


Abstract
A key challenge for the academic and biopharmaceutical communities is the rapid and scalable production of recombinant proteins for supporting downstream applications ranging from therapeutic trials to structural genomics efforts. Here, we describe a novel system for the production of recombinant mammalian proteins, including immune receptors, cytokines and antibodies, in a human cell line culture system, often requiring <3 weeks to achieve stable, high-level expression: Daedalus. The inclusion of minimized ubiquitous chromatin opening elements in the transduction vectors is key for preventing genomic silencing and maintaining the stability of decigram levels of expression. This system can bypass the tedious and time-consuming steps of conventional protein production methods by employing the secretion pathway of serum-free adapted human suspension cell lines, such as 293 Freestyle. Using optimized lentiviral vectors, yields of 20–100 mg/l of correctly folded and post-translationally modified, endotoxin-free protein of up to ~70 kDa in size, can be achieved in conventional, small-scale (100 ml) culture. At these yields, most proteins can be purified using a single size-exclusion chromatography step, immediately appropriate for use in structural, biophysical or therapeutic applications.
INTRODUCTION
The ability to express milligram-to-gram quantities of highly purified, recombinant proteins has become essential to support cutting-edge therapeutic and structural genomics efforts. Available protein production platforms are commonly based on expression in bacterial, yeast, insect and, more recently, mammalian cell culture systems. Systems based on recombinant expression in Escherichia coli, one of the oldest and most widely used expression platforms, allows for the simple, rapid and cost-effective production of target proteins. However, eukaryotic proteins produced in bacteria often suffer from poor solubility, resulting in aggregation or misfolding, or lack proper post-translational modifications necessary for full biological activity (1,2). Yeast-based expression systems, particularly those employing Saccharomyces cerevisiae and Pichia pastoris, offer improvements over bacterial systems in terms of yield, the complexity of proteins successfully expressed and the ability to perform some (but not all) post-translational modifications (3,4). Baculovirus-based or stably transformed insect cell systems using Drosophila (S2) or Spodoptera frugiperda (SF9) derived cell lines have become widely used for routine production of complex recombinant proteins. However, insect cell line-based platforms can be cumbersome to implement and do not correctly recapitulate complex mammalian N-glycans containing galactose or sialic acid residues (5,6).
For these reasons, most recombinant proteins for biomedical use have been produced in mammalian cell lines, such as Chinese hamster ovary (CHO) or human embryonic kidney (HEK) lines. Up to 70% of the recombinant proteins produced commercially are made in CHO cells, including many successful therapeutic biologics (e.g. Etanercept, Trastuzumab and Rituximab) (7). However, CHO-based expression systems may not be ideal for therapeutic protein production due to the addition of terminal galactose-α-1,3-galactose epitopes during N-glycosylation of recombinant glycoproteins. This antigen can be responsible for allergic hypersensitivities leading to adverse clinical events such as anaphylaxis, as seen in the use of the anti-cancer antibody Cetuximab (8). Another major drawback of using mammalian cell line-based platforms is the frequent need to laboriously select stable, high expressing clones which can be tedious, time consuming (often taking months) and costly.
Purification of cytoplasmically targeted recombinant proteins requires cell lysis after culturing to high densities, an inherently piecemeal approach. However, stable transduction of recombinant proteins targeted to the secretion pathway generates producer lines that can be consistently maintained in culture, allowing the protein to be harvested periodically and purified directly from culture supernatants over a period of time. Secretion may also improve the expression of proteins that are toxic to the producer line when produced as cytoplasmically targeted constructs. Recent advances in developing cell lines adapted to serum-free media synergizes with this approach by minimizing serum protein contaminants, dramatically simplifying purification. Indeed, most recombinantly expressed therapeutic proteins, such as cytokines and antibodies, are secreted proteins in their native state.
Introduction of vectors encoding recombinant protein constructs for large-scale protein production in transient expression systems is often accomplished with costly and sometimes unreliable chemical transfection reagents. Alternatively, lentiviral vectors can be used to efficiently transduce cells yielding stable transductants (9,10). However, limitations of lentiviral vectors include constrained packaging size (lentiviral particles are capable of packaging only about 10 kb of DNA efficiently) and genomic silencing of integrated lentiviral transgenes, seen both in vitro and in vivo (11,12). Ubiquitous chromatin opening elements (UCOEs) have shown promise in maintaining high levels of protein expression over extended periods of time in in vitro transfection settings (13,14). UCOEs have also been incorporated into lentiviral vectors in order to prolong the expression of integrated lentiviral transgenes in gene therapy settings (15,16). The smallest element tested in these settings was a 2.0-kb fragment of the HNRPA2B1/CBX3 locus (UCOE2.0); however, given the size constraints of lentiviral packaging, the use of this fragment significantly limits the size of recombinant protein constructs that can be expressed.
Initially to address our own particular need to express a natively glycosylated mammalian protein (Siderocalin) for crystallographic studies, but also to more generally address the limitations of current technologies, we report the development of a generalizable protein expression platform using lentiviral transduction of serum-free adapted human 293 Freestyle (293-F) cells. This system, designated ‘Daedalus’, is capable of producing large quantities of readily purifiable, secreted recombinant protein in a rapid manner. To enhance this system, we have engineered a novel 0.7-kb UCOE fragment (UCOE0.7) that, when incorporated into our lentiviral vector, leads to the stable and enhanced expression of recombinant proteins of sizes approaching 70 kDa. In addition, we utilize a cis-linked fluorescent reporter (GFP) driven by an internal ribosome entry site (IRES) that allows for rapid detection of transduced populations, tracking relative protein expression levels, and obviates time-consuming steps required for drug selection and isolation of high expressing clones. Of the 14 proteins tested with the Daedalus system, 12 were successfully expressed at levels between 20 and 100 mg/l in conventional 100 ml-scale cultures and readily purified via a single chromatography step for use in multiple applications.
MATERIALS AND METHODS
Cell culture
293 Freestyle cells (Invitrogen) were grown in Freestyle 293 Expression media (Gibco) with shaking at 130 rpm, at 37°C and 8% CO2 in vented 125-ml shake flasks (Nalgene). CHO-S cells (Invitrogen) were grown in CHO Expression media (Gibco) under the same conditions.
