

Abstract
Deep sequencing was used to bring high resolution to the human cytomegalovirus (HCMV) transcriptome at the stage when infectious virion production is under way, and major findings were confirmed by extensive experimentation using conventional techniques. The majority (65.1%) of polyadenylated viral RNA transcription is committed to producing four noncoding transcripts (RNA2.7, RNA1.2, RNA4.9, and RNA5.0) that do not substantially overlap designated protein-coding regions. Additional noncoding RNAs that are transcribed antisense to protein-coding regions map throughout the genome and account for 8.7% of transcription from these regions. RNA splicing is more common than recognized previously, which was evidenced by the identification of 229 potential donor and 132 acceptor sites, and it affects 58 protein-coding genes. The great majority (94) of 96 splice junctions most abundantly represented in the deep-sequencing data was confirmed by RT-PCR or RACE or supported by involvement in alternative splicing. Alternative splicing is frequent and particularly evident in four genes (RL8A, UL74A, UL124, and UL150A) that are transcribed by splicing from any one of many upstream exons. The analysis also resulted in the annotation of four previously unrecognized protein-coding regions (RL8A, RL9A, UL150A, and US33A), and expression of the UL150A protein was shown in the context of HCMV infection. The overall conclusion, that HCMV transcription is complex and multifaceted, has implications for the potential sophistication of virus functionality during infection. The study also illustrates the key contribution that deep sequencing can make to the genomics of nuclear DNA viruses.
The genetic repertoire of human cytomegalovirus (HCMV; species Human herpesvirus 5) is incompletely understood. Most bioinformatic investigations have focused on identifying open reading frames (ORFs) that are conserved in other organisms or that exhibit pattern-based similarities (e.g., in nucleotide or codon bias) to recognized protein-coding regions (CRs) (1). Our current map of the wild-type HCMV genome, based on strain Merlin, contains 166 protein-coding genes (2–5). It is entirely possible that additional, small protein-coding genes will be found. Candidates involve ORFs that overlap recognized CRs and for which there is some evidence for expression (6), ORFs highlighted in pattern-based bioinformatic (7) or proteomic analyses (8), and ORFs whose expression is presently unsuspected.
Recognition of many protein-coding genes has been supplemented by information on protein expression and function. However, HCMV also specifies polyadenylated (polyA) transcripts that, because they lack sizeable, conserved ORFs, appear unlikely to function via translation. One class consists of noncoding, nonoverlapping transcripts (NNTs) that do not substantially overlap the designated CRs of other genes. These include an abundant 2.7-kb RNA (β2.7 or RNA2.7) (9), a 1.1-kb spliced RNA and associated 5-kb stable intron (RNA5.0) (10, 11), and a 1.2-kb RNA (RNA1.2) (12). RNA2.7 functions in inhibiting apoptosis (13), the RNA5.0 homolog in murine cytomegalovirus is involved in virulence (14), and the role of RNA1.2 is unknown. Antisense transcripts (ASTs) form a second class of noncoding RNA, and are transcribed antisense to CRs (15, 16). In addition, HCMV specifies several small, nonpolyA RNAs that may have regulatory roles, including microRNAs (miRNAs) (17) processed from longer transcripts and RNAs associated with the origin of DNA replication (18).
To extend the understanding of HCMV genome expression, we used deep sequencing to study viral transcription in human fibroblasts. The focus was on identifying previously unrecognized protein-coding or noncoding polyA RNAs, and on determining the extent of RNA splicing.
Results
Transcription Profiles.
Whole-cell RNA was isolated from human fibroblasts infected with HCMV strain Merlin at 72 h after infection, when infectious virion production was under way. An Illumina DNA sequence dataset (DS) was generated from each of two RNA preparations: a nondirectional DS (NDS; 24,439,691 reads) and a directional DS (DDS; 32,514,724 reads). Transcript orientation could be inferred from the DDS but not the NDS.
The DSs were assembled against the 236-kbp HCMV genome, which has the structure ab-UL-b′a′c′-US-ca, where UL and US are unique regions flanked by inverted repeats (ab/b′a′ and a′c′/ca). Similar percentages (42.24 and 43.03, respectively) of NDS and DDS reads originated from HCMV. The light yellow windows in Fig. 1 depict transcription profiles, calculated from read densities across the genome. In the lower windows, the NDS profile (gray) on a linear scale demonstrates that some regions are transcribed abundantly (a few off-scale), whereas others are expressed at very low levels. The NDS profile (black) on a logarithmic scale makes it clear that almost every region in the genome is transcribed at some level. As anticipated, the DDS profile (green) for rightward and leftward transcripts combined on a logarithmic scale is similar to that for the NDS (black). The plots in the upper windows show the DDS profiles for rightward (magenta) and leftward (cyan) transcripts separately, on a logarithmic scale, and demonstrate that ASTs are produced from many regions. Further features of Fig. 1 are expanded on below.
Fig. 1.

