


Abstract
Post-transcriptional control of mRNA transcript processing by RNA binding proteins (RBPs) is an important step in the regulation of gene expression and protein production. The post-transcriptional regulatory network is similar in complexity to the transcriptional regulatory network and is thought to be organized in RNA regulons, coherent sets of functionally related mRNAs combinatorially regulated by common RBPs. We integrated genome-wide transcriptional and translational expression data in yeast with large-scale regulatory networks of transcription factor and RBP binding interactions to analyze the functional organization of post-transcriptional regulation and RNA regulons at a system level. We found that post-transcriptional feedback loops and mixed bifan motifs are overrepresented in the integrated regulatory network and control the coordinated translation of RNA regulons, manifested as clusters of functionally related mRNAs which are strongly coexpressed in the translatome data. These translatome clusters are more functionally coherent than transcriptome clusters and are expressed with higher mRNA and protein levels and less noise. Our results show how the post-transcriptional network is intertwined with the transcriptional network to regulate gene expression in a coordinated way and that the integration of heterogeneous genome-wide datasets allows to relate structure to function in regulatory networks at a system level.
INTRODUCTION
In response to environmental signals, cells regulate gene expression and protein production in a coordinated fashion at multiple levels, from transcription through translation and post-transcriptional control. Post-transcriptional control of mRNA transcript processing, export, localization, degradation and translation has traditionally recieved less attention than transcriptional control of mRNA production. However, recent genome-wide studies have shown that post-transcriptional regulation at a system level is no less complex or important than transcriptional regulation (1–4). To explain the limited correlation between mRNA and protein abundance (5), the theory of RNA regulons or operons has been proposed, according to which trans-acting factors combinatorially regulate multiple mRNAs to achieve functionally coordinated translation in the face of stochastic gene expression (2). While several examples of RNA regulons have been found at all levels of mRNA processing [reviewed in (2)], recent experimental breakthroughs in particular hint at extensive organization of post-transcriptional control into RNA regulons.
In Saccharomyces cerevisiae, mRNA targets were identified in a genome-wide fashion for a set of 40 RNA binding proteins (RBPs) (6), a major class of proteins mediating post-transcriptional control by binding to structural elements in the UTRs of target transcripts (7). It was found that many of the RNA binding proteins bind functionally related mRNAs and that most mRNAs associate with multiple RNA binding proteins (6). However, while clusters of mRNAs bound by the same RNA binding proteins potentially correspond to RNA regulons, it is unclear from these data alone if and how the network of RBP–mRNA regulatory interactions functions in coordination with the transcriptional regulatory network.
In order to link transcriptional to translational control, transcriptome and translatome expression profiles were measured in S. cerevisiae under the same six stress conditions (8). It was found that severe stress induces a highly coordinated transcriptional and translational response, while mild stress leads to a non-correlated response mainly affecting the translatome profiles (8). However, only global correlations between transcriptome and translatome profiles were analyzed. The involvement of RNA binding proteins in shaping these dynamic responses has remained unexplored and is considered an important challenge in the field (3).
A first study in this direction has shown that RNA binding proteins have distinct expression dynamics compared with other protein coding genes in S. cerevisiae (9). They are less stable at the transcript level and tightly controlled at the protein level. Furthermore, the connectivity of an RBP in the RBP–mRNA binding network correlates with its protein stability and abundance and anticorrelates with its expression noise. These results highlight the central role of RNA binding proteins in controlling gene expression at the post-transcriptional level (9).
In this study, we integrated genome-wide transcriptional and translational expression data (8) with networks of transcriptional (10) and post-transcriptional (6) regulatory interactions in yeast to characterize in a systematic and unbiased way the structural and functional organization of RNA regulons in the post-transcriptional regulatory network. We demonstrate how the post-transcriptional network is intertwined with the transcriptional network to regulate gene expression in a coordinated way and that the integration of heterogeneous sources of high-throughput, genome-wide data allows to relate structure to function in regulatory networks at a system level.
