

Abstract
Fibrosis of the lungs and other organs is characterized by the accumulation of myofibroblasts, effectors of wound-repair that are responsible for the deposition and organization of new extracellular matrix (ECM) in response to tissue injury. During the resolution phase of normal wound repair, myofibroblast apoptosis limits the continued deposition of ECM. Mounting evidence suggests that myofibroblasts from fibrotic wounds acquire resistance to apoptosis, but the mechanisms regulating this resistance have not been fully elucidated. Endothelin-1 (ET-1), a soluble peptide strongly associated with fibrogenesis, decreases myofibroblast susceptibility to apoptosis through activation of phosphatidylinositol 3′-OH kinase (PI3K)/AKT. Focal adhesion kinase (FAK) also promotes myofibroblast resistance to apoptosis through PI3K/AKT-dependent and – independent mechanisms, although the role of FAK in ET-1 mediated resistance to apoptosis has not been explored. The goal of this study was to investigate whether FAK contributes to ET-1 mediated myofibroblast resistance to apoptosis and to examine potential mechanisms downstream of FAK and PI3K/AKT by which ET-1 regulates myofibroblast survival. Here, we show that ET-1 regulates myofibroblast survival by Rho/ROCK-dependent activation of FAK. The anti-apoptotic actions of FAK are, in turn, dependent on activation of PI3K/AKT and the subsequent increased expression of Survivin, a member of the inhibitor of apoptosis protein (IAP) family. Collectively, these studies define a novel mechanism by which ET-1 promotes myofibroblast resistance to apoptosis through upregulation of Survivin.
Keywords: Fibrosis, Rho-kinase, Inhibitor of Apoptosis, Mesenchymal Cell, Focal Adhesion Kinase, AKT
1. INTRODUCTION
Fibroblasts are mesenchymal cells which, in response to tissue injury, differentiate into alpha-smooth muscle actin expressing myofibroblasts. These myofibroblasts function as the effector cells in wound healing through their synthesis, secretion, organization and contraction of new extracellular matrix (ECM) (Hinz and Gabbiani, 2010). Ultimately, the normal resolution of the wound repair response following injury requires the elimination of myofibroblasts by apoptosis. To date, the mechanistic triggers for myofibroblast apoptosis upon the conclusion of successful repair remain undefined (Desmouliere et al., 1995, Hernandez-Gea and Friedman, 2011, Thannickal and Horowitz, 2006). In contrast to normal wound healing, tissue fibrosis is characterized by the accumulation of myofibroblasts and the excessive deposition of ECM (Tomasek et al., 2002). The organization and remodeling of this excessive ECM can result in the progressive destruction of tissue architecture and lead to impaired organ function. Indeed, fibrosis of various organs is estimated to contribute to 45% of deaths in the developed world (Wynn, 2008).
Idiopathic pulmonary fibrosis (IPF) is a fibrotic disease of the lung parenchyma with no effective therapy and a mortality of approximately 50% within 2–3 years of the diagnosis (Horowitz and Thannickal, 2006). Mortality in IPF correlates with the profusion of fibroblastic foci, the active sites of fibrosis in IPF which are composed of accumulated myofibroblasts in close proximity to an abnormal alveolar epithelium (King et al., 2001). These foci represent an apoptosis paradox with evidence of robust apoptosis in the epithelial compartment and a distinct absence of apoptosis in the adjacent myofibroblasts (Korfei et al., 2008, Lepparanta et al., 2010, Maher et al., 2010, Thannickal and Horowitz, 2006). Importantly, the absence of apoptosis in fibroblastic foci is associated with an increased resistance to apoptosis in mesenchymal cells isolated from fibrotic lungs (Buhling et al., 2005, Chang et al., 2010, Hinz and Gabbiani, 2010, Huang et al., 2009, Moodley et al., 2004).
The mechanisms regulating resistance to apoptosis in fibrotic lung mesenchymal cells are poorly understood. Studies have shown that normal lung fibroblasts have basal resistance to Fas-mediated apoptosis that is overcome in the presence of cycloheximide (CHX), an inhibitor of protein translation, or by exposure to the inflammatory cytokines tumor necrosis factor-alpha (TNF alpha) and interferon-gamma (Buhling et al., 2005, Frankel et al., 2006, Huang et al., 2009, Kulasekaran et al., 2009). Endothelin-1 (ET-1) and transforming growth factor beta-1 (TGF-β1) are soluble mediators implicated in the pathogenesis of fibrosis, and each of these has been shown to promote myofibroblast differentiation, cell contraction and collagen synthesis. We have shown that ET-1 and TGF-β1 independently increase myofibroblast resistance to apoptosis induced by the combination of Fas-activation and CHX (Fas/CHX) through activation of phosphatidylinositol 3′OH Kinase/Protein Kinase B (PI3K/AKT) (Kulasekaran et al., 2009). Additional studies show that focal adhesion kinase (FAK), a non-receptor tyrosine kinase that mediates myofibroblast differentiation by TGF-β1, also promotes myofibroblast resistance to apoptosis through PI3K/AKT-dependent and – independent mechanisms (Ding et al., 2008, Horowitz et al., 2007, Thannickal et al., 2003, Xia et al., 2004). Both FAK and AKT are activated in murine lungs following a fibrotic injury and pharmacologic inhibition of these kinases attenuates lung fibrosis in-vivo (Vittal et al., 2005). The downstream mechanisms by which FAK and AKT mediate myofibroblast susceptibility to apoptosis have not been shown.