Transcription and splicing of the HCMV genome. The genome is shown in five sections, with the inverted repeats (ab/b′a′ and a′c′/ca) shaded gray. CRs are shown by color-shaded arrows and NNTs as white-shaded arrows, with gene nomenclature below. Introns connecting CRs or the RNA5.0 exons are shown as narrow white bars. Colors applied to CRs indicate conservation among α-, β-, and γ-herpesviruses (core genes) or between β- and γ-herpesviruses (subcore genes), with subsets of the noncore genes grouped into families of related genes. UL72 is both a member of the deoxyuridine triphosphatase-related protein (DURP) gene family and a core gene. The light yellow windows depict transcription profiles (Methods) as explained in the key in the bottom right corner. The plots in the lower windows show rightward and leftward transcripts combined: the NDS profile on linear and logarithmic scales (right and left, respectively) and the DDS profile on a logarithmic scale. The plots in the upper windows show the DDS profiles for rightward and leftward transcripts separately, on a logarithmic scale (right and left, respectively). The horizontal bar above the gene nomenclature shows regions where rightward (magenta) or leftward (cyan) transcription predominates. The vertical lines in the lower windows show the locations of the subset of splice sites supported by >10 reads, oriented rightward or leftward as explained in the key. The height of the line depicts the number of reads representing the site, plotted on a logarithmic scale (left). The complete list of splice sites is provided in Dataset S1.
Previously Unrecognized Transcripts.
Selected transcribed regions that apparently lack CRs were investigated by using RACE (Fig. S1 and Table S1) and Northern blotting (NB) (Fig. S2). These experiments led to the annotation of one previously unrecognized CR (US33A), further characterization of a recently proposed CR (UL30A) (4), mapping of an NNT (RNA4.9), and investigation of ASTs. US33A, UL30A and RNA4.9 are included in Fig. 1 and Fig. S3.
The size of the US33A transcript (1.3 kb) (Fig. S2) is consistent with the locations of the 5′ and 3′ ends (Table S1 and Fig. S3). A US34A probe also detected this RNA, plus major 0.9- and minor 0.4-kb transcripts (Fig. S2), suggesting that US34 and US34A are 3′-coterminal with US33A. US33A is conserved in chimpanzee cytomegalovirus (CCMV), which is the closest relative of HCMV (19) (Fig. S4).
UL30A is related to the adjacent gene UL30, conserved among primate cytomegaloviruses, and potentially expressed from a nonconventional initiation codon (ACG) (4). The location of the 3′ end in the middle of UL30 (Table S1 and Fig. S3) is consistent with the major 0.55-kb transcript (Fig. S2). Minor RNAs of 5.7 and 3.5 kb were also apparent, the latter perhaps representing a UL32 transcript that is 3′-coterminal with UL30A and antisense to UL31.
The 5′ and 3′ ends of RNA4.9 (Table S1 and Fig. S3) accord with the transcript size of 4.9 kb (Fig. S2). The location of the 3′ end of a major RNA apparently mapped from partial transcripts is the same as that of this NNT (15, 20, 21). The majority (65.1%) of viral polyA RNA transcription is committed to production of NNTs, with 46.8, 7.9, 10.1, and 0.3% of appropriately oriented DDS reads representing RNA2.7, RNA1.2, RNA4.9, and RNA5.0, respectively. The RNA2.7, RNA1.2, RNA4.9, and RNA5.0 populations comprise 65.7, 26.9, 7.2, and 0.2% of NNT molecules, respectively. These statistics prompt the startling observation that only about one-third of viral polyA RNA transcription is protein-coding. The most abundant mRNA is that of UL22A, which encodes an immunoregulatory RANTES (regulated upon activation, normal T cell expressed and secreted) decoy receptor (22) and accounts for 1.7% of appropriately oriented DDS reads, equivalent to 14% of NNT molecules.
Extensive overlap of protein-coding RNAs specified by opposing strands of the HCMV genome is uncommon. Examples examined include the 3′ ends of UL48A and UL53, which are located well inside UL48 and UL54, respectively (Table S1 and Fig. S3). In contrast, ASTs are apparent in most regions (Fig. 1). Overall, they account for 8.7% of transcription from CRs and are thus generally expressed at lower levels than their sense counterparts, although there are exceptions (e.g., UL29). RACE data indicate that one AST (UL115as) initiates at a specific location, and that several terminate downstream from polyA signals, with instances of a tail-to-tail arrangement in relation to other transcripts (Table S1). Examples (followed by the sense gene and the distance between polyA signals on the two strands) are UL16as (putative UL15A, 35 b), UL30as (UL30A, 36 b), UL121as (UL122, overlapping), UL145as (not arranged tail-to-tail), UL142as (UL141, 193 and 245 b), UL139as (UL138, 28 b), and IRS1as (UL150A, 1219 b).
Characterization of Splicing Patterns.
The primary DS used for splicing analysis was the NDS, rather than the DDS, because fewer biochemical manipulations were involved in its generation. Custom software, capable of coping with genomes that have a high transcript density, was developed to identify reads containing potential splice junctions, from which donor and acceptor sites were proposed. These sites were required to be located adjacent to the canonical GT or AG dinucleotides, respectively, for the following reasons. Processing of RNA samples for deep sequencing involved reverse transcription, during which template switching may occur. This process involves transfer of an elongating cDNA molecule from one template RNA molecule to another, by repriming from a short sequence repeated in both templates (23). Template switching has been acknowledged to generate artifactual junctions in experimental studies (24). Also, the vast majority (∼99%) of eukaryotic splice sites comply with the GT/AG rule (25), all previously recognized HCMV sites (described below) comply, and only a single noncompliant HCMV site was identified in ancillary searches (see below). Exerting the GT/AG rule resulted in the identification of 264 potential donor and 178 acceptor sites.