MATERIALS AND METHODS
Expression data
Normalized genome-wide expression profiles of total mRNA [transcriptome (TS)] and ribosome-bound mRNA [translatome (TL)] under six stress conditions with a total of 26 arrays (including controls) were obtained from Ref. (8). No further postprocessing was done on the data except normalizing each gene to mean zero and unit standard deviation.
Regulatory networks
The transcriptional regulatory network with targets for 198 transcription factors was obtained from ChIP-chip data (10), using a P-value cutoff of 0.005. The post-transcriptional regulatory network with targets for 40 RNA binding proteins was obtained from Ref. (6).
Data integration methodology
We developed a stepwise data integration methodology to integrate genome-wide data on transcriptional and translational regulation and to relate structure to function in the post-transcriptional regulatory network (Fig. 1). We first clustered the transcriptome and translatome expression profiles and reconstructed transcriptional and post-transcriptional regulatory networks from expression data using a previously developed module network inference method (11,12) (Figure 1A). Independently, we combined the transcription factor (TF) binding and RBP binding interactions and identified significantly enriched network motifs in the integrated network (Figure 1B). Finally, we mapped network motifs to the three classes of translatome clusters (Figure 1C). Details for each step in this procedure follow below.
Figure 1.

Methodology to integrate genome-wide data on transcriptional and translational regulation. (A) We combined transcriptome and translatome expression data to infer coexpression clusters and regulatory module networks. (B) We integrated known networks of transcriptional and post-transcriptional regulatory interactions and searched for overrepresented network motifs. (C) By mapping network motifs to distinct classes of expression clusters, we found that motifs explain translational coexpression in the absence of transcriptional coexpression and thus characterized the large-scale structural and functional properties of RNA regulons.
Clustering and module network inference
For each dataset, we generated 20 locally optimal clustering solutions, from which tight coexpression clusters were created (11). A list of candidate regulators for network inference was generated from the list of transcription factors and RNA binding proteins with known binding targets (see above), as only predictions for these regulators can be validated against known interactions. Module networks were inferred using LeMoNe (12) (software available at http://bioinformatics.psb.ugent.be/software/details/LeMoNe). LeMoNe predicted a ranked list of regulators for each cluster, based on an ensemble of 10 regulatory program trees with maximum one level per cluster built from five different experiment partitions and significant regulators sampled from 100 candidate regulator-split value pairs for each tree node. Three regulatory programs were generated: TS regulators → TS clusters, where TS profiles of regulators were assigned to TS clusters; TS regulators → TL clusters; and TL regulators → TL clusters.
Translatome cluster classification
We define a measure called Relative Adjacency Score (RAS) for each translatome cluster to assess its expression coherence at transcriptional level. RAS is the ratio of the total adjacency score in TS data and total adjacency score in TL data for a given translatome cluster. The adjacency score Aij for a pair of genes (i, j) is the probability of observing genes i and j clustered together in the ensemble of equivalent clustering solutions, and hence
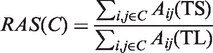 |
(1) |
is the Relative Adjacency Score for cluster C. If this value is close to zero, the genes in cluster C are coexpressed in translatome but not in transcriptome data, and if it is close to 1 then the cluster is also coexpressed in transcriptome data. We defined transcriptionally coregulated clusters (Class I) to be the ones with RAS ≥0.5, dually regulated clusters (Class II) with 0.1 < RAS < 0.5 and translationally coregulated clusters (Class III) with RAS ≤ 0.1.
Network motif calculations
Enriched network motifs in the transcriptional and post-transcriptional network were calculated using the FANMOD algorithm (13), keeping all motifs with P < 0.05 (five motifs, see Supplementary Figure S3). We refer to these motifs respectively as the post-transcriptional feedback loop (ID46), the transcriptional feedforward loop (ID38), the post-transcriptional coregulatory motif (ID6), the RBP-TF cascade (ID12) and the mixed bifan (ID36). The overrepresentation of network motifs in Classes I, II and III clusters was calculated using a hypergeometric test.