Lentiviral vector constructs
Lentiviral vector, pCVL-A (Figure 1A), was constructed from pRRL-SIN-cPPT-PGK-GFP-WPRE (Addgene# 12252) by replacing the WPRE element with a WPRE-O fragment (17), inserting two copies of a polyadenylation (polyA) enhancer (18) into the 3′ SIN LTR, and replacing the native U5-polyA signal with the bovine growth hormone (BGH) polyA signal sequence. The PGK promoter was replaced with the spleen focus forming virus (SFFV) promoter and the GFP sequence was replaced with an IRES-eGFP sequence. The murine Siderocalin sequence insert was isolated by PCR amplification of the expression construct used in E. coli with primers including Xho I and Bam HI sites plus a C-terminal Strep-tag II (SAWSHPQFEK) purification tag (19) and subcloned into the unique Xho I and Bam HI restriction sites in the pCVL-A vector; the resulting construct was designated pCVL-SFFV-muScn-IRES-GFP. The UCOE fragment within the human HNRPA2B1/CBX3 housekeeping gene locus (15) was amplified from Nalm 6 genomic DNA [using primers: UCOE5′Nhe I—GCTAGCggatcctggtacctaaaacagc and UCOE3′Sal I—GTCGACagtcgcttcagcccg (anti-sense)], TA-cloned and verified by sequencing and restriction mapping. The 2.0-kb Acc 65/Xba I fragment of the UCOE construct was blunted using Klenow (NEB) and ligated into Mlu I digested/blunted pCVL-SFFV-muScn-IRES-GFP upstream of the SFFV promoter. The forward orientation of the UCOE was confirmed by restriction mapping; the derived construct was designated pCVL-UCOE2.0-SFFV-muScn-IRES-GFP. A UCOE0.7 fragment (containing the CBX3 promoter of UCOE2.0) was constructed by digesting the UCOE2.0 fragment with Mlu I (internal) and Eco RI, the SFFV promoter was digested with Eco RI and Xho I and both fragments were cloned into the pCVL-SFFV-muScn-IRES-GFP vector (digested with Mlu I and Xho I) via a three-part ligation reaction. The final construct was designated pCVL-UCOE0.7-SFFV-muScn-IRES-GFP. All subsequent candidate protein coding sequences were either amplified with PCR primers (available upon request) containing the required N-terminal signal peptide and a C-terminal STREP-tag II sequence, or gene synthesized (with codon utilization optimized for human expression) and cloned into pCVL-UCOE0.7-SFFV-muScn-IRES-GFP using the unique Xho I and Bam HI restriction sites, replacing the muScn sequence. All restriction enzymes were FastDigest, purchased from Fermentas.
Figure 1.

Maps of the lentiviral vectors used. Schematics are shown of (A) lentiviral construct pCVL-A, (B) non-UCOE containing construct pCVL-SFFV-muScn-IRES-GFP and (C) minimized UCOE containing construct pCVL-UCOE0.7-SFFV-muScn-IRES-GFP. SP, signal peptide; UCOE, ubiquitous chromatin opening element; SFFV, spleen focus-forming virus enhancer/promoter; muScn, murine Siderocalin; IRES, internal ribosome entry site.
Lentivirus production, concentration, titration and transduction
Lentivirus was produced by transient transfection of 293T (ATCC) cells using linear 25-kDa polyethyleneimine (PEI; Polysciences). Briefly, 4 × 106 cells were plated onto 10-cm tissue culture plates. After 24 h, 3 µg of psPAX2, 1.5 µg of pMD2G (Addgene plasmid #12260 and #12259, respectively) and 6 µg of lentiviral vector plasmid were mixed in 500 µl diluent (5 mM HEPES, 150 mM NaCl, pH = 7.05) and 42 µl of PEI (1 mg/ml) and incubated for 15 min. The DNA/PEI complex was then added to the plate drop-wise. Lentivirus was harvested 48 h post-transfection, concentrated 100-fold by low-speed centrifugation at 8000g for 18 h and titered on human Nalm6 cells by flow cytometry and qPCR as previously described (20). Transduction of the target cell line was carried out in six-well plates containing 2 × 106 cells per well in 2 ml of growth media and 4 µg/ml hexadimethrine bromide (Polybrene; SIGMA). Titered virus was added to each well at the designated multiplicity of infection (MOI; four wells per condition) and the cells were incubated with shaking (130 rpm) at 37°C, in 8% CO2 for 4–6 h. The cells were then harvested, pooling the replicate wells, and pelleted at low speed (1000g). Transduction media was removed and replaced with 20 ml fresh media and cells were transferred to a 125-ml vented shake flask (Nalgene). Copy number was estimated using a previously established genomic Q-PCR-based assay with comparison to a housekeeping gene as well as control cell lines with a defined viral integration number based on Southern blotting (21).
Protein expression, purification and analysis
Transduced 293-F cells (at least 1 week post-transduction) were seeded at 5 × 105 cells/ml in a 1-l vented shaker flask (Nalgene) in 100 ml of 293 Expression media (Gibco). Twenty-five milliliters of fresh media was added 2–3 days later, when cells reached densities of 2–3 × 106 cells/ml. The media was harvested after 5 days of total incubation after measuring final cell concentration and viability. Culture supernatants were harvested by low-speed centrifugation to remove cells and filtered through a 0.22-micron Centricon ultrafilter (Millipore). NaCl was added to a final concentration of 250 mM and the supernatants were concentrated to final volumes of ~5 ml using a Vivacell-100 centrifugal concentrator (Sartorius Stedim). Recombinant proteins were separated from media by size-exclusion chromatography (SEC) on a Superdex 75 column (GE Healthsciences). Proteins were transferred to PVDF-FL (Millipore) membranes for western blot analysis with mouse anti-STREP primary (IBA) and an anti-mouse-Alexa 680 (Molecular Probes) secondary antibody, with results visualized using Li-COR fluorescent detection system (Odyssey). Endotoxin levels were measured by the Pyrogene endotoxin detection system (Lonza) following the manufacturer's protocols.