The number of NDS reads supporting every possible junction between these sites (regardless of order and orientation in the genome, a total of 46,992 combinations) was determined. A heuristic approach was taken to exclude junctions that most probably arose from template switching and happened to comply with the GT/AG rule. Excluded junctions were required to have at least two properties consistent with template switching: originating from highly transcribed regions (especially RNA2.7), supported by low read numbers, not involved in alternative splicing (a site that is alternatively spliced is much less likely to have been identified as a result of template switching than one that is not), characterized by repeats at the cognate genome locations, and not matching the wider consensuses for splice site sequences (25). This analysis led to the identification of 389 junctions supported by one or more reads, generated from 229 potential donor and 132 acceptor sites. Dataset S1 provides full information on these sites, and Fig. 1 shows the locations of the subset of sites supported by >10 reads. A total of 63 donor sites is located in CRs, 115 in ASTs, and 51 between CRs or in regions encoding NNTs. The corresponding numbers for the acceptor sites are 69, 31, and 32, respectively. Overall, 58 CRs contain one or more splice sites on the appropriate strand. The notation (e.g., D+27528) used below to denote a splice site is D (donor) or A (acceptor), + (rightward) or – (leftward), and the nucleotide location of the exon end. A solidus (/) indicates alternative sites, and a caret (^) indicates a junction.
To confirm splice junctions identified from the NDS analysis, a large number (96) was tested by RT-PCR and sequencing (Fig. S1). These were represented by 16–131,826 reads (Table S2 and Dataset S1), a range that extends over nearly four orders of magnitude. A total of 89 junctions was confirmed, as indicated by the colored borders of the cells in Dataset S1. For controls, two junctions classed as having arisen from template switching were tested and found negative (Table S2). RACE experiments carried out to map RNA ends (see below) incidentally confirmed many junctions, including two of the seven not detected by RT-PCR (Dataset S1). Moreover, three of the five not detected by RT-PCR or RACE involved alternatively spliced sites that were unlikely to have been identified as a result of template switching. This left 2 of the 96 tested junctions (D+3005^A+3122 and D+97900^A+97993) unsupported by RT-PCR and RACE and not involved in alternative splicing. Overall, only 26 pairs of sites identified by the NDS analysis were neither involved in alternative splicing nor tested by RT-PCR or RACE. From these considerations, it seems likely that the inferred splicing patterns are a close representation of reality.
Ancillary searches aimed at detecting sites that do not conform to the GT/AG rule but are alternatively spliced to conforming sites yielded a single example. This was D+192592 (as D+192592^A+193169/A+193172), in which the GT dinucleotide is substituted by GC, which is the most common noncanonical donor site (25).
Splice sites identified from the DDS accorded generally with those identified from the NDS, although there was variation in supporting read numbers (Dataset S1). Also, each DS yielded a unique set of sites represented by low read numbers (usually 1). These differences may have been due to the precise stage at which RNA was harvested, the conditions used for sample processing, and stochastic effects occurring during reverse transcription of rare RNAs.
Previously Reported Splicing Patterns.
Studies on various HCMV strains have identified splice sites that are crucial for full-length protein expression. They are located in UL22A (10), UL29 (5), UL33 (26), UL36 (27), UL37 (27), UL89 (10), UL111A (28), UL112 (29), UL119 (10), UL122 (30), UL123 (31, 32), UL128 (33), and UL131A (33). All were confirmed in the present study. Moreover, many reported sites that are not crucial were also confirmed. Examples are those within UL112 (29), US3 (34), and UL122 (30, 35), and those linking UL122 and UL119 (10). The sites in RNA5.0 (10) were also identified. However, other reported sites were not detected. Examples are those within RNA2.7 (10), UL37 (36), and UL123 (IE19 and IE17.5) (37, 38), and most instances in the UL115–UL119 region (39). Lack of detection of these sites does not imply that the original identifications were erroneous, as they may have been expressed at very low levels or at other stages of the replication cycle, or they may not be conserved in strain Merlin.
Previously Unrecognized Splicing Patterns.
The NDS analysis detected splice sites sensitively, but was not capable of determining the extent of exons or the ends of RNAs. These features were investigated in selected transcripts by using RACE (Fig. S1 and Table S1) and NB (Fig. S2), which are less sensitive and identified major species only. Key findings are summarized in Fig. 1 and Fig. S3.
Splicing in two genes (UL8 and US27) was revealed, using sites that are conserved in CCMV. These sites had been predicted previously, but experimental confirmation was unsuccessful (10). UL7 and UL8 are members of the RL11 gene family (Fig. 1), and encode predicted membrane glycoproteins (40). An intron (D+16449^A+16552) links UL7 and UL8, thus 5′-extending the original UL8, with UL7 being expressed from the unspliced RNA (Fig. 1 and Dataset S1). The expression ratio (ER) of spliced to total RNA for UL8 was 0.57, the remainder presumably constituting the unspliced UL7 transcript. US27 is a member of the GPCR (G protein-coupled receptor) gene family (Fig. 1). An intron (D+224024^A+224100) is apparent in the 5′ leader (ER = 0.85) (Fig. S3 and Dataset S1). RACE and NB experiments confirmed that the US27 and US28 transcripts (2.7- and 1.3-kb, respectively) are 3′-coterminal (Table S1 and Fig. S2).