Large-scale experimental validation datasets
Functional categories, literature curated protein complexes and cellular localization information, obtained from MIPS (ftp://ftpmips.gsf.de/yeast/catalogues/), were used for assessing functional coherence using a hypergeometric test. Genome-wide data on mRNA level (14,15), protein abundance (16) and protein noise (17) were obtained from the respective publications. To assess whether or not Class I, II and III genes exhibit significant differences with respect to these genome-wide datasets, we used Wilcoxon rank-sum test to compare each pair (Class I versus II, Class II versus III and Class I versus III).
Supplementary website
We created a supplementary website for this article with expression profiles, coclustering figures and functional information for all clusters. This website serves as a resource for anyone wishing to derive detailed hypotheses from our large-scale analysis. The URL is http://omics.frias.uni-freiburg.de/RNAregulon.
RESULTS
Translatome clusters are functionally more coherent than transcriptome clusters
We clustered a transcriptome and translatome dataset with expression profiles measured under the same six stress conditions (8) using a Gibbs sampling two-way clustering procedure (11) (see ‘Materials and Methods’ section for details). The algorithm identified more clusters for a higher total number of genes from translatome data than from transcriptome data (Table 1). A higher fraction of translatome clusters is enriched with respect to MIPS functional categories (Table 1), and moreover they are more functionally coherent (lower P-values) than transcriptome clusters (Figure 2A).
Table 1.
Transcriptome versus translatome clustering statistics
| Transcriptome | Translatome | |
|---|---|---|
| Clustered genes | 1568 | 1924 |
| Clusters | 94 | 112 |
| Enriched clusters (%) | 69 (73) | 91 (81) |
‘Enriched clusters’ is the number of clusters functionally enriched with P < 0.001.
Figure 2.

At different P-value cutoffs (x-axis), the total number of unique MIPS functional categories (A) and cellular compartments (B) functionally enriched in transcriptome clusters (+) and translatome clusters (⋆). At different P-value cutoffs, the total number of clusters enriched for known RBP targets (C) and known TF targets (D) in transcriptome clusters (+) and translatome clusters (⋆).
RNA binding proteins play important roles in the subcellular localization and efficient assembly of protein complexes and functional systems by ensuring that the location in the cell at which mRNAs are translated is not left to chance (18). Genes co-localized in the same cellular compartment and/or belonging to the same protein complex indeed cluster together more often in translatome clusters than transcriptome clusters (Figure 2B and Supplementary Figure S1). Translatome clusters are also better enriched for RBP targets (Figure 2C) as well as TF targets (Figure 2D). We conclude that translatome expression profiles are more biologically informative in every respect than transcriptome expression profiles obtained under the same experimental setup.
Transcriptional and post-transcriptional network inference performs better with translatome than transcriptome data
Expression profiles measured under diverse experimental conditions are commonly used to reconstruct regulatory networks (19). We considered known TFs and RNA binding proteins as candidate regulators to infer transcriptional and post-transcriptional regulatory networks from translatome and transcriptome data. We used a previously developed module network inference method (12) to assign transcriptome profiles of regulators to transcriptome clusters, transcriptome profiles of regulators to translatome clusters and translatome profiles of regulators to translatome clusters (see ‘Materials and Methods’ section for details). We validated the inferred networks against known TF (10) and RBP (6) binding data.
Translatome clusters are more functionally coherent and more enriched for TF and RBP targets (Figure 2) and consequently the overlap between the predicted and known networks is higher for translatome clusters, for the transcriptional as well as the post-transcriptional regulatory network (Table 2, columns 2 and 3 versus column 1). For the transcriptional regulatory network (Table 2, row 1), an additional set of true positives comes from the fact that the transcriptome profiles of some transcription factors match the translatome profiles rather than the transcriptome profiles of their targets. The underlying regulatory mechanism in this case is not evident as the known transcriptional and post-transcriptional networks are largely incomplete.
Table 2.