Crystallization and structure determination
Crystals of muScn were obtained by hanging drop vapor diffusion at 25°C. The protein was concentrated to 12 mg/ml in standard phosphate-buffered saline (PBS) and mixed 1:1 v/v with a reservoir solution of 0.1 M HEPES (pH 7.5), 10% v/v isopropanol and 20% w/w PEG 4000. Crystals grew overnight and were cryoprotected in mother liquor containing 10% v/v glycerol. Diffraction data were collected at the Advanced Light Source 5.0.1 beamline (Lawrence Berkeley National Laboratory). Data were reduced with d*TREK (22). Initial structure factor phase information was determined by molecular replacement, using the program PHASER (23) as a part of the CCP4i program suite (24). The structure of human Siderocalin (PDB code: 1X89, chain A) was used as a search model. Model building and refinement were carried out using COOT (25) and refmac5 (26). The structure was submitted to the TLSMD server (27,28) and TLS refinement using a single group was applied to the final model. Structure validation was carried out with the MolProbity server (29) and the RCSB ADIT server.
Functional assays
Bone marrow was harvested from C57BL/6 mice (Jackson laboratories) and plated in a six-well dish at 1 × 106 cells/ml in 2 ml of lymphocyte media (RPMI 1640 (Hyclone) supplemented with 10% FCS (Omega Scientific), 5 mM l-Alanyl-l-Glutamine (Mediatech), 100 IU/ml penicillin–streptomycin (Mediatech) and 2 mM β-mercaptoethanol (Gibco)) with 5 ng/ml of Daedalus IL-7 or commercial IL-7 (PeproTech). Media with IL-7 was replaced at Day 3 and the resulting cell populations were evaluated by fluorescence-activated cell sorting (FACS) at Day 5. Cells were stained with fluorescein isothiocyanate (FITC) anti-mouse IgM (Jackson Immuno Research) and PE anti-mouse B220 (BD) in FACS staining buffer (PBS with 2.5% FCS) and analyzed on an LSRII flow cytometer (BD) and using FlowJo software (Tree Star, Ashland, OR, USA). Recombinant human LIF was tested for its ability to maintain pluripotency of murine ES cells. Briefly, murine ES cells (AB1 ES cells kindly provided by Steve Jones, University of Massachusetts) were plated on irradiated feeder cells with either commercial human LIF (Millipore) or Daedalus recombinant human LIF at 10 ng/ml. Cells were split every 2 days with fresh media and stained with APC anti-SSEA1 (BD) for analysis by flow cytometry.
RESULTS
Expression of murine Siderocalin using lentiviral transduction of 293 Freestyle cells
The coding sequence of murine Siderocalin (muScn) was cloned into the optimized lentiviral backbone pCVL-A (Figure 1A) resulting in the construct pCVL-SFFV-muScn-IRES-GFP (Figure 1B). The ability of this lentiviral vector to transduce 293-F cells was compared to the more commonly used CHO-S cell line. Compared to CHO-S cells, the 293-F cells were more receptive to viral transduction, at almost all multiplicities of infection (MOI; 0.25–10), as measured by the higher mean fluorescence intensity (MFI) of the GFP reporter. 293-F cells were also more resistant to virus-mediated toxicity at the highest MOI (10; Supplementary Figure S1).
293-F cells were transduced with the muScn lentivirus at three different MOIs (Figure 2A) and GFP levels were measured by flow cytometry at 1 week post-transduction. Essentially 100% of the cells were GFP positive (Figure 2A) and muScn secretion was detectable even by Coomassie blue staining of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) analyses of unconcentrated, 20 µl samples of media supernatants as soon as 7 days post-transduction (Figure 2B). The identity of the protein was confirmed by western blot using an anti-STREP antibody (Figure 2B). The IRES-GFP reporter proved to be a good surrogate for protein expression as the increase in protein levels correlated well with the increase in GFP levels at all three MOIs. Effective protein yield in a 125-ml culture was ~13 mg/l of fully glycosylated muScn; proper folding was confirmed by comparative reduced/non-reduced SDS–PAGE and SEC.
Figure 2.

Transduction of 293-F cells at varying multiplicity of infection (MOI). (A) GFP MFI (mean fluorescence intensity) is shown for three different doses of pCVL-SFFV-muScn-IRES-GFP virus. (B) Analyses of expression of levels of muScn are shown; 20-µl aliquots from media supernatants from each transduction were separated by SDS–PAGE and stained with Coomassie blue (left) or analyzed by western blot with an anti-Strep II tag antibody (right).
Use of a novel, minimized UCOE to stabilize high-level expression of muScn
Consistent with previous observations of silencing of lentivirus integrants in vitro and in vivo (11,12), long-term culture (up to 1 month) of the transduced cells resulted in a steady decline in GFP MFI and corresponding protein levels (Figure 3A). In order to enhance protein production and stably maintain expression levels for extended periods, we incorporated two alternative UCOE elements derived from the CpG island within the human HNRPA2B1/CBX3 locus, either UCOE2.0 or UCOE0.7. Expression levels from vectors incorporating either element were comparable by initial GFP MFI signal and were stable over extended periods of up to 1 month (Supplementary Figure S2). However, expression levels, as measured by GPF MFI, from transductants incorporating the truncated UCOE0.7 were substantially enhanced relative to transductants lacking UCOEs (at matched copy numbers of ~8–10 copies) and were stable over at least 1 month, consistent with resistance to silencing (Figure 3B). The loss of GFP in the transductants lacking the UCOE element was not due to loss of the integrated gene as the viral copy number did not vary significantly over the 1-month period (data not shown). It is possible to further enhance and prolong protein expression by sorting for the highest GFP positive (GFP+) cells (Supplementary Figure S3). This additional step, however, was not required and was omitted for testing the expression/purification of the multiple subsequent proteins reported here.
Figure 3.

Comparison of non-UCOE vs. UCOE0.7 containing constructs expressing murine Siderocalin. (A) GFP MFI measured over a 1-month period for pCVL-SFFV-muScn-IRES-GFP. (B) GFP MFI, for the same period of time, for pCVL-UCOE0.7-SFFV-muScn-IRES-GFP. Cells were matched for copy number and data are representative of three replicate experiments.
