||
| hsa-mir-196a-2 | Chr12: 54385522 [+] | 54380426 | 5096 | 278 | 5 | + | + | |
| hsa-mir-3193 | Chr20: 30194989 [+] | 30161116 | 33873 | 24 | 40 | + | + | |
| hsa-mir-3142~146a | Chr5: 159901409 [+] | 159895244 | 6165 | 686 | 37 | + | + | |
| hsa-mir-130a | Chr11: 57408671 [+] | 57405960 | 2711 | 81 | 54 | + | + | |
| hsa-mir-548o | Chr7: 102046302 [−] | 102074066 | 27764 | 675 | + | + | ||
| hsa-mir-9-2 | Chr5: 87962757 [−] | 87980642 | 17885 | 4171 | + | + | ||
| hsa-mir-190b | Chr1: 154166219 [−] | 154209596 | 43377 | 2198 | + | + | ||
| hsa-mir-3167 | Chr11: 126858438 [−] | 126870487 | 12049 | 1 | + | + | ||
| hsa-mir-124-2 | Chr8: 65291706 [+] | 65285788 | 5918 | 2 | 5 | + | + | |
| hsa-mir-143~145 | Chr5: 148808481 [+] | 148786413 | 22068 | 2689 | 2 | + | + | |
| hsa-mir-1470 | Chr19: 15560359 [+] | 15511768 | 48591 | 251 | 21 | + | + | |
| hsa-mir-193b~365-1 | Chr16: 14397824 [+] | 14396078 | 1746 | 37 | 14 | + | + | |
| hsa-mir-1244-2 | Chr5: 118310281 [+] | 118310172 | 109 | 12 | + | + | ||
| hsa-mir-659 | Chr22: 38243781 [−] | 38273766 | 29985 | 225 | + | + | ||
| hsa-mir-146b | Chr10: 104196269 [+] | 104179511 | 16758 | 2 | 26 | + | + | |
| hsa-mir-324 | Chr17: 7126698 [−] | 7141111 | 14413 | 4544 | + | + | ||
| hsa-mir-200c~141 | Chr12: 7072862 [+] | 7036976 | 35886 | 21 | 5928 | 6 | + | + |
| hsa-mir-142 | Chr17: 56408679 [−] | 56409879 | 1200 | 12 | + | + | ||
| hsa-mir-1305 | Chr4: 183090446 [+] | 183065816 | 24630 | 82 | 1 | + | + | |
| hsa-mir-607 | Chr10: 98588521 [−] | 98592266 | 3745 | 3 | + | + | ||
| hsa-mir-29b-1~29a | Chr7: 130562298 [−] | 130596999 | 34701 | 100 | + | + | ||
| hsa-mir-21 | Chr17: 57918627 [+] | 57915327 | 3300 | 1 | 28 | + | + | |
| hsa-mir-181c~181d | Chr19: 13985513 [+] | 13976434 | 9079 | 2 | 10 | + | + | |
| hsa-mir-122 | Chr18: 56118306 [+] | 56113494 | 4812 | 18 | 1 | + | + | |
| hsa-mir-563 | Chr3: 15915278 [+] | 15901463 | 13815 | 36 | 33 | + | + | |
| hsa-mir-200b~200a~429 | Chr1: 1102484 [+] | 1098321 | 4163 | 15 | 2 | + | + | |
All TSSs are listed in Supplementary Table S4.
Figure 4.

Statistical comparison of intergenic miRNA genes and protein-coding genes. (Upper) The distances between intergenic miRNA TSSs and their precursors significantly fluctuate from <100 bp to 50 kb. (Lower) The 5′-UTR lengths of 7286 protein-coding genes are nearly within 50–100 bp.