Five acceptor sites are notable in being spliced from one or another of a large number of upstream exons: A–8196 (25 exons), A+108854 (46 exons), A+135253 (22 exons), A+174081 (9 exons), and a pair at A+193169/A+193172 (11 exons) (Dataset S1). Excepting one of the pair, these superacceptor sites (SASs) are conserved in CCMV, and each is located upstream from a potential CR (RL8A, UL74A, UL92, UL124, and UL150A, respectively) (Fig. S3). The observation that RL8A, UL92, UL124, and UL150A contain a potential ATG initiation codon suggests that upstream exons may form untranslated leaders, although this does not rule out the possibility that some may 5′-extend the CR. In contrast, UL74A lacks an initiation codon, and must be supplied with one by at least one upstream exon. Because SASs form an unusual class of acceptor site, four of the five examples were investigated in further detail.
The partial transcript arrangement in the vicinity of the SAS at A–8196 has been deduced previously (12, 40, 41), and was extended to include transcripts from previously unrecognized genes RL8A, which is downstream from the SAS, and RL9A (Fig. 1 and Fig. S3). Both predicted proteins contain potential transmembrane regions and are conserved in CCMV (Fig. S4). In RACE experiments, four 5′ ends of upstream exons spliced to RL8A were identified, and two 5′ ends for RL9A, which is not spliced, were mapped (Table S1 and Figs. S1 and S3). Both genes share a 3′ end with RNA1.2 at 6755. They appear to be expressed at low levels, as transcripts of the sizes inferred from RACE were not detected by NB. The 4.2-kb transcript detected using an RL8A probe (Fig. S2) is too large and of unknown provenance, and no RL9A transcripts were detected.
The UL74A CR is preceded by the SAS at A+108854, conserved among β-herpesviruses, and encodes a predicted membrane glycoprotein (3). Four 5′ ends of upstream exons were identified by RACE, and the 3′ end mapped to 109088 (Table S1 and Figs. S1 and S3). These locations are consistent with the major 0.8-kb transcript (UL73; ER = 0.96) and heterogeneous 0.45-kb transcripts (UL74A; ER = 0.28) detected by NB (Fig. S2). The origin of the minor 1.5-kb RNA is unknown. Some of the features of UL74A in strain Merlin are equivalent to those for the corresponding exon (UL73.5) in strain AD169 (3). In particular, UL74A acquires an initiation codon from the upstream exon spliced at D+108155 (Fig. 1 and Fig. S3). It is not known whether initiation codons are also supplied by other upstream exons, thus generating UL74A proteins with heterogeneous N termini. Many of the upstream exons, and others downstream from UL74A, are alternatively spliced to the SAS at A+135253, which is upstream from a recognized ORF, UL92 (Dataset S1). This SAS was not characterized further.
A single 5′ end of an upstream exon spliced to the SAS at A+174081, which precedes UL124, was identified, and the 3′ end mapped to 174649 (Table S1 and Figs. S1 and S3). UL124 is conserved in CCMV and encodes a predicted membrane glycoprotein (19). The major 0.7-kb transcript detected by NB is consistent with these locations (Fig. S2). A minor 5.5-kb transcript of unknown provenance was also apparent.
Four 5′ ends of upstream exons spliced to the SASs at A+193169/A+193172 were identified by RACE, and the 3′ end mapped to 194348 (Table S1 and Figs. S1 and S3). The two protein-coding exons of previously unrecognized gene UL150A (ER = 0.97), separated by an intron (D+193542^A+193654), are located downstream from the SASs (Fig. 1 and Fig. S3). The splice sites between the protein-coding exons are conserved in CCMV, as is the encoded protein (Fig. S4). The intervening intron specifies one of the HCMV miRNAs (miR-UL148D) (17). Sizes at the lower end of the heterogeneous transcripts (1.2–1.6 kb) detected by NB are consistent with the RACE data (Fig. S2). The UL150A CR is unusual in that it is entirely antisense to a larger, unspliced CR (UL150; 7.5-kb RNA) (Fig. 1 and Figs. S2 and S3), which is also conserved in CCMV (19). In the region of overlap, the amino acid sequence of UL150A is better conserved (47% identical) than that of UL150 (40%), and codon bias is marginally more favorable (0.94 and 0.93, respectively, using the UL54, UL82, and UL86 CRs for calibration). The prediction of UL150A as a CR was tested by expressing V5-tagged UL150A proteins in the context of HCMV infection. A doublet at ∼34 kDa and a minor species at 50 kDa were detected at 1, 3, and 6 d after infection (Fig. 2). The predicted mass of the tagged primary translation product is 31 kDa. The detection of more than one protein might reflect protein processing or splicing from upstream exons that 5′-extend the CR.
Fig. 2.
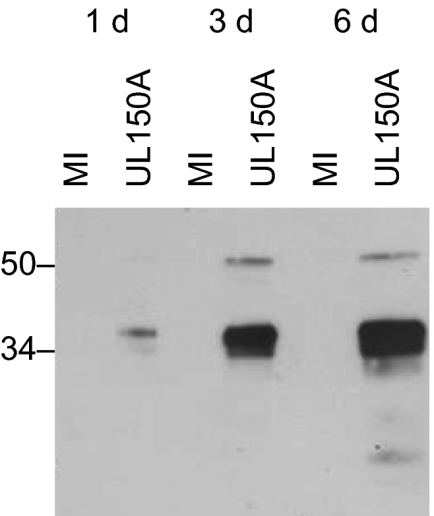
Immunoblot analysis of V5-tagged UL150A proteins expressed in the context of HCMV. Proteins harvested at 1, 3, and 6 d after infection from mock-infected cells (MI) or cells infected with the HCMV recombinant (UL150A) were probed using a V5-Tag antibody. Estimated masses (kDa) are shown.