In this study we investigated the role of FAK and the relationship between FAK and PI3K/AKT in the regulation of myofibroblast resistance to apoptosis by ET-1. Additionally, we examined the downstream mechanism by which these pro-survival kinases regulate susceptibility to apoptosis in myofibroblasts. Herein, we demonstrate that FAK is rapidly activated by ET-1 through a mechanism that is dependent on Rho-associated coiled-coil forming protein kinase (ROCK), that FAK phosphorylation is required for PI3K/AKT activation by ET-1, and that this ET-1/ROCK/FAK/PI3K/AKT signaling cascade regulates myofibroblast susceptibility to apoptosis. Additionally, we show that the expression of Survivin, a member of the inhibitor of apoptosis protein (IAP) family, is increased by ET-1 and that inhibition of Survivin reverses the anti-apoptotic effects of ET-1. Collectively, these studies elucidate a novel mechanism by which ET-1 decreases myofibroblast susceptibility to apoptosis.
2. MATERIALS AND METHODS
2.1. Cells and Cell Culture
Primary normal human fetal lung mesenchymal cells (IMR-90; Institute for Medical Research, Camden, NJ) between passages 8 and 14 were cultured in 5% CO2 at 37° C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (Sigma, St. Louis, MO), penicillin/streptomycin (100 U/ml, Sigma) and fungizone (250 mg/ml, Invitrogen, Carlsbad CA). The growth medium was changed every two days. Unless otherwise indicated, when cells reached approximately 60% confluence they were growth arrested in serum-free DMEM for 24 hours. After 24 hours, cells were treated with/without ET-1 (1 nM or 100 nM) in the presence or absence of the indicated inhibitors. In all cases, pharmacologic inhibitors were added 30 minutes prior to treatment with ET-1 for the indicated time points. Because pharmacologic inhibitors may have unanticipated off target effects, key experiments were done using two different inhibitors of FAK (FAK14 or PF573228; see “Reagents” section) or PI3K (LY294002 or Wortmannin; see “Reagents” section). For apoptosis studies, cells were treated with/without the combination of Fas-activating ligand and CHX (Fas/CHX) in the presence or absence of ET-1 with or without the indicated inhibitor for 16 hours, a time point based on previous studies from our laboratory and others (Buhling et al., 2005, Kulasekaran et al., 2009, Tanaka et al., 2002).
2.2. Reagents
Human Porcine ET-1 and CHX were from Sigma. The ET-A and -B receptor antagonists (BQ123 and BQ788, respectively) and the ROCK inhibitor (Y27632) were from Calbiochem (Gibbstown, NJ). The cell permeable C3 transferase (CT04) was from Cytoskeleton, Inc. (Denver, CO). FAK14 (1,2,4,5-benzenetetraamine tetrahydrochloride), which specifically inhibits FAK activation through blockade of Y397 phosphorylation without impacting PI3K or Src (Golubovskaya et al., 2008), was from Tocris Bioscience (Ellisville, MO). PF573228, which directly blocks FAK kinase activity (subsequently blocking Y397 autophosphorylation) without significantly inhibiting PI3K or Rho-kinase (ROCK) (Slack-Davis et al., 2007) and the PI3K inhibitor Wortmannin were also from Tocris Bioscience (Ellisville, MO). LY294002, (a PI3K inhibitor) was from Cell Signaling Technology (Beverly, MA). Mitogen activated protein kinase (MAPK) inhibitors (p38: SB203580; MEK/ERK1/2: PD98059 and Jnk: SP600125) were from Calbiochem (La Jolla, CA). Y27632, LY294002, Wortmannin, SB203580 and PD98059 have all been shown to have high specificity for their target kinases (Davies et al., 2000).
