

Abstract
Epithelioid malignant peripheral nerve sheath tumors arising in pre-existing schwannomas are extremely rare. We report an unusual example occurring in a patient with multiple schwannomas (schwannomatosis), all but one of which showed “neuroblastoma-like” histology. By immunohistochemistry, both the epithelioid malignant peripheral nerve sheath tumor and the schwannomas showed a complete loss of the Smarcb1 protein. Subsequent genetic evaluation revealed the presence of a novel germline mutation in the SMARCB1/INI1 gene in the patient and three of her children, two of whom were diagnosed with atypical teratoid/rhabdoid tumors of the brain.
Keywords: Malignant peripheral nerve sheath tumor, schwannoma, schwannomatosis, SMARCB1/INI1
Introduction
Schwannomas are benign, relatively common, nearly always solitary peripheral nerve sheath neoplasms composed exclusively of Schwann cells. Multiple schwannomas, frequently showing a plexiform growth pattern, occur in both neurofibromatosis type 2 (NF2) and in schwannomatosis (15). Schwannomatosis, defined by the absence of the vestibular schwannomas, the key feature of NF2, is a familial or sporadic syndrome which may be associated with germline mutations of the SMARCB1/INI1 gene (1, 16). While the genetic basis of schwannomatosis remains to be definitely established, an estimated 30% of cases of inherited schwannomatosis may be associated with SMARCB1 mutations (7) and some individuals with germline SMARCB1 mutations show a predisposition to schwannomatosis (2, 10, 11).
Malignant transformation is extremely rare in schwannomas, with fewer than 20 reported cases (18, 20, 22, 26). Unlike neurofibromas, which most often give rise to conventional spindle cell malignant peripheral nerve sheath tumors, sarcomas arising in schwannomas frequently show epithelioid morphology, including epithelioid malignant peripheral nerve sheath tumor and epithelioid angiosarcoma (18, 24, 26).
Herein we report a highly unusual example of epithelioid malignant peripheral nerve sheath tumor arising in a patient with schwannomatosis, all but one of which showed features of the rare “neuroblastoma-like” variant. Subsequent evaluation showed this patient to carry a novel germline SMARCB1 mutation. To the best of our knowledge this is the first report of this kind.
Clinical history
A previously well 31 year-old woman presented with a several-year history of a painless, palpable mass along the lateral aspect of the left lower leg. During her third pregnancy, the mass rapidly enlarged, causing numbness of the left foot and prompting her to seek medical attention. Magnetic resonance imaging (MRI) revealed multiple masses arising from the superficial branch of the common peroneal nerve. The largest mass measured 6.7 cm and demonstrated heterogeneous internal signal on T1-weighted imaging with mixed areas of isointense and hyperintense signal compared to skeletal muscle (Figure 1A,1B). More inferiorly in the lower leg were several smaller masses with more uniform signal hypointense to skeletal muscle. A needle core biopsy demonstrated features of schwannoma. Based upon MRI findings, the possibility of schwannomatosis was suggested. Further MR imaging of the right upper extremity above the elbow, demonstrated additional masses associated with the right ulnar nerve (Figure 1C). The tumors were subsequently resected in a conservative fashion. Cranio-spinal magnetic resonance imaging was negative for intracerebral lesions, in particular vestibular schwannomas. This patient thus met the clinicopathologic criteria for schwannomatosis (1, 16).
Figure 1.

Sagittal T1-weighted MR image (A) of the lower leg demonstrating several benign schwannomas with homogeneously decreased T1 signal (arrows). More proximally, there was a larger lesion with heterogenous T1 signal, representing the biopsy proven epithelioid malignant peripheral nerve sheath tumor (arrowhead). Axial T1-weighted MR image (B) established the anatomic continuity of the malignant peripheral nerve sheath tumor with the traversing segment of the superficial peroneal nerve. Coronal T2-weighted fat-suppressed MR image of the right upper extremity above the elbow (C) demonstrating two benign schwannomas in continuity with the traversing segment of the ulnar nerve (arrows).
Following the demonstration of a Smarcb1-negative epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma (see Pathologic Findings, below), the patient was referred for adjuvant radiation therapy. Further discussion revealed that 6 years earlier, her then 6-month old son had been found to have a 3.7 cm cerebellar mass, ultimately diagnosed as an atypical teratoid/ rhabdoid tumor, without ancillary Smarcb1 analysis. In light of the mother's diagnosis, this tumor was re-reviewed and shown to be Smarcb1-negative (data not shown). Despite aggressive treatment, including chemoradiation and autologous stem cell transplantation, the child died at 18 months of age.