Overlap between predicted and true transcriptional (TRN) and post-transcriptional (PRN) regulatory networks using various combinations of (TS) and translatome (TL) data, given by the F-measure and the total number of true positives (in braces)
| TS regulators | TS regulators | TL regulators | |
|---|---|---|---|
| ↓ | ↓ | ↓ | |
| TS clusters | TL clusters | TL clusters | |
| TRN | 0.0084 (119) | 0.0110 (182) | 0.0099 (163) |
| PRN | 0.0084 (109) | 0.0167 (255) | 0.0252 (384) |
For the post-transcriptional, but not the transcriptional, regulatory network, we observe a further improvement in performance by assigning translatome instead of transcriptome profiles of regulators to translatome clusters (Table 2, column 3 versus column 2). This suggests that RNA binding proteins are themselves more often post-transcriptionally regulated, in agreement with previous results (9) and with the fact that RBPs have a significantly higher in-degree than randomly selected nodes in the real post-transcriptional regulatory network (P < 10−11). In contrast, transcription factors are not significantly more often regulated at post-transcriptional level. Moreover, in the real regulatory networks, 41% of the RBPs associate with their own mRNA, but only 6% of TFs bind to their own promoter, suggesting extensive autoregulation at the post-transcriptional level (20). Several functionally important examples of autoregulation of post-transcriptional regulators such as microRNAs, splicing factors, etc. have been found in other systems as well (4).
The results in Table 2 can be illustrated with some specific examples. Translatome cluster 41 and transcriptome cluster 33 are both enriched for MET32 targets, but translatome cluster 41 has four known targets while transcriptome cluster 33 has two. Hence, although LeMoNe predicted MET32 correctly as regulator for both clusters, the translatome cluster gives rise to a higher number of true positives. Translatome cluster 98 contains four genes, PHO11, PHO12, SPL2 and VTC3, which are also part of transcriptome cluster 54. Two of these genes are known targets of the transcription factor ARO80. ARO80 is predicted as the second best regulator for translatome cluster 98, but not predicted for transcriptome cluster 54. Translatome clusters 2, 5 and 60 are enriched for ribosomal proteins, most of which are targets of the RNA binding protein PAB1. Only the translatome profile, and not the transcriptome profile, of PAB1 matches with its targets.
Translatome profiles are less noisy and more coherent than corresponding transcriptome profiles
Next we asked whether the increased functional coherence of translatome data compared with transcriptome data as observed in the previous sections is an overall effect over all clusters or can be attributed to individual clusters enriched for specific functional categories. Translatome clusters emerge due to the convolution of transcriptional and post-transcriptional regulatory signals. Hence, if two genes share coexpression in translatome but not transcriptome data, we hypothesize that they are under active post-transcriptional control, while if they are coexpressed in transcriptome as well as translatome data, we hypothesize that they are regulated mainly at transcriptional level. To characterize the extent to which transcriptional and post-transcriptional signals can be decoupled, we defined a metric called RAS as the ratio of transcriptional to post-transcriptional expression coherence (see ‘Materials and Methods’ section for details). A RAS of value 1 indicates that the genes in a given translatome cluster cocluster perfectly in the transcriptome data as well. The lower the RAS, the less the transcriptional coherence of that translatome cluster.
Based on the RAS values, translatome clusters can be partitioned into three classes with distinct regulatory patterns (see Figure 3 and ‘Materials and Methods’ section for details). Genes in Class I clusters (high RAS) cocluster in transcriptome and translatome data and are mainly regulated at the transcriptional level. Genes in Class III clusters (low RAS) cocluster only in translatome data and are mainly regulated at post-transcriptional level. Class II clusters have intermediate RAS values and are dually regulated. We hypothesize that post-transcriptional regulation filters noise from weakly coexpressed but transcriptionally coregulated genes (Class II clusters) and equalizes expression profiles of genes not coregulated at transcriptional level (Class III clusters) and thus results in a more informative translatome output signal.
Figure 3.

Translatome clusters are classified in three classes using a RAS, which compares the sum of coclustering frequencies Ai,j (cfr. color scale) between genes in translatome data (upper triangular part in each matrix) and transcriptome data (lower triangular part) for a given translatome cluster. Missing values (white lower triangular entries) indicate that those gene pairs never clustered together in the transcriptome data.