The anti-Fas (activating) antibody (clone CH11) was from Millipore (Billerica, MA). Antibodies to tyrosine-397 (Y397) phospho-FAK, total FAK, serine-473 (S473) phospho-AKT, total AKT, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and poly-(ADP-ribose) polymerase (PARP) were from Cell Signaling. The mouse monoclonal antibody to Survivin was from Abnova. CAY10625, which inhibits Survivin function by disrupting the interaction between Survivin and pro-apoptotic Smac/Diablo (second mitochondrial activator of caspases/direct IAP binding protein with low isoelectric point), thereby allowing Smac/Diablo to carry out its pro-apoptotic function, was from Cayman Chemical Company (Ann Arbor, MI) (Oikawa et al., 2010). YM155, which inhibits Survivin by binding to promoter and blocking transription (Ryan et al., 2009), was from Selleck Chemicals (Houston, TX). Horseradish peroxidase-conjugated secondary antibodies were from Pierce (Rockford, IL).
2.3. Adenoviral infections
To knock down expression of FAK, we infected cells with adenoviral vectors expressing green fluorescent protein (GFP) or FRNK (FAK-related non-kinase) as previously described (Xia et al., 2004). Cells were cultured as described above and, when 50% confluent, infected with either the GFP-expressing or FRNK-expressing adenovirus at a multiplicity of infection (MOI) of 40 in DMEM containing 10% FBS. 24 hours after infection, the media was changed to serum free DMEM for 24 hours prior to cell treatments.
2.4. Western Immunoblotting
At the time points indicated for each experiments, cells were washed with phosphate-buffered saline and whole-cell lysates were collected in ice-cold RIPA buffer (1% nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 0.15 M NaCl, 0.01 M NaH2PO4, 2 mM EDTA, 0.5 mM NaF) containing 2 mM sodium orthovanadate and 1:100 dilution of protease inhibitor mixture III (Calbiochem, San Diego, CA). Protein estimation was done with the Bio-Rad DC protein assay (Bio-Rad, Hercules, CA). For each experiment, RIPA buffer was used to adjust the volume so that each sample had an equal protein concentration. Cell lysates were reduced by mixing with a 1:5 vol/vol ratio of 6x electrophoresis sample buffer (0.2 M EDTA, 40 mM dithiothreitol, 6% SDS, 0.06 mg/ml pyronin, pH 6.8) and boiling for 7 minutes. Next, equal volumes of the reduced lysates were subjected to SDS-PAGE electrophoresis and Western blotting as previously described (Kulasekaran et al., 2009). All Western blots were stripped and re-probed for a loading control (GAPDH, total AKT or total FAK).
2.5. Apoptosis assessments
Apoptosis was assessed by Western blotting for cleaved PARP as the primary assay (Huang et al., 2009) and by ELISA-based detection of histone-associated DNA fragments using the Cell Death Detection ELISA Kit (Roche Applied Science, Indianapolis IN) according to the manufacturer’s protocol. To assess cell morphology, cells were fixed with 10% formaldehyde and phase-contrast images were obtained at 16 or 48 hours with a Nikon Eclipse E400 microscope (Melville, NY). Images were captured with a Diagnostic instruments Camera and SPOT Basic version 4.0.8 software.
2.6. Densitometry and Statistical Analyses
Densitometric analysis of Western blots was done using the public domain NIH ImageJ program available at http://rsbweb.nih.gov/ij/. To allow for statistical comparisons, each Western blot was stripped and probed for a loading control (GAPDH, Total FAK or Total AKT). The band density of each target protein and the corresponding loading control was measured and adjusted for the background density of the developed film. Next, the ratio of each target protein to its corresponding loading control was determined. This ratio was then normalized such that the untreated control condition was equal to 1 (for apoptosis assessments by cleaved PARP, the ratio was normalized to Fas/CHX-treated cells).
Statistical analysis was completed using ANOVA with Newman-Keuls post-test (Graphpad Prism software version 5.01 for Windows, GraphPad Software, San Diego CA) with a minimum of three replicates of each experiment. A p < 0.05 was considered significant.
3. RESULTS
3.1. ET-1 induces FAK activation in normal lung fibroblasts
FAK activation by phosphorylation at tyrosine-397 (Y397) is critical for TGF-β1 induced myofibroblast differentiation and inhibition of apoptosis (Thannickal et al., 2003, Golubovskaya et al., 2008, Slack-Davis et al., 2007, Horowitz et al., 2007, Xia et al., 2004). To determine if ET-1 similarly induces FAK activation, normal lung fibroblasts were treated with/without escalating concentrations of ET-1 for durations between 15 minutes and 24 hours (Figure 1). FAK phosphorylation increased significantly within 30 minutes of exposure to ET-1 with concentrations as low as 1.0 nM (Figure 1A–D). The increased FAK phosphorylation was maintained for up to 24 hours, although there was a decline in phosphorylation between 6 and 16 hours (Figure 1C and D). FAK phosphorylation by ET-1 preceded the activation of PI3K/AKT, which we have observed to be maximal at 3 hours following ET-1 treatment (Kulasekaran et al., 2009). These findings suggest that either ET-1 activation of FAK lies upstream of PI3K/AKT, or that these two pro-survival pathways are independently regulated by ET-1.