Given the possibility that this family might harbor a germ-line SMARCB1 mutation genetic counseling was initiated. During this process, the patient's younger son presented at 18 months of age with symptoms of raised intracranial pressure, and was subsequently found to have a 3.5 cm atypical teratoid/rhabdoid tumor of the cerebellum. This child is currently undergoing treatment according to the Children's Oncology Group protocol (ACNS0333).
Materials and Methods
Tissues were fixed in buffered 10% formalin, embedded in paraffin, sectioned into 4 micron slices and stained with hematoxylin and eosin as per routine protocol. Immunohistochemistry was performed at the University of Alberta on deparaffinized, rehydrated tissue sections with antibody-specific antigen retrieval techniques using the Ventana Benchmark XT (Ventana Medical Systems, Tucson, USA) automated system for detection of the following primary antigens: vimentin (pre-diluted, Ventana), S100 protein (Dako, Glostrup, Denmark, 1:300), and low-molecular weigh cytokeratins (CAM5.2, Becton Dickinson, Franklin Lakes, N.J., U.S.A. pre-diluted). Immunohistochemistry for Smarcb1 was performed at Mayo Clinic (clone 25, 1:40, BD Transduction Laboratories, Franklin Lakes, NJ, USA) using steam heat induced epitope retrieval and the Dako Envision detection system (Dako Corp., Carpinteria, CA).
Pathologic Findings
Histologic sections of the largest peroneal nerve mass demonstrated in part features of schwannoma, including encapsulation, thick-walled blood vessels, Verocay bodies, and a single small “neuroblastoma-like” rosette (Figures 2A and B). However, much of this specimen consisted of a clearly malignant-appearing neoplasm with epithelioid to rhabdoid cytomorphology, frequent mitotic activity and geographic necrosis (Figure 2C). By immunohistochemistry both the schwannoma and the sarcomatous component were diffusely positive for S100 protein (Figure 2D) and vimentin, characteristic of epithelioid malignant peripheral nerve sheath tumor arising within a schwannoma. Other markers were negative. Because of the partially rhabdoid features of the epithelioid malignant peripheral nerve sheath tumor (Figure 2E), immunohistochemistry for Smarcb1 protein was performed, showing complete loss of expression, with retained expression in adjacent non-neoplastic tissues (Figure 2F). The smaller tumors arising from the common peroneal and ulnar nerves demonstrated typical features of “neuroblastoma-like” schwannoma as described (5), including giant rosette-like structures with a central collagenous core surrounded by amitotic, moderately hyperchromatic “small round blue cells” (Figures 3A-C). The neuroblastoma-like schwannomas were similarly positive for S100 protein (Figure 3D) and negative for Smarcb1 protein expression (Figure 3E).
Figure 2.

Low-power view of epithelioid malignant peripheral nerve sheath tumor arising in schwannoma (A). Hypocellular schwannoma (upper left hand corner), showing a single small rosette-like structure, juxtaposed to epithelioid malignant peripheral nerve sheath tumor (B). High-power view of epithelioid malignant peripheral nerve sheath tumor (C). S100 protein-expression in epithelioid malignant peripheral nerve sheath tumor (D). Rhabdoid cytomorphology in epithelioid malignant peripheral nerve sheath tumor (E). Complete absence of Smarcb1 protein expression, with retained expression in normal endothelial cells (F).
Figure 3.

Low-power view of neuroblastoma-like schwannoma, arising adjacent to a normal nerve (A). Antoni A and B zones in neuroblastoma-like schwannoma (B). Giant collagenous rosette surrounded by small, moderately hyperchromatic Schwann cells (C). The neuroblastoma-like schwannoma was diffusely S100 protein-positive (D) and showed complete loss of Smarcb1 expression (E).