Moreover, this work compared putative TSSs identified by our SVM model with experimentally verified TSSs from previous efforts. First, the miRNA cluster hsa-miR-23a~27a~24-2 was examined and the putative TSS located 1821 bp upstream of the hsa-miR-23a precursor was obtained with a score of 0.94935. This observation markedly differs from the position verified experimentally in a previous study (18). Although the SVM model identified a putative TSS near the position reported by Lee et al. (47), that TSS is not included in the list of ten TSS candidates. Next, putative TSSs of miR-146a and miR-146b in miRStart were compared with the reported loci. miRStart identified miR-146a TSS located 17 115 bp upstream of its precursor, which perfectly matches the experimentally verified TSS. With regard to miR-146b, the TSS candidate located 813 bp upstream of its precursor is quite near the verified one. Another intergenic miRNA examined in this work is hsa-miR-21. Cai et al. (48) indicated that the TSS of hsa-miR-21 is located 2445 bp upstream of its precursor, whereas a different TSS was identified of the longer distance about 3300 bp. According to our results, the putative TSS is located 3300 bp upstream of the precursor and overlaps with the protein-coding gene, TMEM49. Notably, many positive regions have a high probability ranging from 1 to 4500 bp upstream of the hsa-mir-21 precursor, as identified by the SVM model. This finding reveals that hsa-mir-21 gene may have multiple TSSs. Supplementary Table S5 summarizes more putative TSSs overlap with annotated genes for reference.
In addition to the putative miRNA TSSs suggested in miRStart, the user-friendly web resource allows scientists to customize their preferable miRNA TSSs based on a straightforward display of CAGE tags, TSS Seq tags and H3K4me3 modification. After the representative TSS for each miRNA gene is selected, miRStart offers the 5000 bp-long upstream sequence of that TSS. Users can download miRNA promoter sequences and search for possible cis- and trans-elements within them in a relevant database such as JASPAR (49).
Experimental validation of putative miR-122 TSS/promoter
To estimate the reliability of putative miRNA TSSs from miRStart resource, hsa-mir-122 was selected to perform the validation process. Investigations have been shown that this liver-specific miRNA is significantly down-regulated in hepatocellular carcinoma and profoundly impacts carcinogenesis (38). Figure 5A illustrates the occurrence distribution of experimental evidence within the 50 kb upstream region of pre-miR-122 in miRStart web interface. The putative miR-122 TSS identified by SVM model is located at 56113494 (4812 bp upstream of the precursor).
Figure 5.

Identification of hsa-miR-122 TSS. (A) CAGE tags, TSS Seq tags, H3K4me3 modification, ESTs and conservation patterns are clearly displayed in the 50 kb upstream region of hsa-miR-122. The red line in the block of putative TSS denotes the representative TSS, whereas the blue lines denote other TSS candidates of hsa-miR-122. (B) Only liver-specific CAGE tags and TSS Seq tags (overlapped in dotted blue circle) are shown in the upstream region of miR-122 precursor.
Hsa-mir-122 is an intergenic miRNA located at 18q21.31. Previously we successfully ectopically expressed mature miR-122 from a 562-bp cDNA fragment encompassing 54 269 034–54 269 595 bp of 18q21.31 (UCSC Genome Browser NCBI36/hg18 Assembly) subcloned in the lentiviral expression vector (38). In order to determine the full-length pri-mir-122, we first enriched the abundance of the primary transcripts by reducing the endogenous level of DGCR8 with RNAi approach. As shown in Figure 6, knockdown of DGCR8 resulted in the accumulation of pri-miR-122 and reduction of mature miR-122. In the upstream region from this 562-bp fragment, an EST clone R01863 which was derived from a cDNA library of human fetal liver and spleen origin (Soares fetal liver spleen 1NFLS) was identified. Using the primers R01863_F′ and 122_R′, a distinct 2 kb fragment from the total RNA prepared from DGCR8-knockdown HuH7 cells was obtained (Figure 6D). Nucleotide sequencing results showed that this 2 kb fragment contains R01863 and pre-miR-122 sequences. We then performed the 5′RLM-RACE with poly+ RNA derived from DGCR8-knockdown HuH7 cells with a gene-specific primer R01863_R′. A DNA fragment of approximate 350 bp in length was cloned. The nucleotide sequences revealed the potential TSS and an intron of 2969 bp in length (Figure 6E). We further performed 3′ RLM-RACE reactions and revealed three transcripts of 2770 nucleotides, 2944 nucleotides and 3078 nucleotides in length. Three poly adenylation sites were mapped. The gene structure of full-length pri-miR-122 is illustrated in Supplementary Figure S5.
Figure 6.