Figure 1. ET-1 induces FAK activation in normal lung fibroblasts.

(A) Growth-arrested normal human lung fibroblasts (IMR-90) were treated with ET-1 at doses ranging from 1 pM (0.001 nM) to 1 μM (103 nM). At 30 minutes, cell lysates were assessed for FAK activation by Western blotting for phosphorylation at tyrosine-397 (Y397 p-FAK). The blot was stripped and probed for total FAK. (B) Densitometric analysis of (A) normalized to untreated controls. * p < 0.01 compared to untreated controls; n = 3. (C) IMR-90 fibroblasts were treated with ET-1 (1.0 nM) for the time periods indicated and assessed for FAK activation by Western blotting for Y397 p-FAK. The blots were stripped and probed for total FAK. (D) Densitometric analysis of (C) normalized to untreated controls. * p < 0.01 compared to untreated controls; n = 3.
ET-1 signals via two G- protein-coupled receptors, designated ET-A and -B (Fonseca et al., 2011). These receptors are expressed on both fibroblasts and myofibroblasts and the pro-fibrotic effects of ET-1, including activation of PI3K/AKT, have been primarily attributed to the ET-A receptor (Hafizi et al., 2004, Shi-Wen et al., 2004, Simonson and Ismail-Beigi, 2011, Tostes et al., 2002, Wendel et al., 2004). Dual receptor blockade, however, is necessary for inhibition of ET-1 induced collagen synthesis, suggesting that ET-B also contributes to fibrogenesis (Shi-Wen et al., 2001). To determine which of these receptors was necessary for FAK activation, we treated normal lung fibroblasts with ET-1 (1.0 nM) in the presence/absence of inhibitors of ET-A (BQ123, 10 μM) and/or ET-B (BQ788, 10 μM). As shown in Figure 2, blockade of either receptor was sufficient to inhibit FAK activation. This finding suggests that simultaneous binding of the A and B receptors, as can occur with heterodimerization, is required for FAK activation by ET-1 (Evans and Walker, 2008, Fonseca et al., 2011, Gregan et al., 2004).
Figure 2. ET-1 activation of FAK is inhibited by blockade of either ET-A or ET-B.

Growth-arrested IMR-90 fibroblasts were treated with/without ET-1 (1.0 nM) for 1 hour in the presence/absence of inhibitors of ET-A (BQ123, 10 μM) and/or ET-B (BQ788, 10 μM). (A) Y397 p-FAK was assessed by Western blotting and blots were stripped and probed for total FAK. (B) Densitometric analysis normalized to untreated controls. * p < 0.01 vs. untreated control. # p < 0.01 vs. ET-1 treated cells. n = 4.
3.2. FAK activation by ET-1 is mediated by Rho/ROCK
Rho-GTPases and their downstream effector, ROCK, are necessary for FAK activation by mechanical stimuli and by ET-1 (Koyama et al., 2004, Torsoni et al., 2005). Rho/ROCK signaling also mediates the contraction and migration of hepatic and pancreatic stellate cells, the proliferation of astrocytes and the survival of endothelial cells and cardiac myocytes (Kawada et al., 1999, Masamune et al., 2005, Rankin et al., 1994, Wu et al., 2007, Del Re et al., 2008). We found that inhibition of ROCK (with Y27632) or RhoA, RhoB, and RhoC GTPases (with a cell permeable C3 transferase) abolished FAK activation by ET-1, supporting a necessary role for Rho/ROCK signaling in ET-1 induced FAK activation (Figure 3A–C). Next, we compared the effect of a ROCK inhibitor on FAK phosphorylation with two different small molecule inhibitors of FAK (FAK14 and PF573228), finding that each FAK inhibitor effectively blocked ET-1 mediated FAK phosphorylation (Figure 3D–G) (Golubovskaya et al., 2008, Slack-Davis et al., 2007). Densitometric analysis confirmed that there was no significant difference in the inhibition of Y397 FAK phosphorylation by ET-1 when cells were treated with Y27632, FAK14 or PF573228 (Figure 3H). In contrast to Rho/ROCK inhibition, blockade of p38 MAPK, which mediates ET-1 activation of PI3K/AKT, and other MAPKs (ERK1/2 and c-Jun N-terminal kinase), had no effect on ET-1 activation of FAK (Figure 3I and data not shown) (Kulasekaran et al., 2009).
Figure 3. ET-1 activation of FAK is mediated by Rho/ROCK.