Molecular Analysis
Genetic analysis was performed on formalin fixed tumor tissue derived from both the mother and her older (deceased) son. DNA was isolated using a Qiagen Gentra PureGene kit (Germantown, MD) and the samples were sequenced after PCR amplification, and screened for copy number using multiplex ligation-dependent probe amplification (MLPA), according to previously reported methods (3, 4, 12). Both tumor samples demonstrated a deletion of the long arm of chromosome 22, as well as a two base pair insertion in exon 3 of the SMARCB1 gene (c.245_246insAT). DNA from the blood sample of the mother was also sequenced for exon 3, and demonstrated the same two base pair insertion (Figure 4). Subsequent testing on blood samples from the mother's parents and two other children (the younger son and his sister) was then performed. Neither of the mother's parents was found to carry the mutation, suggesting it was a de novo germline abnormality in the mother, and an inherited germline mutation in her children (Figure 5). All three of the children were found to carry the mutation. Thus far, the daughter is unaffected and is being regularly screened by MRI imaging of the brain. Additionally, blood samples from the mother's brother and sister were analyzed, and found to lack the mutation.
Figure 4.

SMARCB1 exon 3 sequencing on the schwannoma from patient 1 demonstrates a 2 bp insertion (c.245_246insAT). B. SMARCB1 exon 3 sequencing on the rhabdoid tumor from patient 2 demonstrates the same 2 bp insertion as his mother. C. SMARCB1 exon 3 sequencing on the blood from the parents of patient 1 is normal, indicating that patient 1 has a de novo mutation that was inherited by her son.
Figure 5.
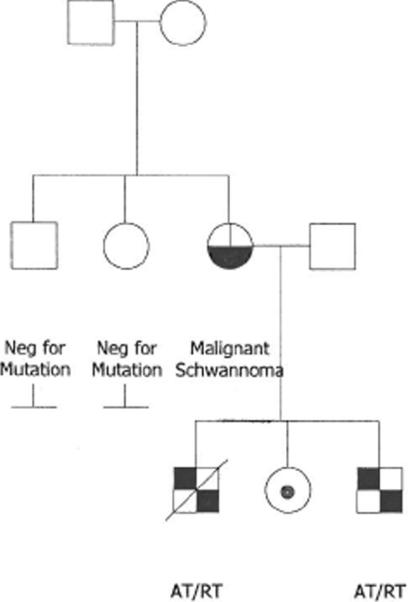
The mother (black and white circle) demonstrated schwannomatosis with malignant transformation. All of her children inherited her SMARCB1 mutation. Her older son died of AT/RT and her younger son is currently being treated for AT/RT (black and white squares). Her daughter is currently asymptomatic. Her parents and siblings were negative for the mutation.
Discussion
Schwannomatosis, defined by the presence of multiple non-dermal schwannomas in the absence of vestibular schwannomas or of a known constitutional NF2 mutation, is very rare, having an annual incidence of 0.58 cases/ 1 million persons (6). Approximately 20% of cases are familial (2). The overwhelming majority of previously reported schwannomatosis-associated schwannomas have been benign (1, 15, 17). However, a very small number of malignant peripheral nerve sheath tumors arising in this clinical setting have recently been reported, including 2 examples (in 14 patients) reported by Gonzalvo and co-workers (6) and one other case reported by Hadfield et al (7). Regrettably, neither of these papers illustrates or describes in detail the pathological features of these tumors, and it is unclear whether these tumors arose within pre-existing schwannomas. Swensen and colleagues have also recently described the familial occurrence of malignant rhabdoid tumors in a family with schwannomatosis and germline duplication of exon 6 of SMARCB1 (23). Several additional families in which a germline mutation in the SMARCB1 gene predisposed carriers to schwannomas or rhabdoid tumors have also recently been described (4,5).
Only a very small number of malignant peripheral nerve sheath tumors have been previously reported as arising within schwannomas (18, 26). This phenomenon was first described in detail by Woodruff and colleagues in 1994, who reported 2 cases of malignant peripheral nerve sheath tumor arising in schwannoma and performed a comprehensive review of all previously reported cases, accepting 7 as bona fide (26). Interestingly, epithelioid morphology was seen in 7 of 9 (78%) of these tumors; the epithelioid variant accounts for 5% or less of all malignant peripheral nerve sheath tumors (25) . This subject was revisited in 2001 by McMenamin and Fletcher (18), who reported 4 cases of epithelioid malignant peripheral nerve sheath tumor arising in schwannoma, including 2 new cases and 2 cases previously reported by Nayler et al (20) and Mikami et al (19). McMenamin and Fletcher also described the presence within other schwannomas of small clusters of atypical epithelioid cells, a phenomenon they termed “epithelioid malignant change”. The significance of “epithelioid malignant change” in schwannoma remains to be established. We did not observe this in any of the schwannomas in our case.