Class I consists of transcriptionally coregulated translatome clusters and contains nine clusters. These clusters are functionally enriched for basic metabolic processes such as catabolism and anabolism of sugars, metabolism of vitamins and energy reserves (Supplementary Data) and they are enriched in genes located in extracellular space and vacuole. As an example, translatome cluster 78 contains four genes involved in the galactose pathway GAL1, GAL2, GAL7 and GAL10, and is identical to transcriptome cluster 75. It is overrepresented for GAL4 targets, but not for any RBP targets and thus is likely to be regulated only at transcriptional level.
Class II consists of dually controlled or combinatorially regulated translatome clusters and contains 42 clusters. We mapped the clusters in this class to corresponding transcriptome clusters with a significant overlap in gene content (P < 0.001). In line with our previous results, translatome clusters are functionally more coherent than their corresponding transcriptome clusters, suggesting that post-transcriptional regulation fine-tunes the transcriptional output signal by filtering noise from the expression profiles of transcriptionally coregulated genes (tighter coexpression in translatome than trascriptome data). For instance, translatome cluster 146 maps to transcriptome cluster 56. Both clusters are involved in sugar transport, but the genes in translatome cluster 146 are only weakly coexpressed in transcriptome data compared with translatome data (Figure 3). Translatome cluster 146 overlaps significantly with the targets of the RNA binding protein VTS1 (P < 10−6). Although this overlap concerns only 3 of 122 known VTS1 targets, regulation of translatome cluster 46 by VTS1 is functionally supported by the fact that VTS1 is one of the predicted regulators by LeMoNe for this cluster.
Class III consists of translatome clusters which are coregulated predominantly at the post-transcriptional level and contains 61 clusters. The genes in these clusters belong to protein synthesis, translational elongation, RNA binding and ATP binding functional categories and are preferentially localized in endoplasmic reticulum (ER) and nucleus (Supplementary Data). For instance, translatome cluster 24 is enriched for translational elongation and for targets of the RNA binding protein SCP160, but the genes in cluster 24 do not cluster together at the transcriptional level (Supplementary Figure S2). Several protein complexes, such as the SAGA complex, 20S proteasome and nucleosomal complex, map to this class of clusters (Supplementary Data) and are thus found to be preferentially coregulated at post-transcriptional level.
Analysis of additional genome-wide data on mRNA level (14, 15), protein abundance (16) and protein noise (17) in yeast yielded further support for the classification of translatome clusters (Table 3). We found that combinatorially coregulated clusters (Class II) are significantly higher expressed at the mRNA and protein level, while purely transcriptionally coregulated clusters (Class I) are expressed with significantly higher noise. This is consistent with previous results that noisy genes (Class I) are typically enriched for energy production and response to environmental changes (17,21) while quiet genes (Classes II and III) are more generally enriched for RNA binding and ribosomal proteins (9,21). Moreover, Class I (membrane, vacuole) and Class III (Golgi, nucleus) genes are enriched for different cellular locations in agreement with previously described relationships between noise and cellular location (17). The average number of TFs or RNA binding proteins regulating each cluster is not significantly different between the three classes and thus cannot explain the variation in protein expression (Table 3).
Table 3.
Median values of diverse parameters with respect to three classes of translatome clusters based on their RAS score
| Class I | Class II | Class III | |
|---|---|---|---|
| Number of TFs | 2.09 | 2.10 | 1.95 |
| Number of RBPs | 1.44 | 1.85 | 1.64 |
| mRNA level (1) | 1.18 | 1.72 | 1.32 |
| mRNA level (2) | 1.18 | 2.81 | 1.72 |
| Protein noise | 23.52 | 17.27 | 18.46 |
| Protein abundance | 3190 | 5670 | 2605 |
Post-transcriptional network motifs associate with Classes II and III translatome clusters
Since the number of known regulators does not distinguish between the three classes of translatome clusters, we turned our attention to network motifs, small wiring patterns occuring significantly more often than expected by chance (22). We searched for all three-node motifs in the integrated network of known transcriptional and post-transcriptional regulatory interactions using the FANMOD algorithm (13) and found five statistically significant overrepresented motifs (see ‘Materials and Methods’ section and Supplementary Figure S3). We then mapped network motifs to translatome expression clusters if a significant fraction (P < 0.001) of genes in a cluster is regulated by a certain motif and computed the overlap between the number of clusters regulated by each motif and the number of clusters in each class. Two motifs involving post-transcriptional regulatory interactions are of most interest (Figure 4).