Expression, purification and crystallization of 293-expressed muScn
The goal of this effort was to produce large quantities of muScn to support crystallographic studies, successful completion of which would confirm the proper folding of the recombinant protein. At MOIs of 10–20, pCVL-UCOE0.7-SFFV-muScn-IRES-GFP efficiently transduced 293-F cells leading to a 100% GFP+ population, obviating any requirement for single-cell cloning or sorting. A 200-ml culture incubated for 5 days yielded ~6 mg of protein (~37 mg/l) after purification by preparative SEC (Table 1 and Figure 4A). This yield was almost 3-fold higher than the yield obtained using the lentiviral vector lacking UCOE. The protein obtained was glycosylated and ≥99% pure (Figure 4A, inset). Final culture density in this experiment was only about 3–4 × 106 cells per ml, compared to typical endpoint densities of 8–10 × 106 cells per ml for CHO cell culture, highlighting the efficiency of recombinant protein expression in this 293-F-based system. The fully glycosylated, 293-F-expressed muScn was readily crystallizable (Figure 4B) where non-glycosylated, recombinant versions of muScn had previously been recalcitrant to crystallization. The crystal structure (dmin = 1.8 Å) of 293-F-expressed muScn showed that the protein was properly folded, with expected N-linked glycosylation and intrachain disulfide bond formation readily apparent (Table 2 and Figure 4C). The time required to go from viral transduction to structure determination in this example was only 18 days. Even though it had been successfully crystallized with protein recombinantly expressed from baculovirus and bacterial systems, human Siderocalin (huScn) was also expressed (at a matched copy number) using the same protocols for comparison. The effective yield of huScn after purification and concentration was ~47 mg/l under similar culture conditions, a modest increase over muScn (Table 1).
Table 1.
List of Daedalus target proteins
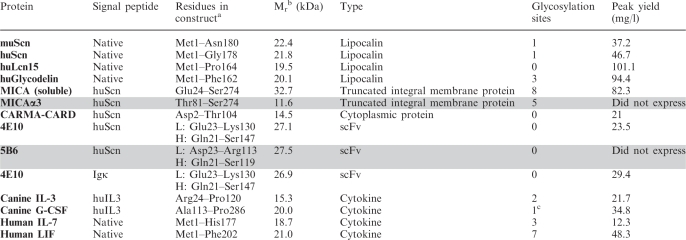 |
aResidues for constructs with a non-native signal peptide correspond to sequence that was cloned downstream of the heterologous signal peptide.
bMolecular weights are listed according to the estimated molecular weight of the mature protein, lacking the signal peptide and any modifications.
cCanine G-CSF contains a single O-linked glycosylation site.
Figure 4.

Purification and crystallization of murine Siderocalin. (A) Preparative Superdex 75 SEC trace for muScn expressed using the optimized lentiviral construct pCVL-UCOE0.7-SFFV-muScn-IRES-GFP. SDS–PAGE analysis of the peak fraction, before and after PNGase digestion, is shown (inset). (B) Crystals obtained using the PEGs II Suite (Qiagen) sparse matrix screen, condition 63 [0.1 M HEPES (pH 7.5), 10% v/v isopropanol and 20% w/w PEG 4000]. (C) Crystal structure of muScn (red) superimposed on that of huScn (1X89 in black). Electron density from a refined Fo–Fc omit map contoured at 2.0 σ showing the presence of the N-linked glycan and the intrachain disulfide bond.
Table 2.
Data collection and refinement statistics (molecular replacement)
| Data collection | |
| Space group | P212121 |
| Lattice constants (Å) | a = 42.89 b = 43.96 c = 101.92 |
| Resolution (Å) | 42.89–1.8 (1.86–1.80) |
| Unique reflections | 18 574 |
| Average redundancy | 6.87 (7.09) |
| Completeness (%) | 99.9 (100.0) |
| Rmerge (%) | 4.6 (39.4) |
| I/σ(I) | 14.7 (3.5) |
| Refinement | |
| Rwork (%) | 20.08 |
| Rfree (%) | 22.61 |
| Number of atoms | |
| Protein | 1341 |
| Ligand | 50 |
| Water | 89 |
| RMSD from ideal values | |
| Bond lengths (Å) | 0.015 |
| Bond angles (°) | 1.50 |
| Chiral volume (Å3) | 0.07 |
| Ramachandran | |
| Most favored (%) | 92.6 |
| Additionally allowed (%) | 6.1 |
| Generously allowed (%) | 0.0 |
| Disallowed (%) | 1.4 |
Expression of other secreted proteins
In order to assess the general applicability of the Daedalus system, a panel of secreted proteins was then tested for expressibility: two additional human lipocalins, Lipocalin 15 (Lcn15) and glycodelin (Gd); the soluble ectodomain of the human major histocompatibility complex class I homolog MICA; the isolated, immunoglobulin-like α3 domain of MICA (MICAα3; Thr81-Ser274); and two antibody single-chain Fv (scFv) constructs, derived from either the anti-HIV antibody 4E10 or the anti-canine CD28 antibody 5B6. All proteins were analyzed by analytical SEC (Supplementary Figure S5A–S5C) and SDS–PAGE under reducing and non-reducing conditions (Supplementary Figure S5D); yields were calculated using the BCA protein assay (Table 1).
Lcn15 has, as yet, no known physiological function; periplasmic expression of Lcn15 in E. coli had previously produced only modest yields of ~1 mg of purified protein per liter of culture. Gd can be isolated in multiple, functionally relevant glycoforms (GdA, GdC, GdF and GdF) from the female reproductive tract and seminal plasma and primarily modulates sperm function (30,31). It had been shown previously that HEK293 cells are capable of producing functional GdA (32). Lcn15 and Gd were cloned into the UCOE0.7-containing vector and expressed and purified following the same methodology used for Scn, resulting in yields of 94 and 103 mg/l respectively, a 100-fold improvement in Lcn15 yield over periplasmic bacterial expression.
MICA is a conditionally expressed self-antigen recognized by the widely expressed, activating immunoreceptor NKG2D (33). MICA, solubilized by truncation prior to the transmembrane-spanning segment at residue 274, had previously been produced as a secreted protein in a baculovirus expression system (34), yielding up to 7 mg/l after harvesting 5 days post infection, or by refolding denatured bacterial inclusion bodies in vitro (35), an arduous and expensive (typically >$900 per milligram of purified protein) process that requires considerable experimental finesse. Yields of purified MICA range up to 1.1 mg/l of input bacterial culture by in vitro refolding. Refolded MICA, lacking N-linked oligosaccharides, tended to aggregate over time. Daedalus expression of MICA yielded up to 82 mg/l of purified, natively glycosylated protein that was stable in solution over at least 3 months following purification, a 74-fold improvement over the bacterial system. MICAα3, a species that expressed poorly by every other technique tried, was targeted to the secretion pathway by fusion with a heterologous huScn signal peptide for testing in the Daedalus system, but failed to yield measurable amounts of protein.