Growth-arrested IMR-90 fibroblasts were treated with/without ET-1 (1.0 nM) for one hour in the presence/absence of the indicated inhibitors Y397p-FAK was assessed by Western blotting. In each experiment, p-FAK blots were stripped and probed for total FAK, with representative blots shown. (A) Cells were treated with/without ET-1 (1.0 nM) in the presence of 0.2, 2.0 or 4.0 μM of cell permeable C3 transferase (C3 Trans), an inhibitor of RhoA, RhoB and RhoC GTPases. (B) Cells were treated with ET-1 (1.0 nM) with/without cell permeable C3 transferase (2.0 μM) or the ROCK inhibitor Y27632 (15 nM). (C) Densitometric analysis of (B) normalized to untreated controls. * p < 0.01 vs. untreated controls. # p < 0.01 vs. ET-1 treated cells; n = 3. (D) Cells were treated with/without ET-1 (1.0 nM) in the presence/absence of 0.1, 1.0 or 10 μM FAK14. (E) Cells were treated with/without ET-1 (1.0 nM) and inhibitors of ROCK (Y27632, 15 nM) or FAK (FAK14, 10 μM). After re-probing the blot for total FAK, we observed a decrease in total FAK in cells treated with FAK14. To ensure equal protein loading, this blot was then stripped and probed again for GAPDH. (F) Cells were treated with/without ET-1 (1.0 nM) in the presence/absence of PF573228 at doses of 0.1, 1.0 and 10 μM. (G) Cells were treated with/without ET-1 (1.0 nM) and inhibitors of ROCK or FAK (PF573228, 10 μM). (H) Densitometric analysis of FAK phosphorylation by ET-1 in the presence/absence of Y27632 (Y), FAK14 (14) or PF573228 (PF). * p < 0.05 vs. untreated control. # p < 0.05 vs. ET-1-treated cells. NS: p is non-significant for comparisons between untreated controls and ET-1 with Y27632, FAK14 or PF573228. (I) Fibroblasts were treated with/without ET-1 (1.0 nM) in the presence/absence of a p38 MAPK inhibitor (SB203580, 10 μM).
3.3. FAK activation by ET-1 decreases myofibroblast susceptibility to apoptosis
Both ET-1 and TGF-β1 decrease myofibroblast susceptibility to apoptosis induced by the combination of Fas/CHX (Kulasekaran et al., 2009). Because each of these soluble mediators induces FAK activation and FAK activity regulates myofibroblast susceptibility to apoptosis, we next sought to determine if the anti-apoptotic effects of ET-1 on myofibroblasts required FAK activation (Horowitz et al., 2007, Xia et al., 2004). Normal lung fibroblasts and ET-1 (1.0 nM) differentiated myofibroblasts were exposed to Fas/CHX in the presence/absence of inhibitors of ROCK or FAK for 16 hours. Cell lysates were assessed for biochemical evidence of apoptosis by immunoblotting for cleaved PARP, which was then quantified by densitometry (Figure 4A and B). Compared to cells treated with Fas/CHX alone, ET-1 treatment reduced cleaved PARP by 42% (mean ± SEM of 1.0 ± .003 without ET-1 compared to 0.58 ± 0.064 with ET-1, p < 0.001). The reduction in cleaved PARP seen in ET-1 treated cells was reversed by inhibition of ROCK (1.383 ± 0.285 for Fas/CHX with ET-1 and Y27632; p = 0.015 compared to Fas/CHX and ET-1). Similarly, FAK inhibition reversed the ET-1 mediated reduction in apoptosis, (1.64 ± .708 for cells treated with Fas/CHX and ET-1 with FAK14;p < 0.05 compared to Fas/CHX and ET-1). There was no statistically significant difference between the band density of cleaved PARP in cells treated with Fas/CHX alone and those treated with Fas/CHX with ET-1 and Y27632 or FAK14. In the absence of Fas/CHX, neither Y27632 nor FAK14 induced apoptosis as assessed by cleaved PARP (data not shown).
Figure 4. Pharmacologic inhibitors of ROCK and FAK attenuate ET-1-mediated protection from apoptosis.