The NDS analysis also yielded evidence for long-range splicing, some of which was supported by RACE data, for example, between US9 and the RL8A SAS (D–204854^A–8196) (Dataset S1). Moreover, splicing was detected between exons that are located out of order in, or on different strands of, the genome. For example, some of the exons spliced to the RL8A or UL150A SASs were alternatively spliced to the UL74A SAS (Dataset S1). These junctions may have resulted from long-range transcription of concatemeric genomes, in which UL and US are present in either orientation.
Discussion
A number of technical limitations apply to the deep-sequencing approach used. Certain spliced polyA RNAs would not have been detected: those containing very short exons, and those not originating via alternative splicing and either consisting of exons mapping <50 b or >32 kb apart in the genome or using noncanonical splicing. Moreover, the data did not reliably facilitate the characterization of intact transcripts, including the locations of 5′ and 3′ ends. Points of interest in this regard were tested by RT-PCR, RACE, and NB. Also, nonpolyA transcripts (including miRNAs) would not have been represented.
The information arising from the study is as follows. Transcripts encoded by four previously unrecognized genes (RL8A, RL9A, UL150A, and US33A) were mapped, and the UL150A CR, which completely overlaps that of UL150, was shown to be expressed as protein during virus infection. The number of NNTs was expanded to four (RNA2.7, RNA1.2, RNA4.9, and RNA5.0). ASTs were detected throughout the genome, some having discrete 5′ and 3′ ends and some mapping tail-to-tail with other RNAs. Splicing is more common than recognized previously, and there is evidence for long-range transcription. UL8 and UL150A were added to the genes comprising multiple coding exons. Alternative splicing is frequent, with four genes (RL8A, UL74A, UL124, and UL150A) and perhaps one other (UL92) expressed via SASs.
These findings demonstrate that HCMV transcription is more complex and multifaceted than recognized previously, with more CRs, splicing, NNTs, and ASTs having become apparent. This has implications for the potential sophistication of HCMV functionality during infection. Approximately one-third (58/170) of HCMV CRs contain splice sites, and the extent to which this affects viral protein-coding capacity is now a question of great interest. Some spliced RNAs are clearly crucial for expression of full-length proteins, and others that are not so obviously in this category may nonetheless have important roles. However, other spliced RNAs may be nonfunctional, representing transcripts that undergo a degree of splicing merely because their synthesis happens to traverse splice sites. This may apply to spliced RNAs that are very rare in comparison with their unspliced counterparts (e.g., ER = 0.003 for D–181199^A–181096 in UL146). Members of one class of noncoding RNAs, NNTs, are encoded by dedicated regions of the genome and likely to have important roles, and more examples probably await discovery. A second class of noncoding RNAs, ASTs, has its historical origins in antiparallel (symmetric) transcripts, which were first reported several decades ago in herpes simplex virus (42). The most extensive study of HCMV to date (15) used cDNA cloning of strain AD169 transcripts to detect ASTs in 34 of the CRs annotated in strain Merlin, and three (UL24as, UL36as, and UL102as) were characterized further by NB. The DDS analysis indicates that ASTs are associated with most CRs (Fig. 1), and that complex splicing is evident in some (e.g., the UL114-UL121 region, which contains UL115as) (Table S1 and Figs. S1 and S3). The functions of ASTs are unknown, and it is possible that some ASTs result from transcriptional profligacy and have none. However, the observation that the 3′ ends of some ASTs appear to have been under evolutionary constraint in relation to neighboring genes implies that they may be important. If so, roles can only be guessed at this stage, with one obvious possibility being to regulate sense strand expression. Some ASTs may exert their effects via cleavage into miRNAs (which represent another layer of genomic complexity) or translation into short oligopeptides (43). However, in these models only a very minor proportion of AST content would be functional, leaving the role of the major proportion in question. One practical outcome of HCMV transcript complexity is that studies using mutants should be conducted with a heightened degree of circumspection.
Deep sequencing represents a stage in HCMV genomics that complements and extends previous approaches, such as microarray-based analyses (44). We have demonstrated the capability of this approach to provide high-resolution information on HCMV transcription and splicing. It can be extended readily to studies of HCMV transcription in other strains and cell types, expression kinetics during lytic and latent infections, comparative properties of protein-coding and noncoding RNAs, and transcription in vivo. It can also be applied to the genomics of other nuclear DNA viruses.
Methods
Preparation of RNA.
HCMV strain Merlin, which is stable in vitro by virtue of point mutations in two genes (RL13 and UL128) (45), was grown at 37 °C in human fetal foreskin fibroblast cells (HFFFs; HFFF2, European Collection of Cell Cultures 86031405) in DMEM containing 10% FCS. HFFF2 monolayers (75-cm2) were infected with 5 pfu per cell of HCMV and incubated for 72 h. Total infected cell RNA was isolated using TRI reagent (Sigma) or an RNeasy Midi Kit (Qiagen). Three preparations were made, one in 2005 and two in 2010.
Deep Sequencing.