Antibody scFv constructs recapitulate the binding properties of the mature immunoglobulin (except bivalent avidity); the 4E10 scFv was refolded from bacterial inclusion bodies at yields up to 0.8 mg per liter of culture (36); Daedalus expression, using either huScn or human Igκ signal peptides, yielded up to 25 mg/l of 4E10 scFv (Table 1), a 31-fold improvement, binding properties of Daedalus-expressed and refolded 4E10 scFv were identical in surface plasmon resonance interaction analyses (data not shown). The heavy and light variable regions of the 4E10 were joined by a thrombin-cleavable linker (sequence: LVPRGSGGGGLVPRGS), yielding pure monobodies after cleavage; Daedalus-expressed scFv cleaved as readily as refolded scFv, with thrombin added directly to culture supernatants after concentration and prior to purification. A second scFv, based on antibody 5B6, was not purifiable from any conventional recombinant system and was available only as sequence information, as the parent hybridoma proved to be unstable. Unfortunately, the 5B6 scFv also failed to produce measurable yields in the Daedalus system.
Expression of functional, endotoxin-free, cytokines
Recombinant cytokines have become widely used in the treatment of cancer (37), primary immunodeficiency diseases (38), anemia and in conditioning patients receiving hematopoietic stem cell transplants (HSCTs) (39). However, recombinant cytokine therapeutics generally display limited plasma half-lives due to their innate instability and rapid in vivo clearance, at least partially accounted for by a lack of glycosylation, which can be overcome by expression with native glycans, improving efficacy and stability (40,41).
Therefore, we also evaluated the capacity of the Daedalus system to produce a panel of candidate cytokines and assessed whether these recombinant proteins exhibited normal levels of glycosylation and activity. We evaluated human interleukin-7 (IL-7), human leukemic inhibitory factor (LIF), canine interleukin-3 (IL-3) and canine granulocyte colony-stimulating factor (G-CSF). IL-7 is essential for lymphocyte development, proliferation and activity; and therapeutically studied for immune reconstitution following stem cell transplantation and chemotherapy, as a vaccine adjuvant, and, more recently, to treat sepsis (42). LIF is a multifunctional growth factor that acts on several cell types. It is heavily glycosylated (LIF has seven putative sites) and it has been suggested that the glycosylation pattern, at least of rat LIF, is essential for its function (43). LIF has more recently become an important cytokine for maintaining embryonic stem cells (ESCs) in an undifferentiated state for their use in regenerative therapies (44) as well as for developing induced pluripotent stem cells (iPSCs) (45). Canine orthologs of IL-3 and G-CSF have been useful for studying hematopoietic HSCT in a canine model (46). Canine IL-3 displays low sequence identity to orthologs from other species (47) necessitating the use of species-matched reagents in the canine model.
Coding sequences for expression constructs for all four cytokines were synthesized, with codon utilization optimized for human expression, and inserted into the UCOE0.7 containing vector; IL-7, LIF and IL-3 were expressed with their native signal peptide. G-CSF is endogenously expressed as a pro-peptide that is further processed to its mature form; for Daedalus expression, the coding sequence for the mature protein was placed downstream from the IL-3 signal peptide sequence that scored higher than the native signal peptide by the SignalP 3.0 algorithm (48). All four proteins were successfully expressed and purified with substantial yields (Table 1), were glycosylated as seen by a shift in molecular weight by SDS–PAGE (Supplementary Figure S5D) and PNGase treatment of IL-7 and LIF (Supplementary Figure S5E). All four proteins were also tested for endotoxin levels and were on average ≤10 EU/mg and IL-7 and LIF were tested for activity. IL-7 is known to promote B-cell development in culture and was tested using mouse bone marrow cells (49). The IL-7 produced using the Daedalus system performed identically to commercial IL-7 (Supplementary Figure S4A) in its ability to promote and maintain B-cell (B220+IgM+) development of mouse bone marrow cells in culture. The recombinant LIF was also functionally tested in its ability to maintain mouse ES cells in an undifferentiated state, as measured by maintenance of the cell-surface marker SSEA1 in culture (Supplementary Figure S4B) (50); there was no difference compared to commercial LIF.
Expression of cytoplasmic proteins
One advantage of the Daedalus system is the ease with which secreted proteins can be purified from the serum-free supernatants of transduced cells. To determine whether the Daedalus system could be adapted to cytoplasmically targeted proteins, the expression of a cytoplasmic protein engineered with a heterologous signal peptide (from huScn) was tested for efficient secretion into media supernatants. Caspase-recruitment domain (CARD)-membrane-associated guanylate kinase protein 1 (CARMA1) is essential for lymphocyte activation and immune function (51). The N-terminal CARD domain of CARMA1 (CARMA-CARD) interacts with that of B-cell lymphoma 10 (Bcl-10) and is essential for lymphocyte activation via nuclear factor-κB (NF-κB) (52). CARMA-CARD was successfully expressed as a cytoplasmically targeted construct in E. coli (Bandanarayake et al., manuscript in preparation) using a combination His6/maltose binding protein purification tag, fused to CARMA-CARD through a tobacco etch virus (TEV) protease recognition sequence. Typical yields of CARMA-CARD after tag cleavage and purification from E. coli were ~10 mg/l (Supplementary Figure S6B). Despite successful expression, this recombinant form failed to crystallize despite exhaustive screening. For testing Daedalus expressibility, the huScn signal peptide sequence was fused in-frame with the coding sequence of human CARMA-CARD (Asp2-Thr104) and the fusion was inserted into the pCVL-UCOE0.7-SFFV-IRES-GFP vector backbone. A 100-ml culture of 293-F cells, transduced with recombinant lentivirus produced as described above, yielded 2.5 mg (25 mg/l) of protein after final SEC purification (~95% pure by PAGE). Interestingly, SEC analysis of the Daedalus-expressed CARMA-CARD protein showed a mixture of monomeric and dimeric forms (Supplementary Figure S6A), whereas the SEC analysis of bacterially expressed protein had shown only the monomeric form (Supplementary Figure S6B). The Daedalus CARMA-CARD homodimer was isolated by preparative SEC and found to be a stable, interchain disulfide-linked species as shown by comparative reduced/non-reduced SDS–PAGE analysis (Supplementary Figure S6C). Another CARD domain protein, NOD1, also homodimerizes by forming a strand-swapped dimer, stabilized by an adventitious interchain disulfide when purified (53), suggesting that the Daedalus system allows CARMA-CARD to access a folding pathway not available in the cytoplasm of bacteria. While possibly not physiological, the CARMA-CARD interchain disulfide may also represent an adventitious linkage, potentially in context of an analogous strand-swapped dimer. Preliminary crystals of the Daedalus-expressed CARMA-CARD dimer were obtained, but require further optimization.