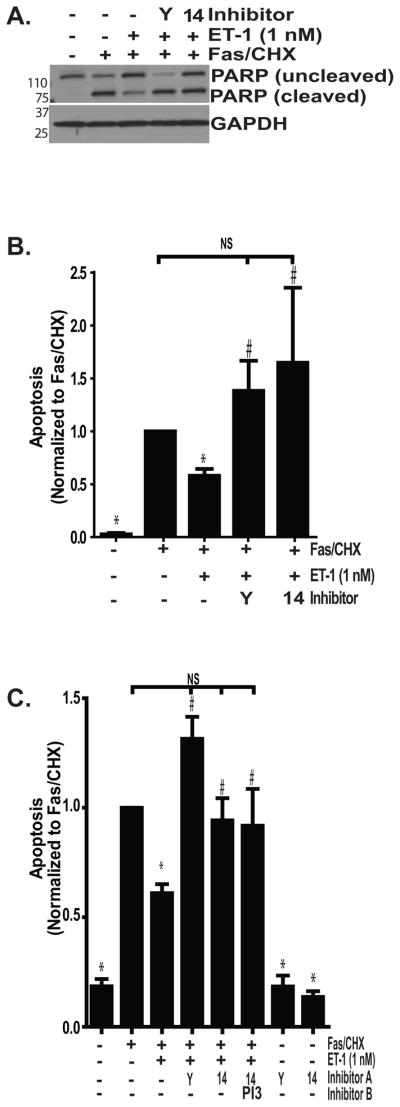
Growth-arrested IMR-90 fibroblasts were treated with/without the combination of Fas-activating antibody (CH11; 250 ng/ml) and CHX (500 ng/ml) for 16 hours with/without ET-1 (1.0 nM) and/or inhibitors of ROCK (Y; Y27632, 15 nM), FAK (14; FAK14, 10 μM), or PI3K (PI3; LY294002; 10 μM). (A) Apoptosis was assessed by immunoblotting for cleaved PARP. Blots were stripped and probed for GAPDH. (B) Densitometric analysis of cleaved PARP/GAPDH, normalized to the Fas/CHX-treated groups. * p < 0.01 vs. Fas/CHX-treated cells. # p < 0.05 vs. Fas/CHX with ET-1. NS = non-significant diffrences; n = 6 replicates per group. (C) Apoptosis was assessed by ELISA for detection of histone-associated DNA and data were normalized to the Fas/CHX-treated groups. * p < 0.01 vs. Fas/CHX-treated. # p < 0.01 vs. Fas/CHX with ET-1. NS = non-significant diffrences; n = 3.
The roles of Rho/ROCK and FAK signaling in ET-1 mediated resistance to apoptosis were confirmed in similar experiments using an ELISA to detect histone-associated DNA fragments as an indicator of apoptosis (Figure 4C). Consistent with the data from cleaved PARP, ET-1 reduced apoptosis induced by Fas/CHX by 39% (mean ± SEM of 1.0 ± 0.001 for Fas/CHX compared to 0.61 ± .04 for Fas/CHX with ET-1, p < 0.001). The decreased susceptibility to apoptosis conferred by treatment with ET-1 was lost in the presence of ROCK inhibition with Y27632 (1.31 ± .1, p < 0.001 compared to Fas/CHX with ET-1) or inhibition of FAK (0.942 ± .102, p < 0.05). Again, there was no significant difference between apoptosis in the Fas/CHX group compared with the Fas/CHX with ET-1 and Y27632 or FAK14 groups. Moreover, neither FAK nor ROCK inhibition was sufficient to induce apoptosis in the absence of Fas/CHX.
We have shown that PI3K/AKT activation by either ET-1 or TGF-β1 mediates myofibroblast resistance to apoptosis (Horowitz et al., 2004, Kulasekaran et al., 2009). In the case of TGF-β1, PI3K/AKT activation and protection from apoptosis was independent of FAK phosphorylation. To determine if ET-1 similarly reduced myofibroblast apoptosis through independent activation of PI3K/AKT and FAK, we simultaneously blocked FAK and PI3K prior to treatment with Fas/CHX (Figure 4C). This combination had no greater effect than inhibition of FAK alone, suggesting that the anti-apoptotic effects of FAK activation by ET-1 might be mediated by activation of PI3K/AKT.
The critical role of FAK in ET-1 mediated protection from apoptosis was then confirmed by infecting normal lung fibroblasts with adenoviral vectors that overexpress either GFP (as a control) or FRNK, which functions as a dominant-negative FAK (Figure 5). Using an MOI of 40, we found robust expression of FRNK which effectively knocked down endogenous basal and stimulated levels of phosphorylated FAK (Figure 5A). Next, the GFP and FRNK infected cells were treated with Fas/CHX in the presence/absence of ET-1 and apoptosis was assessed by Western blotting to detect cleaved PARP (Figure 5B) and by ELISA to detect histone-associated DNA fragments (Figure 5C). Additionally, in some experiments (as shown in Figure 5B), we also treated cells with Fas-activating ligand alone to determine if CHX remained necessary to induce apoptosis in the FRNK-infected cells. Consistent with a necessary role for FAK, ET-1 reduced apoptosis by 41% in the GFP-infected cells (mean ± SEM of 1.0 compared to 0.59 ± 0.099 in Fas/CHX with ET-1 treated GFP-infected cells; p < 0.01). However, ET-1 failed to reduce apoptosis in the FRNK infected cells (1.094 ± 0.11 vs. 0.91 ± 0.05).