Mapping of TSS of pri-miR-122 gene by 5′ RLM-RACE. (A) Suppression of DGCR8 mRNA expression in HuH7 cells by RNA interference. Five shDGCR8 lentiviral preparations were tested. #1 (TRCN0000159003) which showed the best knockdown efficiency was chosen for this work. Knockdown of DGCR8 resulted in an enrichment of pri-miR-122 (B) and drastic depletion of mature miR-122. (C) Expression of pri-miR-122 and miR-122 was detected by RT–PCR and northern blotting, respectively. The sequences of the RT–PCR primers were listed in Supplementary Table S2. (D) Total RNA from HuH7 cells with or without DGCR8 knockdown (shDG) was reverse transcribed and amplified by PCR using the primers, R01863_F' and R01863_R'. A distinct 2 kb fragment (arrow) was detected in DGCR8-knockdown HuH7 cells. Marker (M) sizes in base pairs are indicated. Small arrow in (E) marked the 5′ end of R01863_F' primer. (E) The transcription unit of pri-miR-122 at 18q21.31 has a small exon 1 of 78 bp, a large intron of 2969 bp and exon 2. (F) The TSS (+1) based on 5′ RLM-RACE data. Two putative TATA boxes and two CCAAT boxes were marked in the upstream region.
Characterization of the pri-mir-122 promoter
To functionally characterize the pri-mir-122 promoter, the genomic fragments containing the putative core promoter region and the upstream regions were subcloned into pGL3-basic vector (Figure 7A). The reporter constructs were subsequently transfected into two HCC cell lines, HuH7 and Hep3B, as well as human embryonic kidney cell line, HEK293T. As a positive control for the promoter reporter assay, the construct containing the core promoter of neuropilin-1 (pGL3-NRP1) (37) was used. As shown in Figure 7B and C, significant increase of luciferase activity was detected in the constructs containing U fragment (−1 to −182), U1 fragment (−1 to −391), U2 fragment (−375 to −1358) and U12 fragment (−1 to −1358) in both HuH7 cells and Hep3B cells but not in the U3 fragment (−1329 to −2221). The U fragment elicited strongest activation of 45-fold and 5-fold in HuH7 and Hep3B cells, respectively. The difference in induction is due to the poor transfection efficiency of Hep3B cells. Notably, none of the pri-mir-122 promoter constructs directed luciferase gene activity in HEK293T cells, suggesting a preferential activation of the pri-mir-122 promoter in the context of hepatocytes (Figure 7D). Within the core promoter region (−1 to −182), two TATA boxes were identified. We mutated each of the TATA boxes (Figure 8A) and measured luciferase activity following transfection to HuH7 cells. Mutation of TATA1 and TATA2 led to reductions of 20% and 44% of activity, respectively (Figure 8B). This result further confirmed the core promoter of pri-mir-122 gene.
Figure 7.

The activity of the pri-mir-122 promoter in cultured HCC cells. (A) Genomic segments (U1, U2, U3 and U12) of the upstream region were subcloned in pGL3-basic construct (grey bar) for promoter analysis in (B) HuH7, (C) Hep3B and in (D) 293T cells. The luciferase activity was measured 48 h after transfection. NRP1 promoter was used as a positive control. The transfection efficiency was normalized against pRL-TK activity. Normalized luciferase activity from triplicate samples is presented relative to that of cells transfected with the pGL3-basic construct (white bar). The experiment was repeated twice with same results. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 8.

Deletion analysis of the pri-mir-122 core promoter. (A) Schematic representation of the deletion mutants of TATA boxes, mTATA1 and mTATA2. (B) Luciferase activity of the promoter constructs transfected into HEK293T cells. Data shown represent three independent experiments The transfection efficiency was normalized against pRL-TK activity. Normalized luciferase activity from triplicate samples is presented relative to that of cells transfected with the pGL3-basic construct (white bar). *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
Precisely identifying miRNA TSSs is essential for facilitating the discovery of TF-miRNA regulatory relationships and for further elucidating the transcriptional regulation of miRNA expressions. Owing to this significance, an increasing number of investigations have attempted to identify the miRNA promoter by using either a computational or experimental approach. Although chromatin signature is normally used to locate miRNA promoters, numerous miRNA promoters still remain unclear (25–28). Rather than using a single TSS signature, miRStart successfully integrates three next-generation sequencing (NGS) datasets derived from TSS-related experiments to determine the TSS of human miRNAs. Additionally, a SVM-based model is developed to determine the TSS candidates for each miRNA gene systematically, thus providing users the most probable miRNA TSSs with experimental evidences for deciphering the transcriptional regulation of miRNA expressions.