Data were generated by using an Illumina Genome Analyzer IIx in The Sir Henry Wellcome Functional Genomics Facility (University of Glasgow). For the NDS, an Illumina mRNA-Seq prep kit was used: polyA RNA was selected from 1 μg of the 2005 RNA preparation, fragmented, and subjected to first- and second-strand cDNA synthesis by using random primers. The DNA was then end-repaired, 3′-adenylated, ligated to adapters, PCR-amplified, and size-selected (mode = 245 bp). For the DDS, Illumina mRNA-Seq prep and small RNA sample prep kits were used: polyA RNA was selected from 2 μg of one of the 2010 RNA preparations, fragmented, phosphatased, phosphorylated, ligated to RNA adapters, subjected to first-strand cDNA synthesis and PCR-amplified by using adapter-specific primers, and size-selected (mode = 188 bp).
Transcription Levels.
Unaligned, 76-b reads with associated phred quality scores were generated by using SCS v2.6 or v2.8, RTA v1.12, and CASAVA v1.7 (Illumina), and stored in fastq files. The NDS and DDS were then assembled against a reference sequence by using Maq v0.7.1 under default alignment settings (46). The assembly was visualized by using Tablet v1.10.05.21 (47). The reference consisted of the strain Merlin genome sequence (GenBank accession no. AY446894.2) (2) or a truncated version lacking all but 50 b of the terminal repeats (ab and ca). DDS reads were also separated into DSs representing rightward and leftward transcripts and assembled independently. Transcription profiles were calculated from the number of reads commencing in 10-b windows and summing these over 100-b windows (10-b increment for each).
Splice Site Detection.
A primary DS (NDS or DDS) was processed to gather reads containing potential splice junctions by the following steps. The rationale was that at least 21 b of the 5′ and 3′ parts of a junction-containing read must match two regions at least 50 b apart in the HCMV genome. Perl scripts are available from the authors.
i) Duplicate reads were removed, and a regular expression (regexp) search was conducted for reads that contain exact matches between the 21 b at the 5′ and 3′ ends (positions 1–21 and 56–76) and two different regions in the reference, and that meet the following criteria: (i) matches on the same strand of the reference, (ii) order of matching sequences in the read preserved in the reference, and (iii) matching sequences separated in the reference by 50 b to 32 kb.
ii) Each read was divided into two after position 21, and a regexp search was conducted for exact matches of the entire 5′ and 3′ parts to the reference. If criteria i–iii were met and the intervening genome sequence began with GT and ended with AG, a pair of 100 b exon–end sequences was generated. This procedure was repeated iteratively until position 55 was reached, to ensure that sequence redundancy at exon–ends did not result in failure to identify precise locations. Two sets of exon–end sequences were generated from the reads, one corresponding to donor sites and the other to acceptor sites.
iii) Step ii was repeated for the reverse complement of the reference, and the sets of exon–end sequences were combined with those sets from step ii.
iv) To avoid problems associated with lower-quality data toward the 3′ ends of reads, they were trimmed to 60 and 50 b and steps i–iii were repeated with appropriate modifications. The sets of exon–end sequences were combined with those from step iii, and duplicates were removed.
v) The exon–end sequences from step iv were joined to give a set of 200-b junctions comprising every donor site joined to every acceptor site in pairwise combinations. The central 20 b was extracted from each, and a sub-DS of reads specific to that sequence was generated by screening the primary DS for exact matches. Each DS was then assembled against the cognate 200-b sequence by using Maq. Thus, reads supporting each junction were required to match the reference exactly in the 20-b sequence (i.e., 10 b from each exon) and substantially in the flanking regions (<3 b mismatch in 56 b).
ERs for selected spliced RNAs were calculated as described in step v, by extracting 20-b sequences (10 b each side of the spliced exon–exon junction or unspliced exon–intron junctions) from the DDS, generating read sub-DSs, assembling them against the cognate spliced or unspliced 200-b sequence, and counting the numbers of appropriately oriented reads. Complete splicing would yield an ER of 1.
RNA Mapping.
RT-PCR, RACE, and NB were conducted using the 2010 RNA preparations. Titanium One-Step RT-PCR and SMARTer RACE Kits (Clontech) were used with primers listed in Tables S2 and S3, and products were sequenced directly or from plasmid clones. For NB, RNA was transferred from 1% formaldehyde-agarose gels to membranes, and hybridized with biotin-labeled, single-stranded RNA probes produced from plasmids (checked by sequencing) using a DIG Northern Starter Kit (Roche) and primers listed in Table S4.
Tagged Protein Expression.
Published methods (45) were used to introduce a V5 epitope tag (checked by sequencing) at the C terminus of the UL150A protein by modifying bacterial artificial chromosome pAL1111, which contains the strain Merlin genome, and generating a recombinant virus (Table S5). HFFFs were mock-infected or infected at 5 pfu per cell and incubated in FCS-free DMEM for 1 or 2 d, or in DMEM containing 10% FCS for 3 d and then FCS-free DMEM for 3 d. Cell extracts were electrophoresed in 10% SDS/polyacrylamide gels, transferred to membranes, probed with a V5-Tag mouse monoclonal antibody (MCA1360; AbD Serotec), and reprobed with horseradish peroxidase-conjugated goat anti-mouse IgG (170-6516; Bio-Rad). Signal was detected by using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Supplementary Material
Acknowledgments
We thank Wai Kwong Lee and Stewart Laing (British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow) for DNA sequencing services and Elaine Douglas and Fiona Jamieson for technical assistance. This work was funded by the UK Medical Research Council and The Wellcome Trust.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence reported in this paper has been deposited in the European Nucleotide Archive (www.ebi.ac.uk/ena/; accession no. ERA031126).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115861108/-/DCSupplemental.