Figure 4.

Statistically overrepresented post-transcriptional regulatory network motifs found in the integrated network of transcriptional (dashed edges) and post-transcriptional (full edges) regulatory interactions, with an example of a higher order motif cluster. (A) Post-transcriptional FBL; (B) mixed bifan.
The first one is a feedback loop (FBL) where two RNA binding proteins regulate each other and a set of common targets (Figure 4A). This motif is not overrepresented in transcriptional networks, but it has been found before in the form of a composite FBL between miRNAs and TFs in C. elegans (23) and human data (24), and important examples of feedback loops between post-transcriptional regulators in human have been studied as well (4). Feedback in regulatory networks plays an important role in noise reduction (25,26). Accordingly, post-transcriptional FBLs mainly regulate Class II and III clusters (Table 4, row 5). As an example, SCP160 and BFR1 regulate each other and translatome cluster 7, whose genes are involved in rRNA processing and RNA binding (Figure 4A). Moreover, we found that genes regulated by post-transcriptional FBLs have higher mRNA and protein expression with lower noise (Supplementary Table S2). These genes are overrepresented for rRNA processing, ribosomal proteins and translational initiation. RNA binding proteins themselves are also known to be among the genes with lowest expression noise (9,21). The RNA binding proteins involved in FBLs have a siginificantly higher in- and out-degree than other RNA binding proteins, a property that is associated with having a higher information flow (23).
Table 4.
Number of clusters for each translatome cluster class regulated by a given network motif
| Class I | Class II | Class III | |
|---|---|---|---|
| TF direct | 7 | 33 | 43 |
| RBP direct | 4 | 28 | 38 |
| RBP–TF cascade | 4 | 24 | 25 |
| TF FFL | 1 | 12 | 11 |
| RBP FBL | 0 | 5 | 9 |
| Mixed Bifan | 3 | 23 | 27 |
The bold numbers are significantly lower than expected by chance and TF/RBP direct refers to clusters significantly overlapping with the targets of a TF/RBP.
The second motif of interest is a composite network motif, namely the mixed bifan (Figure 4B). This agrees with the result that most translatome clusters are dually regulated (Classes II and III; Table 4, row 6), such as the transcription factor RGM1 and the RNA binding protein PUB1 together regulate multiple COS genes (Figure 4B). Other examples are translatome clusters 2, 5, 16 and 31 which are all overrepresented for ribosomal proteins and are all regulated by two TFs, FHL1 and RAP1, and one RNA binding protein, PAB1. FHL1 is known to be involved in the expression of ribosomal protein genes (27). PAB1 is a general RBP that promotes translation initiation and interacts with CRM1, a major karyopherin, involved in export of proteins, RNAs, and ribosomal subunits from the nucleus and is required for efficient mRNA export to cytoplasm (28).
We further observed that Class I clusters are significantly less often than expected by chance regulated by the transcriptional feedforward loop (FFL) (Table 4, row 4), a well-known network motif which has been found in all transcriptional regulatory networks studied to date, including a network of TF binding interactions in yeast (29). Since this motif does not contain any post-transcriptional links, the mechanism of association to translatome clusters is unclear. We hypothesize that Class I clusters, which are strongly coexpressed, are transcriptionally regulated in a very simple manner and therefore devoid of more complex regulatory motifs like the FFL.
The assigment of the two remaining motifs, the post-transcriptional coregulatory motif and the RBP-TF cascade (cfr. ‘Materials and Methods’ section and Supplementary Figure S3) did not differ significantly between classes (Table 4, rows 2 and 3).
In summary, Class I clusters are significantly devoid of any regulation through network motifs (Table 4, column 1). These clusters are expressed with significantly higher noise (Table 3), suggesting that network motifs might be important in filtering noise. Classes II and III clusters often overlap with known targets of RNA binding proteins and mixed bifans (Table 3) and are thus regulated mainly at post-transcriptional level, in agreement with the results derived from expression data.