Although miRStart identified most human miRNA TSSs, 92 intergenic miRNAs still have no putative TSS, which is attributed to the following reasons. First, their TSSs may be outside the 50 kb upstream regions from precursors. miRStart did not analyze the range beyond 50 kb because a previous study surveyed microarray expression profiles of 175 human miRNAs, indicating that two miRNAs <50 kb-long apart are co-expressed and share the common primary transcript (30). Second, although most intergenic miRNAs are transcribed by RNA polymerase II, Borchert et al. (20) found that Alu elements upstream of C19MC miRNAs retain sequences deemed essential for Pol III activity, concluding that RNA polymerase III can also transcribe human miRNAs. Interestingly, rather than Pol III transcription, a recent study demonstrated that C19MC miRNAs are processed from introns of large Pol II, non-protein-coding transcripts, thus contradicting the previous finding (21). In fact, miRStart failed to obtain putative TSSs for most of C19MC miRNAs because of sparse tag signals in their upstream regions. Evan if the TSSs are determined, the SVM scores are still very low, i.e. miR-515-1 and miR-517a. It reflects the limitation that derived from the 5′-end sequences after cap structures, CAGE tags and TSS Seq tags cannot detect TSS signals if miRNAs are merely transcribed by RNA polymerase III. Third, pri-miRNA has too low of a concentration for detection when performing NGS, for example, miR-187. Low-concentration pri-miRNAs lack a TSS-relevant signal unless the gene encoded Drosha is eliminated to obtain more primary transcripts of miRNAs for NGS. Finally, the TSS signals in the upstream region of miRNA precursors are not identified by the SVM model even if they are obvious, for example, miR-1179. According to the occurrence distribution of TSS evidence, the putative TSS of miR-1179 may be located 3000 bp upstream of its precursor (Supplementary Figure S6).
In addition to officially defining intragenic miRNA TSSs by the transcription initiation of their host genes, miRStart provides novel TSSs for intragenic miRNA genes because previous investigations indicated that several miRNAs do not share common promoters with their host genes due to their inconsistent expression patterns (26,28,45). Those studies further demonstrated that some intragenic miRNA genes may possess individual TSS based on the experimental TSS signals near the upstream of miRNA precursors. For instance, the TSS of miR-99a~let-7c is defined by their host gene C21orf34 (Genomic coordinates Chr21: 17442842 [+]) and is far from the miR-99a precursor (Genomic coordinates Chr21: 17911409-17911489 [+]). According to the occurrence distribution of TSS evidence in the upstream region of miR-99a precursor, CAGE tags and TSS Seq tags are aggregated at 17907551, where 5 ESTs are nearby and the conservation score is extremely high (Supplementary Figure S7). Rather than the TSS of C21orf34, the genomic site located 3858 bp upstream of the miR-99a precursor is most likely the genuine TSS of miR-99a~let-7c.
As is well known, CAGE tags and TSS Seq tags are obtained from various human tissues and cell lines. miRStart generally identifies miRNA TSSs based on the tag occurrence distributions to obtain the entire view of TSS candidates. To estimate how much of an effect of datasets from different tissues/cell lines has on the SVM classifier, we compared the output of the original SVM model with a tissue-specific SVM model. The most typical example is miR-122, a liver-specific miRNA significantly down-regulated in hepatocellular carcinoma (38). No matter what SVM models are used, the putative TSS of miR-122 is identified at the same genomic locus. Even if miR-122 is liver-specific, the original SVM model still performs well. However, the putative TSS selected by SVM model is more obvious and authentic if only liver-specific CAGE tags and TSS Seq tags distributed in the upstream region of miR-122 precursor are considered (Figure 5B). This is the reason why the function displaying specific CAGE tags or TSS Seq tags in miRStart was designed for users.