DISCUSSION
Lipocalin 2 (also known as Siderocalin, NGAL or 24p3) is a bacteriostatic, innate immune system defense, secreted, disulfide-containing glycoprotein that interferes with siderophore mediated iron uptake in bacteria (54). huScn has been successfully expressed recombinantly in both bacterial (≤15 mg/l) and baculovirus (≤5 mg/l) expression systems in order to support successful crystallographic analyses (55,56). However, murine Siderocalin (muScn), despite being successfully expressed in analogous bacterial expression systems (yielding ≤15 mg/l), proved refractory to crystallization. Since the presence or absence of N-glycans can affect crystallizability, an alternate expression system was sought that could produce readily purifiable, natively glycosylated muScn at the multi-milligram levels needed to support crystallization trials. Using the Daedalus system, muScn was expressed at decigram levels and purified (in a single SEC step) to crystallizability within 18 days. Daedalus expression levels for muScn (~37 mg/l) and huScn (~46 mg/l) dramatically exceeded yields from alternate systems.
One dozen additional expression targets were tested, including additional lipocalins, truncated cell-surface glycoproteins, an antibody scFv construct, cytokines and cytoplasmic CARMA-CARD; 10 of these 12 targets were successfully expressed at yields up to 100 mg/ml, demonstrating the wide potential utility and high potential expression levels of the Daedalus platform, even when limited to conventional shaking-flask cell densities. The 293-F secretion system also ensured that only properly folded and stable proteins were exported (57), adding an additional internal quality control that could also be applied to cytoplasmically targeted proteins by fusion with heterologous signal peptide sequences (huScn). Avoidance of bacteria during protein expression also yields recombinant protein free of contaminating endotoxins without additional purification steps.
Due to the packing limitations of lentiviruses, the maximum construct that can be expressed in the UCOE0.7 vector is ~2.0 kb, corresponding to Mrs about 70 kDa. Further optimization of the vector may increase this limit, but larger inserts may be packaged by deletion of the IRES-GFP cassette, which, although useful for rapid selection, is not necessary for viral or protein production. This would increase the insert size by ~1 kb which would enable the production of, for instance, full-length antibody chains.
In summary, we have developed a rapid and scalable expression system in 293-F cells by stabilizing the expression of recombinant proteins using a lentiviral delivery vector. A key element of this invention was the incorporation of a minimized UCOE to prevent genomic silencing of integrated expression cassettes. While previous studies have utilized large UCOE elements (up to 8 kb) within plasmid constructs to obtain high expression clones in mammalian cells (58,59), the Daedalus system exhibits a number of significant advantages including increased speed, no requirement for producer cell selection or cloning, and demonstrated capacity to generate a broad range of functional proteins. The Daedalus system takes advantage of both the more efficient initial transduction rates in 293-F cells and viral elements that permit sustained high-level protein expression without a requirement for cell cloning. The Daedalus platform is simple and robust enough to enable academic labs to rapidly produce recalcitrant proteins for research and may be adaptable enough for therapeutic manufacturing and other biotechnology applications.
ACCESSION NUMBERS
Structure coordinates and structure factors have been deposited in the Protein Data Bank under the ID code 3S26.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Institutes of Health (grant numbers AI48675, AI59432, AI084803, DK073462, DK56465, HD037091, HD075453 and HL092557); the J. Orin Edson Foundation; and the Bill and Melinda Gates Foundation Collaboration for AIDS Vaccine Discovery (CAVD). Funding for open access charge: National Institutes of Health (grant DK56465).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Iram Khan (Viral Core, Seattle Children's Research Institute, Seattle, WA, USA) for technical assistance.