DISCUSSION
The RNA regulon theory posits that post-transcriptional regulators such as RNA binding proteins combinatorially regulate mRNA stability and translation, compensating for transcriptional noise and guaranteeing coordinated translation of functionally related mRNA molecules. Here, we performed a systematic computational validation of RNA regulon theory, by integrating genome-wide datasets on transcriptional and translational regulation in yeast.
Essential for our study was the availability of a dataset measuring transcriptome and translatome expression data under the same six stress conditions. This enabled a direct functional comparison between the two levels of expression to assess the influence of post-transcriptional regulation on gene expression. We found that translatome expression profiles are more informative and functionally coherent than transcriptome profiles measured under the same experimental setup. Based on this observation, we classified translatome coexpression clusters into distinct groups according to their level of coexpression in the transcriptome data. We hypothesize that translatome clusters which also cluster in transcriptome data are regulated at transcriptional level only. Such clusters are indeed rarely enriched for known RBP targets and are expressed with significantly higher noise levels. Post-transcriptional regulation manifests itself in translatome clusters which are not coexpressed in transcriptome data. This is supported by the fact that genes in such clusters are expressed with higher mRNA and protein levels and less noise. RNA binding proteins, ATP binding proteins, ribosomal proteins and several protein complexes are typical examples of post-transcriptionally regulated clusters.
It is known that coexpression clusters derived from transcriptome data often overlap with TF binding target sets. Likewise, we assessed the overlap of transcriptome and translatome clusters with TF and RBP binding targets. Although the binding data were measured in a single condition (rich media) that is different from the stress conditions in which expression was measured, we found a significant overlap between expression clusters and TF and RBP binding targets. However, this overlap did not explain the classification of translatome clusters. Hence, we searched for overrepresented network motifs in the integrated network of transcriptional and post-transcriptional regulatory interactions. We found that translatome clusters which are not coexpressed at transcriptional level are more often regulated by post-transcriptional FBLs, known to suppress noise in regulatory networks, and mixed transcriptional – post-transcriptional bifan motifs. In view of the RNA regulon theory, the enrichment of the mixed bifan and its association to a well-defined class of translatome clusters is particularly interesting. It shows that RNA binding proteins often partner with specific TFs to filter noise and equalize expression profiles of transcriptionally coregulated, but only weakly coexpressed genes.
Our results provide a first genome-wide view on how the transcriptional and post-transcriptional regulatory network are intertwined to regulate gene expression in a coordinated way, but this view is necessarily incomplete. In particular, the number of available translatome expression profiles is small compared with the tens to hundreds of transcriptome profiles for different treatments, genetic perturbations or time courses that are normally used for cluster and network inference analysis. As a result, many processes in the cell may be poorly or not represented by the current set of traslatome clusters. Furthermore, binding data are only available for 40 of possibly several hundreds of RBPs, and only in a single condition, such that many potential overlaps between translatome expression clusters and post-transcriptional interactions are not captured by the current datasets. Despite these limitations, our results clearly show that translatome expression data are better suited for tasks such as clustering or network inference than transcriptome data and that the integration of diverse genome-wide datasets allows to relate structural to functional information on the post-transcriptional regulation of many biological processes.