Based on the 5′RACE procedure and luciferase reporter assay, this work verifies the genuine miR-122 TSS located 4812 bp upstream of its precursor, which definitely matches the putative one. However, the TSS markedly differs from that in a previous study (27). Barski et al. indicated that the TSS of miR-122 gene is located at 56105891 (−12415 from precursor), whereas our TSS is at 56113494 (−4812 from precursor). Actually, that study identified miR-122 promoters by only using chromatin signitures from ChIP-seq data. In addition to using the chromatin signitures (H3K4me3), we also combine CAGE tags and TSS Seq tags to identify miR-122 TSS. This work also differs from Barski et al. in that the latter performed 5′RACE and promoter–reporter assay of miR-122 promoter using total CD4+ T cells and detected the acceptable luciferase activity. Nevertheless, miR-122 is a liver-specific miRNA and is not expressed in other tissues. Conversely, in this work, human HCC cell line, HuH7 was used to perform 5′RACE and luciferase reporter assay, subsequently obtaining a reliable and uncontroversial miR-122 TSS while satisfying promoter activity.
In conclusion, miRStart is a valuable resource for biologists in advanced research in miRNA-mediated regulatory networks. The main contribution of miRStart is to integrate three experimental-based datasets including CAGE tags, TSS Seq tags and H3K4me3 chromatin signature and define miRNA TSSs according to the distribution of tags derived from high-throughput sequencing analysis (because each tag represents a possible TSS signal). The SVM is a strategy to automatically identify putative miRNA TSSs instead of manually selecting by users. Limited to no relevant dataset derived from CAGE, TSS Seq, and ChIP-Seq (H3K4me3) is available for other organisms, miRStart can provide miRNA TSSs in the human genome currently. Although FANTOM4 has published the mouse CAGE datasets, DBTSS offers only one mouse TSS Seq dataset (mouse 3T3 solexa tag mapping data) in the database. It is inadequate to define miRNA TSSs for the SVM. We believe that more and more high-throughput sequencing data will be generated and render miRStart more complete in the near future.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Council of the Republic of China (Contract No. NSC 98-2311-B-009-004-MY3 and NSC 99-2627-B-009-003); UST-UCSD International Center of Excellence in Advanced Bio-engineering sponsored by the Taiwan National Science Council I-RiCE Program (NSC-99-2911-I-010-101, in part); MOE ATU (in part). Funding for open access charge: National Science Council of the Republic of China.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENT
Ted Knoy is appreciated for his editorial assistance.
REFERENCES
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 3.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 4.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 7.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 9.Papadopoulos GL, Reczko M, Simossis VA, Sethupathy P, Hatzigeorgiou AG. The database of experimentally supported targets: a functional update of TarBase. Nucleic Acids Res. 2009;37:D155–D158. doi: 10.1093/nar/gkn809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–D104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu SD, Lin F-M, Wu W-Y, Liang C, Huang W-C, Chan W-L, Tsai W-T, Chen G-Z, Lee C-J, Chiu C-M, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu X, Lin J, Zack DJ, Mendell JT, Qian J. Analysis of regulatory network topology reveals functionally distinct classes of microRNAs. Nucleic Acids Res. 2008;36:6494–6503. doi: 10.1093/nar/gkn712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandyopadhyay S, Bhattacharyya M. Analyzing miRNA co-expression networks to explore TF-miRNA regulation. BMC Bioinformatics. 2009;10:163. doi: 10.1186/1471-2105-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Re A, Cora D, Taverna D, Caselle M. Genome-wide survey of microRNA-transcription factor feed-forward regulatory circuits in human. Mol. Biosyst. 