Figure 5. FRNK overexpression attenduates ET-1 mediated protection from apoptosis.

IMR-90 fibroblasts were infected with adenoviral vectors to overexpress either green fluorescent protein (GFP) or FAK-related non-kinase (FRNK) with a multiplicity of infection (MOI) of 40. (A) Infected cells were treated with/without ET-1 (1.0 nM) for 1 hour, and whole cell lysates were assessed for FAK phosphorylation (upper blot). The blot was stripped and probed for Total FAK (lower blot). In the lower blot, the truncated protein (FRNK) can be appreciated as the low molecular weight band. (B) Infected cells were treated for 16 hours with/without ET-1 (1.0 nM) and/or Fas-activating ligand (CH11; 250 ng/ml) in the presence/absence of CHX (500 ng/ml). Apoptosis was assessed by Western blotting to detect cleaved PARP. (C) GFP-infected cells (white bars) and FRNK-infected cells (black bars) were treated with/without the combination of Fas/CHX for 16 hours in the presence/absence of ET-1. Apoptosis was assessed by ELISA detection of histone-associated DNA fragments, and the data were normalized to the GFP-infected cells treated with Fas/CHX. * p < 0.01 vs. Fas/CHX-treated GFP-infected cells. # = p < 0.01 vs. GFP infected cells treated with Fas/CHX with ET-1. NS = non-significant differences between the indicated groups; n = 3.
In addition to the biochemical indicators of apoptosis, cell morphology was assessed by phase-contrast microscopy at 16 and 48 hours following treatment with/without Fas-activating ligand, CHX, or the combination of Fas/CHX with/without ET-1 in the presence/absence of inhibitors of ROCK or FAK (Supplemental Figure 1A and B, respectively). Neither Fas nor CHX alone had a significant impact on cell morphology at 16 hours, but cells treated with the combination (Fas/CHX) showed marked morphologic changes consistent with apoptosis including membrane blebs, cell rounding, and retractions of cell-cell contacts. These morphologic changes were reduced in cells treated with ET-1, but cells treated with ET-1 and ROCK or FAK inhibitors were qualitatively no different than cells which had not received ET-1. At the 48 hour time-point, untreated cells and cells treated with Fas or CHX alone appeared no different than the controls at 16 hours. Cells treated with Fas/CHX, however, were non-viable. At this late time point, ET-1 conferred minimal resistance to Fas/CHX-induced apoptosis, although qualitatively there were an increased number of normal-appearing cells than were seen without ET-1. In the presence of ROCK or FAK inhibitors, any decrease in cell death conferred by ET-1 was lost at the 48 hour time point.
3.4. PI3K/AKT activation by ET-1 is mediated by FAK
Our time-course experiments showed that maximal FAK phosphorylation, which occurred within 30 minutes of ET-1 treatment, precedes maximal AKT phosphorylation, which is seen between 1 and 3 hours following ET-1 treatment (Figure 1 and (Kulasekaran et al., 2009). This, together with the finding that the simultaneous inhibition of FAK and PI3K had no greater impact on apoptosis induced by Fas/CHX than did inhibition of FAK alone, suggested that PI3K/AKT activation by ET-1 might be mediated by FAK. To define the relationship between FAK and AKT activation by ET-1, normal lung fibroblasts were treated with/without ET-1 (1 or 100 nM) for 3 hours in the presence/absence of an inhibitor of ROCK (Figure 6A) or an inhibitor of FAK (Figure 6B) and AKT activation was assessed by immunoblotting. Consistent with our previous report, ET-1 induced AKT activation in a dose-dependent manner (Figure 6C). Regardless of the ET-1 dose used, inhibition of ROCK with Y27632 or FAK with PF573228 attenuated ET-1 induced AKT activation (Figure 6C), demonstrating that that AKT activation requires Rho/ROCK and FAK activation. Conversely, blockade of PI3K using two different inhibitors failed to reduce FAK phosphorylation, confirming the position of PI3K/AKT downstream of ROCK and FAK (Figure 6D).
Figure 6. ROCK and FAK mediate ET-1 activation of PI3K/AKT.

IMR-90 fibroblasts were treated with ET-1 (1.0 or 100 nM) for 3 hours in the presence of (A) ROCK inhibitor Y27623 (15 nM) or (B) FAK inhibitor PF573228 (10 μM). Activation of AKT was assessed by immunoblotting with antibodies specific for S473 p-AKT. Blots were stripped and probed for Total AKT. (C) Densitometric analysis of AKT phosphorylation under the conditions shown in A and B. Y; Y27632, FAK; PF573228, 10 μM. * p < 0.01 vs. untreated controls. ** p < 0.001 vs. untreated controls. # p < 0.01 vs. ET-1 treated cells (1 and 100 nM). NS = no significant difference compared to untreated controls. (D) Cells were treated with ET-1 (1.0 nM) for one hour in the presence/absence of inhibitors of ROCK (Y; Y27632, 15 nM), FAK (PF573228, 10 μM), or PI3K (LY; LY294002; 10 μM or WM; Wortmannin; 50 nM). FAK activation was determined by immunoblotting for Y397 phospho-FAK. Blots were stripped and probed for Total FAK. Each blot is representative of at least 3 separate experiments.