REFERENCES
- 1.Hannig G, Makrides SC. Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol. 1998;16:54–60. doi: 10.1016/s0167-7799(97)01155-4. [DOI] [PubMed] [Google Scholar]
- 2.Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004;22:1399–408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- 3.Holz C, Prinz B, Bolotina N, Sievert V, Büssow K, Simon B, Stahl U, Lang C. Establishing the yeast Saccharomyces cerevisiae as a system for expression of human proteins on a proteome-scale. J. Struct. Funct. Genomics. 2003;4:97–108. doi: 10.1023/a:1026226429429. [DOI] [PubMed] [Google Scholar]
- 4.Daly R, Hearn MTW. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J. Mol. Recogn. 2005;18:119–138. doi: 10.1002/jmr.687. 10.1002/jmr.687. [DOI] [PubMed] [Google Scholar]
- 5.Kost TA, Condreay JP, Jarvis DL. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 2005;23:567–575. doi: 10.1038/nbt1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang G. Improvement of glycosylation in insect cells with mammalian glycosyltransferases. J. Biotechnol. 2003;102:61–71. doi: 10.1016/s0168-1656(02)00364-4. 10.1016/S0168-1656(02)00364-4. [DOI] [PubMed] [Google Scholar]
- 7.Chu L, Robinson DK. Industrial choices for protein production by large-scale cell culture. Curr. Opin. Biotechnol. 2001;12:180–187. doi: 10.1016/s0958-1669(00)00197-x. [DOI] [PubMed] [Google Scholar]
- 8.Bosques C, Collins B, Meador J, III, Sarvaiya H, Murphy J, DelloRusso G, Bulik D, Hsu I, Washburn N, Sipsey S, et al. Chinese hamster ovary cells can produce galactose-α-1,3-galactose antigens on proteins. Nat. Biotechnol. 2010;28:1153–1156. doi: 10.1038/nbt1110-1153. 10.1038/nbt1110-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberbek A, Matasci M, Hacker DL, Wurm FM. Generation of stable, high-producing CHO cell lines by lentiviral vector-mediated gene transfer in serum-free suspension culture. Biotechnol. Bioeng. 2010;41:10. doi: 10.1002/bit.22968. 1002/bit.22968. [DOI] [PubMed] [Google Scholar]
- 10.Gaillet B, Gilbert R, Broussau S, Pilotte A, Malenfant F, Mullick A, Garnier A, Massie B. High-level recombinant protein production in CHO cells using lentiviral vectors and the cumate gene-switch. Biotechnol. Bioeng. 2010;106:203–215. doi: 10.1002/bit.22698. 10.1002/bit.22698. [DOI] [PubMed] [Google Scholar]
- 11.Hino S, Fan J, Taguwa S, Akasaka K, Matsuoka M. Sea urchin insulator protects lentiviral vector from silencing by maintaining active chromatin structure. Gene Ther. 2004;11:819–828. doi: 10.1038/sj.gt.3302227. 10.1038/sj.gt.3302227. [DOI] [PubMed] [Google Scholar]
- 12.Hofmann A, Kessler B, Ewerling S, Kabermann A, Brem G, Wolf E, Pfeifer A. Epigenetic regulation of lentiviral transgene vectors in a large animal model. Mol. Ther. J. Am. Soc. Gene Ther. 2006;13:59–66. doi: 10.1016/j.ymthe.2005.07.685. 10.1016/j.ymthe.2005.07.685. [DOI] [PubMed] [Google Scholar]
- 13.Antoniou M, Harland L, Mustoe T, Williams S, Holdstock J, Yague E, Mulcahy T, Griffiths M, Edwards S, Ioannou P, et al. Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing. Genomics. 2003;82:269–279. doi: 10.1016/s0888-7543(03)00107-1. 10.1016/S0888-7543(03)00107-1. [DOI] [PubMed] [Google Scholar]
- 14.Williams S, Mustoe T, Mulcahy T, Griffiths M, Simpson D, Antoniou M, Irvine A, Mountain A, Crombie R. CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells. BMC Biotechnol. 2005;5:17. doi: 10.1186/1472-6750-5-17. 10.1186/1472-6750-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang F, Thornhill SI, Howe SJ, Ulaganathan M, Schambach A, Sinclair J, Kinnon C, Gaspar HB, Antoniou M, Thrasher AJ. Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells. Blood. 2007;110:1448–1457. doi: 10.1182/blood-2006-12-060814. 10.1182/blood-2006-12-060814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang F, Frost A, Blundell M, Bales O. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation–mediated silencing of lentiviral vectors. Mol. Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schambach A, Bohne J, Baum C, Hermann FG, Egerer L, von Laer D, Giroglou T. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006;13:641–645. doi: 10.1038/sj.gt.3302698. 10.1038/sj.gt.3302698. [DOI] [PubMed] [Google Scholar]
- 18.Schambach A, Galla M, Maetzig T, Loew R, Baum C. Improving transcriptional termination of self-inactivating gamma-retroviral and lentiviral vectors. Mol. Ther. J. Am. Soc. Gene Ther. 2007;15:1167–1173. doi: 10.1038/sj.mt.6300152. 10.1038/sj.mt.6300152. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt TGM, Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 20.Kerns HM, Ryu BY, Stirling BV, Sather BD, Astrakhan A, Humblet-Baron S, Liggitt D, Rawlings DJ. B cell-specific lentiviral gene therapy leads to sustained B-cell functional recovery in a murine model of X-linked agammaglobulinemia. Blood. 2010;115:2146–2155. doi: 10.1182/blood-2009-09-241869. 10.1182/blood-2009-09-241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sather BD, Ryu BY, Stirling BV, Garibov M, Kerns HM, Humblet-Baron S, Astrakhan A, Rawlings DJ. Development of B-lineage predominant lentiviral vectors for use in genetic therapies for B cell disorders. Mol. Ther. J. Am. Soc. Gene Ther. 2010;19:515–525. doi: 10.1038/mt.2010.259. 10.1038/mt.2010.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. 10.1107/S090744499900935X. [DOI] [PubMed] [Google Scholar]
- 23.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. S0907444903008126 [pii] [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. S0907444904019158 [pii] 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. 10.1107/S0907444996012255 S0907444996012255 [pii] [DOI] [PubMed] [Google Scholar]
- 27.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 28.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006;39:109–111. 10.1107/S0021889805038987. [Google Scholar]
- 29.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. gkm216 [pii] 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seppala M, Koistinen H, Koistinen R. Glycodelins. Trends Endocrinol. Metab. 2001;12:111–117. doi: 10.1016/s1043-2760(00)00365-9. S1043-2760(00)00365-9 [pii] [DOI] [PubMed] [Google Scholar]
- 31.Yeung WS, Lee KF, Koistinen R, Koistinen H, Seppala M, Ho PC, Chiu PC. Roles of glycodelin in modulating sperm function. Mol. Cell. Endocrinol. 2006;250:149–156. doi: 10.1016/j.mce.2005.12.038. S0303-7207(05)00484-3 [pii] 10.1016/j.mce.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 32.Van den Nieuwenhof IM, Koistinen H, Easton RL, Koistinen R, Kamarainen M, Morris HR, Van Die I, Seppala M, Dell A, Van den Eijnden DH. Recombinant glycodelin carrying the same type of glycan structures as contraceptive glycodelin-A can be produced in human kidney 293 cells but not in Chinese hamster ovary cells. Eur. J. Biochem. 2000;267:4753–4762. doi: 10.1046/j.1432-1327.2000.01528.x. ejb1528 [pii] [DOI] [PubMed] [Google Scholar]