2009;5:854–867. doi: 10.1039/b900177h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Lu M, Qiu C, Cui Q. TransmiR: a transcription factor-microRNA regulation database. Nucleic Acids Res. 2010;38:D119–D122. doi: 10.1093/nar/gkp803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006;13:1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 21.Bortolin-Cavaille ML, Dance M, Weber M, Cavaille J. C19MC microRNAs are processed from introns of large Pol-II, non-protein-coding transcripts. Nucleic Acids Res. 2009;37:3464–3473. doi: 10.1093/nar/gkp205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saini HK, Griffiths-Jones S, Enright AJ. Genomic analysis of human microRNA transcripts. Proc. Natl Acad. Sci. USA. 2007;104:17719–17724. doi: 10.1073/pnas.0703890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou X, Ruan J, Wang G, Zhang W. Characterization and identification of microRNA core promoters in four model species. PLoS Comput. Biol. 2007;3:e37. doi: 10.1371/journal.pcbi.0030037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saini HK, Enright AJ, Griffiths-Jones S. Annotation of mammalian primary microRNAs. BMC Genomics. 2008;9:564. doi: 10.1186/1471-2164-9-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corcoran DL, Pandit KV, Gordon B, Bhattacharjee A, Kaminski N, Benos PV. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS ONE. 2009;4:e5279. doi: 10.1371/journal.pone.0005279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barski A, Jothi R, Cuddapah S, Cui K, Roh TY, Schones DE, Zhao K. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009;19:1742–1751. doi: 10.1101/gr.090951.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, Zhang X, Song JS, Fisher DE. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22:3172–3183. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flicek P, Aken BL, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Coates G, Fairley S, et al. Ensembl's 10th year. Nucleic Acids Res. 2010;38:D557–D562. doi: 10.1093/nar/gkp972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawaji H, Severin J, Lizio M, Waterhouse A, Katayama S, Irvine KM, Hume DA, Forrest AR, Suzuki H, Carninci P, et al. The FANTOM web resource: from mammalian transcriptional landscape to its dynamic regulation. Genome Biol. 2009;10:R40. doi: 10.1186/gb-2009-10-4-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamashita R, Wakaguri H, Sugano S, Suzuki Y, Nakai K. DBTSS provides a tissue specific dynamic view of Transcription Start Sites. Nucleic Acids Res. 2010;38:D98–D104. doi: 10.1093/nar/gkp1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. 2001 http://www.csie.ntu.edu.tw/~cjlin/libsvm (16 April 2011, date last accessed) [Google Scholar]
- 37.Liao YL, Sun YM, Chau GY, Chau YP, Lai TC, Wang JL, Horng JT, Hsiao M, Tsou AP. Identification of SOX4 target genes using phylogenetic footprinting-based prediction from expression microarrays suggests that overexpression of SOX4 potentiates metastasis in hepatocellular carcinoma. Oncogene. 2008;27:5578–5589. doi: 10.1038/onc.2008.168. [DOI] [PubMed] [Google Scholar]
- 38.Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, Lin CD, Liao YL, Wang JL, Chau YP, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571–1582. doi: 10.1002/hep.22806. [DOI] [PubMed] [Google Scholar]
- 39.Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl Acad. Sci. USA. 2003;100: 15776–15781. doi: 10.1073/pnas.2136655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D, Lu M, Miao J, Li T, Wang E, Cui Q. Cepred: predicting the co-expression patterns of the human intronic microRNAs with their host genes. PLoS ONE. 2009;4:e4421. doi: 10.1371/journal.pone.0004421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vapnik VN. The Nature of Statistical Learning Theory. 175 Fifth Avenue, New York, NY 10010, USA: Springer-Verlag New York, Inc.; 1995. [Google Scholar]
- 44.Burges CJC. A tutorial on support vector machines for pattern recognition. Data Mining Knowl. Discov. 1998;2:121–127. [Google Scholar]
- 45.Monteys AM, Spengler RM, Wan J, Tecedor L, Lennox KA, Xing Y, Davidson BL. Structure and activity of putative intronic miRNA promoters. RNA. 2010;16:495–505. doi: 10.1261/rna.1731910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki Y, Ishihara D, Sasaki M, Nakagawa H, Hata H, Tsunoda T, Watanabe M, Komatsu T, Ota T, Isogai T, et al. Statistical analysis of the 5' untranslated region of human mRNA using "Oligo-Capped" cDNA libraries. Genomics. 2000;64:286–297. doi: 10.1006/geno.2000.6076. [DOI] [PubMed] [Google Scholar]
- 47.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl Acad. Sci. USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujita S, Ito T, Mizutani T, Minoguchi S, Yamamichi N, Sakurai K, Iba H. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 2008;378:492–504. doi: 10.1016/j.jmb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 49.Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102–106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.