Finally, we note that a top–down systems analysis such as presented here does not in itself establish detailed biological mechanisms. Rather it should be seen as an unbiased generator of novel hypotheses, statistically supported by high-throughput data, for molecular biologists working on a specific regulatory pathway or process. To facilitate such follow-up analysis, we have created a Supplementary website with easy access to all our results, which will be upgraded as more translatome and RNA binding protein interaction data becomes available.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
IWT (SBO-BioFrame); IUAP P6/25 (BioMaGNet); Ghent University (Multi-disciplinary Research Partnership ‘Bioinformatics: from nucleotides to networks’). Funding for open access charge: Freiburg Institute for Advanced Studies.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Mata J, Marguerat S, Bähler J. Post-transcriptional control of gene expression: a genome-wide perspective. Trends Biochem Sci. 2005;30:506–514. doi: 10.1016/j.tibs.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Keene JD. RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 3.Halbeisen RE, Galgano A, Scherrer T, Gerber AP. Post-transcriptional gene regulation: from genome-wide studies to principles. Cell Mol. Life Sci. 2008;65:798–813. doi: 10.1007/s00018-007-7447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kishore S, Luber S, Zavolan M. Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief. Func. Gen. 2010;9:391–404. doi: 10.1093/bfgp/elq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotech. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 6.Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255. doi: 10.1371/journal.pbio.0060255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halbeisen RE, Gerber AP. Stress-dependent coordination of transcriptome and translatome in yeast. PLoS Biol. 2009;7:e105. doi: 10.1371/journal.pbio.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mittal N, Roy N, Madan Babu M, Janga SC. Dissecting the expression dynamics of RNA-binding proteins in post-transcriptional regulatory networks. Proc. Natl Acad. Sci. USA. 2009;106:20300–20305. doi: 10.1073/pnas.0906940106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi A, Van de Peer Y, Michoel T. Analysis of a Gibbs sampler for model based clustering of gene expression data. Bioinformatics. 2008;24:176–183. doi: 10.1093/bioinformatics/btm562. [DOI] [PubMed] [Google Scholar]
- 12.Joshi A, De Smet R, Marchal K, Van de Peer Y, Michoel T. Module networks revisited: computational assessment and prioritization of model predictions. Bioinformatics. 2009;25:490–496. doi: 10.1093/bioinformatics/btn658. [DOI] [PubMed] [Google Scholar]
- 13.Wernicke S, Rasche F. FANMOD: a tool for fast network motif detection. Bioinformatics. 2006;22:1152–1153. doi: 10.1093/bioinformatics/btl038. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc. Natl Acad. Sci USA. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miura F, Kawaguchi N, Yoshida M, Uematsu C, Kito K, Sakaki Y, Ito T. Absolute quantification of the budding yeast transcriptome by means of competitive PCR between genomic and complementary DNAs. BMC Genomics. 2008;9:574. doi: 10.1186/1471-2164-9-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghaemmaghami S, Huh W-K, Bowel K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 17.Newman JRS, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 18.Gerber AP, Herschlag D, Brown PO. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2004;2:E79. doi: 10.1371/journal.pbio.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;308:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- 20.Mittal N, Scherrer T, Gerber AP, Chandra Janga S. Interplay between post-transcriptional and post-transcriptional interactions of RNA-binding proteins. J. Mol. Biol. 2011;409:466–479. doi: 10.1016/j.jmb.2011.03.064. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Min R, Vizeacoumar FJ, Jin K, Xin X, Zhang Z. Exploiting the determinants of stochastic gene expression in Saccharomyces cerevisiae for genome-wide prediction of expression noise. Proc. Natl Acad. Sci. USA. 2010;107:10472–10477. doi: 10.1073/pnas.0914302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 23.Martinez NJ, Ow MC, Barrasa MI, Hammell M, Sequerra R, Doucette-Stamm L, Roth FP, Ambros VR, Walhout AJ. A C. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev. 2008;22:2535–2549. doi: 10.1101/gad.1678608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architexture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hornung G, Barkai N. Noise propagation and signaling sensitivity in biological networks: a role for positive feedback. PLoS Comput. Biol. 2008;4:e8. doi: 10.1371/journal.pcbi.0040008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruggeman FJ, Blüthgen N, Westerhoff HV. Noise management by molecular networks. PLoS Comput. Biol. 2009;5:e1000506. doi: 10.1371/journal.pcbi.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inoki K, Guan KL. Complexity of the TOR signaling network. Trends Cell Biol. 2006;16:206–212. doi: 10.1016/j.tcb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Brune C, Munchel SE, Fischer N, Podtelejnikov AV, Weis K. Yeast poly(A)-binding protein Pab1 shuttles between the nucleus and the cytoplasm and functions in mRNA export. RNA. 2005;11:517–531. doi: 10.1261/rna.7291205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.