We are not aware of a previous report that has examined Smarcb1 expression in epithelioid malignant peripheral nerve sheath tumor arising in schwannoma, although loss of Smarcb1 expression has been reported by Hornick et al in 50% of epithelioid malignant peripheral nerve sheath tumors (9). Sporadic schwannomas show retained expression of Smarcb1, whereas familial schwannomatosis-associated schwannomas show a distinctive pattern of partial or “mosaic” loss (21). In the present case, we observed complete loss of Smarb1 expression in both the epithelioid malignant peripheral nerve sheath tumor and in the neuroblastoma-like schwannomas, most likely reflecting the presence of both the germline SMARCB1 mutation and a second somatic event.
An additional interesting aspect of this case is the presence of multiple “neuroblastoma-like” schwannomas. Neuroblastoma-like schwannoma, described by Goldblum and colleagues in 1994, is a very rare variant of schwannoma defined by the presence of large, collagenous, rosette-like structures surrounded by small, round, somewhat hyperchromatic Schwann cells with scant cytoplasm (5). We are not aware of a prior report of multiple neuroblastoma-like schwannomas, or an association with schwannomatosis.
Finally, this is the first report of a germline SMARCB1 mutation in a patient with an epithelioid malignant peripheral nerve sheath tumor arising within a schwannoma, specifically a two base pair insertion in exon 3 of the SMARCB1 gene (c.245_246insAT). This mutation differs from those SMARCB1 mutations previously reported in 2 cases of malignant extrarenal rhabdoid tumor arising in schwannomatosis, including a large duplication involving exon 6 (23) and a point mutation in exon 4 (4). In all 3 cases Smarcb1 protein expression was lost. Smarcb1, an obligate component of the chromatin-remodeling SWI/SNF complex, functions as a tumor suppressor (8). Thus, mutations in familial SMARCB1-associated tumors typically confer bi-allelic loss-of-function, with a germline “first hit” of one SMARCB1 allele, followed by a somatic “second hit”. In the present case, loss of the long arm of chromosome 22 was present in both the mother and her older (deceased) son, representing the somatic allelic loss. It is unclear how loss of functional Smarcb1 results in tumor formation, although a recent report suggests that loss of functional Smarcb1results in aberrant signaling through the Hedgehog pathway, leading to tumorigenesis (13). Individuals with germline SMARCB1 mutations have a variable risk of malignant rhabdoid tumor development (14).
In summary, we have reported an extraordinary example of epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline mutation in the SMARCB1 gene. Cases such as this may provide insight into the mechanisms underlying the rare phenomenon of malignant transformation in schwannomas, in particular the critical role of SMARCB1. In the light of the present report, as well as prior reports of malignancy arising in schwannomatosis, patients with this rare syndrome should be carefully followed for change in tumor size or imaging characteristics, with the hope that early detection of malignant change may improve clinical outcome. Germline testing of such individuals is also critical, since carriers have a theoretical 50% risk of passing an altered allele to their children. It is clear that the same mutation in the family can predispose to both schwannomas, as in the mother in this family, as well as to malignant rhabdoid tumors, as seen in her children.