References
- 1.Davison AJ. In: Human Herpesviruses: Biology, Therapy and Immunoprophylaxis. Arvin A, et al., editors. Cambridge, UK: Cambridge University Press; 2007. pp. 10–26. [PubMed] [Google Scholar]
- 2.Dolan A, et al. Genetic content of wild-type human cytomegalovirus. J Gen Virol. 2004;85:1301–1312. doi: 10.1099/vir.0.79888-0. [DOI] [PubMed] [Google Scholar]
- 3.Scalzo AA, Forbes CA, Smith LM, Loh LC. Transcriptional analysis of human cytomegalovirus and rat cytomegalovirus homologues of the M73/M73.5 spliced gene family. Arch Virol. 2009;154(1):65–75. doi: 10.1007/s00705-008-0274-8. [DOI] [PubMed] [Google Scholar]
- 4.Davison AJ. Herpesvirus systematics. Vet Microbiol. 2010;143(1):52–69. doi: 10.1016/j.vetmic.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell DP, Savaryn JP, Moorman NJ, Shenk T, Terhune SS. Human cytomegalovirus UL28 and UL29 open reading frames encode a spliced mRNA and stimulate accumulation of immediate-early RNAs. J Virol. 2009;83:10187–10197. doi: 10.1128/JVI.00396-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bego M, Maciejewski J, Khaiboullina S, Pari G, St Jeor S. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J Virol. 2005;79:11022–11034. doi: 10.1128/JVI.79.17.11022-11034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy E, Shenk T. Human cytomegalovirus genome. Curr Top Microbiol Immunol. 2008;325:1–19. doi: 10.1007/978-3-540-77349-8_1. [DOI] [PubMed] [Google Scholar]
- 8.Varnum SM, et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J Virol. 2004;78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonough SH, Staprans SI, Spector DH. Analysis of the major transcripts encoded by the long repeat of human cytomegalovirus strain AD169. J Virol. 1985;53:711–718. doi: 10.1128/jvi.53.3.711-718.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rawlinson WD, Barrell BG. Spliced transcripts of human cytomegalovirus. J Virol. 1993;67:5502–5513. doi: 10.1128/jvi.67.9.5502-5513.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kulesza CA, Shenk T. Human cytomegalovirus 5-kilobase immediate-early RNA is a stable intron. J Virol. 2004;78:13182–13189. doi: 10.1128/JVI.78.23.13182-13189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutchinson NI, Tocci MJ. Characterization of a major early gene from the human cytomegalovirus long inverted repeat; predicted amino acid sequence of a 30-kDa protein encoded by the 1.2-kb mRNA. Virology. 1986;155(1):172–182. doi: 10.1016/0042-6822(86)90177-7. [DOI] [PubMed] [Google Scholar]
- 13.Reeves MB, Davies AA, McSharry BP, Wilkinson GWG, Sinclair JH. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science. 2007;316:1345–1348. doi: 10.1126/science.1142984. [DOI] [PubMed] [Google Scholar]
- 14.Kulesza CA, Shenk T. Murine cytomegalovirus encodes a stable intron that facilitates persistent replication in the mouse. Proc Natl Acad Sci USA. 2006;103:18302–18307. doi: 10.1073/pnas.0608718103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang G, et al. Antisense transcription in the human cytomegalovirus transcriptome. J Virol. 2007;81:11267–11281. doi: 10.1128/JVI.00007-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma YP, et al. Novel transcripts of human cytomegalovirus clinical strain found by cDNA library screening. Genet Mol Res. 2011;10:566–575. doi: 10.4238/vol10-2gmr1059. [DOI] [PubMed] [Google Scholar]
- 17.Pfeffer S, et al. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 18.Prichard MN, et al. Identification of persistent RNA-DNA hybrid structures within the origin of replication of human cytomegalovirus. J Virol. 1998;72:6997–7004. doi: 10.1128/jvi.72.9.6997-7004.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davison AJ, et al. The human cytomegalovirus genome revisited: Comparison with the chimpanzee cytomegalovirus genome. J Gen Virol. 2003;84(Pt 1):17–28. doi: 10.1099/vir.0.18606-0. [DOI] [PubMed] [Google Scholar]
- 20.Scott GM, Barrell BG, Oram J, Rawlinson WD. Characterisation of transcripts from the human cytomegalovirus genes TRL7, UL20a, UL36, UL65, UL94, US3 and US34. Virus Genes. 2002;24(1):39–48. doi: 10.1023/a:1014033920070. [DOI] [PubMed] [Google Scholar]
- 21.Davis MG, Huang ES. Nucleotide sequence of a human cytomegalovirus DNA fragment encoding a 67-kilodalton phosphorylated viral protein. J Virol. 1985;56(1):7–11. doi: 10.1128/jvi.56.1.7-11.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D, Bresnahan W, Shenk T. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc Natl Acad Sci USA. 2004;101:16642–16647. doi: 10.1073/pnas.0407233101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Temin HM. Retrovirus variation and reverse transcription: Abnormal strand transfers result in retrovirus genetic variation. Proc Natl Acad Sci USA. 1993;90:6900–6903. doi: 10.1073/pnas.90.15.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houseley J, Tollervey D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010;5:e12271. doi: 10.1371/journal.pone.0012271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheth N, et al. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006;34:3955–3967. doi: 10.1093/nar/gkl556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margulies BJ, Browne H, Gibson W. Identification of the human cytomegalovirus G protein-coupled receptor homologue encoded by UL33 in infected cells and enveloped virus particles. Virology. 1996;225(1):111–125. doi: 10.1006/viro.1996.0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kouzarides T, Bankier AT, Satchwell SC, Preddy E, Barrell BG. An immediate early gene of human cytomegalovirus encodes a potential membrane glycoprotein. Virology. 1988;165(1):151–164. doi: 10.1016/0042-6822(88)90668-x. [DOI] [PubMed] [Google Scholar]