- 33.Bahram S, Bresnahan M, Geraghty DE, Spies T. A second lineage of mammalian major histocompatibility complex class I genes. Proc. Natl Acad. Sci. USA. 1994;91:6259–6263. doi: 10.1073/pnas.91.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bauer S, Willie ST, Spies T, Strong RK. Expression, purification, crystallization and crystallographic characterization of the human MHC class I related protein MICA. Acta Crystallogr. D Biol. Crystallogr. 1998;54:451–453. doi: 10.1107/s0907444997015229. [DOI] [PubMed] [Google Scholar]
- 35.Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat. Immunol. 2001;2:443–451. doi: 10.1038/87757. 10.1038/87757 87757 [pii] [DOI] [PubMed] [Google Scholar]
- 36.Xu H, Song L, Kim M, Holmes MA, Kraft Z, Sellhorn G, Reinherz EL, Stamatatos L, Strong RK. Interactions between lipids and human anti-HIV antibody 4E10 can be reduced without ablating neutralizing activity. J. Virol. 2010;84:1076–1088. doi: 10.1128/JVI.02113-09. 10.1128/JVI.02113-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellegrini M, Mak TW, Ohashi PS. Fighting cancers from within: augmenting tumor immunity with cytokine therapy. Trends Pharmacol. Sci. 2010;31:356–363. doi: 10.1016/j.tips.2010.05.003. 10.1016/j.tips.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Roy-Ghanta S, Orange JS. Use of cytokine therapy in primary immunodeficiency. Clin. Rev. Allergy Immunol. 2010;38:39–53. doi: 10.1007/s12016-009-8131-4. 10.1007/s12016-009-8131-4. [DOI] [PubMed] [Google Scholar]
- 39.Huang C-J, Lowe AJ, Batt CA. Recombinant immunotherapeutics: current state and perspectives regarding the feasibility and market. Appl. Microbiol. Biotechnol. 2010;87:401–410. doi: 10.1007/s00253-010-2590-7. 10.1007/s00253-010-2590-7. [DOI] [PubMed] [Google Scholar]
- 40.Mahmood I, Green MD. Pharmacokinetic and pharmacodynamic considerations in the development of therapeutic proteins. Clin. Pharmacokinetics. 2005;44:331–347. doi: 10.2165/00003088-200544040-00001. [DOI] [PubMed] [Google Scholar]
- 41.Solá RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs: Clin. Immunotherap. Biopharm. Gene Ther. 2010;24:9. doi: 10.2165/11530550-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat. Rev. Immunol. 2011;11:330–342. doi: 10.1038/nri2970. 10.1038/nri2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasai K, Aikawa JI, Saburi S, Tojo H, Tanaka S, Ogawa T, Shiota K. Functions of the N-glycans of rat leukemia inhibitory factor expressed in Chinese hamster ovary cells. J. Biochem. 1998;124:999–1003. doi: 10.1093/oxfordjournals.jbchem.a022219. [DOI] [PubMed] [Google Scholar]
- 44.Imsoonthornruksa S, Noisa P, Parnpai R, Ketudat-Cairns M. A simple method for production and purification of soluble and biologically active recombinant human leukemia inhibitory factor (hLIF) fusion protein in Escherichia coli. J. Biotechnol. 2011;151:295–302. doi: 10.1016/j.jbiotec.2010.12.020. 10.1016/j.jbiotec.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 45.Cotterman R, Knoepfler PS. N-Myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PLoS ONE. 2009;4:e5799. doi: 10.1371/journal.pone.0005799. 10.1371/journal.pone.0005799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lupu M, Storb R. Five decades of progress in haematopoietic cell transplantation based on the preclinical canine model. Cancer Res. 2007;5:14–31. doi: 10.1111/j.1476-5829.2006.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin IS, Kim HR, Nam MJ, Youn HY. Studies of cocktail therapy with multiple cytokines for neoplasia or infectious disease of the dog I. cDNA cloning of canine IL-3 and IL-6. J. Vet. Sci. 2001;2:115–120. [PubMed] [Google Scholar]
- 48.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 49.Corfe SA, Gray AP, Paige CJ. Generation and characterization of stromal cell independent IL-7 dependent B cell lines. J. Immunol. Methods. 2007;325:9–19. doi: 10.1016/j.jim.2007.05.010. 10.1016/j.jim.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 50.Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885–896. doi: 10.1242/dev.01670. 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- 51.Rawlings DJ, Sommer K, Moreno-Garcia ME. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat. Rev. Immunol. 2006;6:799–812. doi: 10.1038/nri1944. 10.1038/nri1944. [DOI] [PubMed] [Google Scholar]
- 52.Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Ovechkina YL, Moreno-garcı ME, Rawlings DJ. Phosphorylation of the CARMA1 linker controls NF-kB activation. Immunity. 2005;23:561–574. doi: 10.1016/j.immuni.2005.09.014. 10.1016/j.immuni.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 53.Srimathi T, Robbins SL, Dubas RL, Hasegawa M, Inohara N, Park YC. Monomer/dimer transition of the caspase-recruitment domain of human Nod1. Biochemistry. 2008;47:1319–1325. doi: 10.1021/bi7016602. 10.1021/bi7016602. [DOI] [PubMed] [Google Scholar]
- 54.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 55.Strong RK, Bratt T, Cowland JB, Borregaard N, Wiberg FC, Ewald AJ. Expression, purification, crystallization and crystallographic characterization of dimeric and monomeric human neutrophil gelatinase associated lipocalin (NGAL) Acta Crystallogr. Sect. D, Biol. Crystallogr. 1998;54:93–95. doi: 10.1107/s0907444997010615. [DOI] [PubMed] [Google Scholar]
- 56.Goetz DH, Willie ST, Armen RS, Bratt T, Borregaard N, Strong RK. Ligand preference inferred from the structure of neutrophil gelatinase associated lipocalin. Society. 2000;39:1935–1941. doi: 10.1021/bi992215v. [DOI] [PubMed] [Google Scholar]
- 57.Otero JH, Lizák B, Hendershot LM. Life and death of a BiP substrate. Semin. Cell Dev. Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams S, Mustoe T, Mulcahy T, Griffiths M, Simpson D, Antoniou M, Irvine A, Mountain A, Crombie R. CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells. BMC Biotechnol. 2005;5:17. doi: 10.1186/1472-6750-5-17. 10.1186/1472-6750-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benton T, Chen T, McEntee M, Fox B, King D, Crombie R, Thomas TC, Bebbington C. The use of UCOE vectors in combination with a preadapted serum free, suspension cell line allows for rapid production of large quantities of protein. Cytotechnology. 2002;38:43–46. doi: 10.1023/A:1021141712344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.