Footnotes
Disclosures: The author(s) have no conflicts of interest or funding to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baser ME, Friedman JM, Evans DG. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology. 2006;66:730–732. doi: 10.1212/01.wnl.0000201190.89751.41. [DOI] [PubMed] [Google Scholar]
- 2.Boyd C, Smith MJ, Kluwe L, et al. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clinical genetics. 2008;74:358–366. doi: 10.1111/j.1399-0004.2008.01060.x. [DOI] [PubMed] [Google Scholar]
- 3.Bruggers CS, Bleyl SB, Pysher T, et al. Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatric blood & cancer. 2011;56:1026–1031. doi: 10.1002/pbc.22757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eaton KW, Tooke LS, Wainwright LM, et al. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatric blood & cancer. 2011;56:7–15. doi: 10.1002/pbc.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldblum JR, Beals TF, Weiss SW. Neuroblastoma-like neurilemoma. The American journal of surgical pathology. 1994;18:266–273. doi: 10.1097/00000478-199403000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalvo A, Fowler A, Cook RJ, et al. Schwannomatosis, sporadic schwannomatosis, and familial schwannomatosis: a surgical series with long-term follow-up. Clinical article. Journal of neurosurgery. 2011;114:756–762. doi: 10.3171/2010.8.JNS091900. [DOI] [PubMed] [Google Scholar]
- 7.Hadfield KD, Newman WG, Bowers NL, et al. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. Journal of medical genetics. 2008;45:332–339. doi: 10.1136/jmg.2007.056499. [DOI] [PubMed] [Google Scholar]
- 8.Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420. doi: 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. The American journal of surgical pathology. 2009;33:542–550. doi: 10.1097/PAS.0b013e3181882c54. [DOI] [PubMed] [Google Scholar]
- 10.Hulsebos TJ, Kenter SB, Jakobs ME, et al. SMARCB1/INI1 maternal germ line mosaicism in schwannomatosis. Clinical genetics. 2010;77:86–91. doi: 10.1111/j.1399-0004.2009.01249.x. [DOI] [PubMed] [Google Scholar]
- 11.Hulsebos TJ, Plomp AS, Wolterman RA, et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. American journal of human genetics. 2007;80:805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson EM, Sievert AJ, Gai X, et al. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:1923–1930. doi: 10.1158/1078-0432.CCR-08-2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jagani Z, Mora-Blanco EL, Sansam CG, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nature medicine. 2010;16:1429–1433. doi: 10.1038/nm.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janson K, Nedzi LA, David O, et al. Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatric blood & cancer. 2006;47:279–284. doi: 10.1002/pbc.20622. [DOI] [PubMed] [Google Scholar]
- 15.Lu-Emerson C, Plotkin SR. The neurofibromatoses. Part 2: NF2 and schwannomatosis. Rev Neurol Dis. 2009;6:E81–86. [PubMed] [Google Scholar]
- 16.MacCollin M, Chiocca EA, Evans DG, et al. Diagnostic criteria for schwannomatosis. Neurology. 2005;64:1838–1845. doi: 10.1212/01.WNL.0000163982.78900.AD. [DOI] [PubMed] [Google Scholar]
- 17.MacCollin M, Woodfin W, Kronn D, et al. Schwannomatosis: a clinical and pathologic study. Neurology. 1996;46:1072–1079. doi: 10.1212/wnl.46.4.1072. [DOI] [PubMed] [Google Scholar]
- 18.McMenamin ME, Fletcher CD. Expanding the spectrum of malignant change in schwannomas: epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: a study of 17 cases. The American journal of surgical pathology. 2001;25:13–25. doi: 10.1097/00000478-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 19.Mikami Y, Hidaka T, Akisada T, et al. Malignant peripheral nerve sheath tumor arising in benign ancient schwannoma: a case report with an immunohistochemical study. Pathol Int. 2000;50:156–161. doi: 10.1046/j.1440-1827.2000.01019.x. [DOI] [PubMed] [Google Scholar]
- 20.Nayler SJ, Leiman G, Omar T, et al. Malignant transformation in a schwannoma. Histopathology. 1996;29:189–192. doi: 10.1046/j.1365-2559.1996.d01-491.x. [DOI] [PubMed] [Google Scholar]
- 21.Patil S, Perry A, Maccollin M, et al. Immunohistochemical analysis supports a role for INI1/SMARCB1 in hereditary forms of schwannomas, but not in solitary, sporadic schwannomas. Brain pathology. 2008;18:517–519. doi: 10.1111/j.1750-3639.2008.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robson DK, Ironside JW. Malignant peripheral nerve sheath tumour arising in a schwannoma. Histopathology. 1990;16:295–297. doi: 10.1111/j.1365-2559.1990.tb01118.x. [DOI] [PubMed] [Google Scholar]
- 23.Swensen JJ, Keyser J, Coffin CM, et al. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. Journal of medical genetics. 2009;46:68–72. doi: 10.1136/jmg.2008.060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trassard M, Le Doussal V, Bui BN, et al. Angiosarcoma arising in a solitary schwannoma (neurilemoma) of the sciatic nerve. American Journal of Surgical Pathology. 1996;20:1412–1417. doi: 10.1097/00000478-199611000-00014. [DOI] [PubMed] [Google Scholar]
- 25.Weiss SW, Goldblum JR, editors. Enzinger and Weiss Soft Tissue Tumors. Mosby Elsevier; 2008. [Google Scholar]
- 26.Woodruff JM, Selig AM, Crowley K, et al. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. The American journal of surgical pathology. 1994;18:882–895. doi: 10.1097/00000478-199409000-00003. [DOI] [PubMed] [Google Scholar]