- 28.Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10) Proc Natl Acad Sci USA. 2000;97:1695–1700. doi: 10.1073/pnas.97.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright DA, Spector DH. Posttranscriptional regulation of a class of human cytomegalovirus phosphoproteins encoded by an early transcription unit. J Virol. 1989;63:3117–3127. doi: 10.1128/jvi.63.7.3117-3127.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenberg RM, Witte PR, Stinski MF. Multiple spliced and unspliced transcripts from human cytomegalovirus immediate-early region 2 and evidence for a common initiation site within immediate-early region 1. J Virol. 1985;56:665–675. doi: 10.1128/jvi.56.3.665-675.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stenberg RM, Thomsen DR, Stinski MF. Structural analysis of the major immediate early gene of human cytomegalovirus. J Virol. 1984;49(1):190–199. doi: 10.1128/jvi.49.1.190-199.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akrigg A, Wilkinson GWG, Oram JD. The structure of the major immediate early gene of human cytomegalovirus strain AD169. Virus Res. 1985;2(2):107–121. doi: 10.1016/0168-1702(85)90242-4. [DOI] [PubMed] [Google Scholar]
- 33.Akter P, et al. Two novel spliced genes in human cytomegalovirus. J Gen Virol. 2003;84:1117–1122. doi: 10.1099/vir.0.18952-0. [DOI] [PubMed] [Google Scholar]
- 34.Weston K. An enhancer element in the short unique region of human cytomegalovirus regulates the production of a group of abundant immediate early transcripts. Virology. 1988;162:406–416. doi: 10.1016/0042-6822(88)90481-3. [DOI] [PubMed] [Google Scholar]
- 35.Kerry JA, et al. Isolation and characterization of a low-abundance splice variant from the human cytomegalovirus major immediate-early gene region. J Virol. 1995;69:3868–3872. doi: 10.1128/jvi.69.6.3868-3872.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adair R, Liebisch GW, Colberg-Poley AM. Complex alternative processing of human cytomegalovirus UL37 pre-mRNA. J Gen Virol. 2003;84:3353–3358. doi: 10.1099/vir.0.19404-0. [DOI] [PubMed] [Google Scholar]
- 37.Shirakata M, et al. Novel immediate-early protein IE19 of human cytomegalovirus activates the origin recognition complex I promoter in a cooperative manner with IE72. J Virol. 2002;76:3158–3167. doi: 10.1128/JVI.76.7.3158-3167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Awasthi S, Isler JA, Alwine JC. Analysis of splice variants of the immediate-early 1 region of human cytomegalovirus. J Virol. 2004;78:8191–8200. doi: 10.1128/JVI.78.15.8191-8200.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leatham MP, Witte PR, Stinski MF. Alternate promoter selection within a human cytomegalovirus immediate-early and early transcription unit (UL119–115) defines true late transcripts containing open reading frames for putative viral glycoproteins. J Virol. 1991;65:6144–6153. doi: 10.1128/jvi.65.11.6144-6153.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davison AJ, et al. Homology between the human cytomegalovirus RL11 gene family and human adenovirus E3 genes. J Gen Virol. 2003;84:657–663. doi: 10.1099/vir.0.18856-0. [DOI] [PubMed] [Google Scholar]
- 41.Greenaway PJ, Wilkinson GWG. Nucleotide sequence of the most abundantly transcribed early gene of human cytomegalovirus strain AD169. Virus Res. 1987;7(1):17–31. doi: 10.1016/0168-1702(87)90055-4. [DOI] [PubMed] [Google Scholar]
- 42.Kozak M, Roizman B. RNA synthesis in cells infected with herpes simplex virus. IX. Evidence for accumulation of abundant symmetric transcripts in nuclei. J Virol. 1975;15(1):36–40. doi: 10.1128/jvi.15.1.36-40.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu Y, Ganem D. Making sense of antisense: Seemingly noncoding RNAs antisense to the master regulator of Kaposi's sarcoma-associated herpesvirus lytic replication do not regulate that transcript but serve as mRNAs encoding small peptides. J Virol. 2010;84:5465–5475. doi: 10.1128/JVI.02705-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chambers J, et al. DNA microarrays of the complex human cytomegalovirus genome: Profiling kinetic class with drug sensitivity of viral gene expression. J Virol. 1999;73:5757–5766. doi: 10.1128/jvi.73.7.5757-5766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanton RJ, et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest. 2010;120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milne I, et al. Tablet—Next generation sequence assembly visualization. Bioinformatics. 2010;26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.