3.5. ET-1 increases expression of Survivin
The downstream mechanisms by which FAK and AKT regulate myofibroblast susceptibility to apoptosis have not been elucidated. We have recently shown that the anti-fibrotic lipid mediator prostaglandin E2 (PGE2) sensitizes fibroblasts to Fas-mediated apoptosis while reducing expression of Survivin, a member of the Inhibitor of Apoptosis (IAP) family of proteins which is involved in the regulation of cell survival and proliferation (Altieri, 2010, Huang et al., 2009). To determine if Survivin had a role in ET-1 mediated protection from apoptosis, we first examined the effects of ET-1 on Survivin expression (Figure 7). Treatment of normal lung fibroblasts with ET-1 (1.0 nM) led to an increase in Survivin protein expression that was first appreciable at one hour, significant at four hours, and maintained at increased levels through sixteen hours (Figure 7A and B). At 24 hours, Survivin protein levels had returned to baseline. Supporting a role for increased Survivin expression in ET-1 mediated regulation of myofibroblast survival, inhibition of ROCK, FAK, and PI3K significantly blocked the ability of ET-1 to induce increased Survivin expression (Figure 7C, D and E). Consistently, FRNK overexpression completely blocked Survivin upregulation by ET-1 (Figure 8).
Figure 7. Survivin upregulation by ET-1 is mediated by ROCK, FAK and PI3K/AKT.
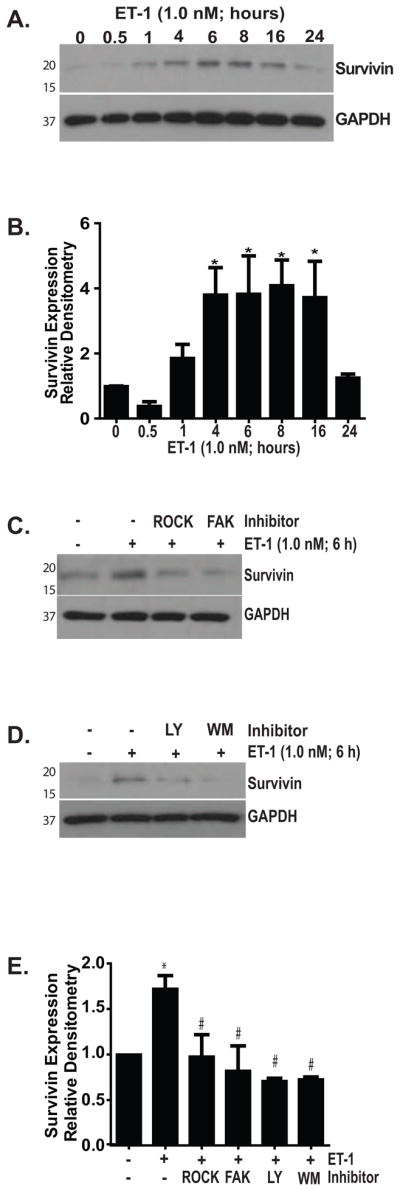
(A) IMR-90 fibroblasts were treated with ET-1 (1.0 nM) for the indicated time points and Survivin expression was measured by Western blotting. Blots were stripped and probed for GAPDH. (B) Densitometric analysis of Survivin expression.* p < 0.01 vs. untreated controls; n = 4. (C) Fibroblasts were treated with/without ET-1 (1.0 nM) for 6 hours in the presence/absence of inhibitors of ROCK (Y27632, 15 nM) or FAK (PF573228, 10 μM). Survivin expression was assessed by Western blotting. Blots were stripped and probed for GAPDH. (D) Fibroblasts were treated with ET-1 (1.0 nM) for 6 hours in the presence/absence of two different PI3K inhibitors (LY294002; 10 μM or Wortmannin 50 nM). Survivin expression was assessed by Western blotting and blots were stripped and probed for GAPDH. (E) Densitometric analysis of Survivin expression in the presence/absence of the inhibitors used in (C and D), normalized to untreated controls. * p < 0.001 vs. untreated controls; # p < 0.01 vs. ET-1 treated cells. n = 3 per condition.
Figure 8. FRNK overexpression inhibits the upregulation of Survivin by ET-1.

IMR-90 fibroblasts were infected with adenoviral vectors expressing GFP or FRNK with an MOI of 40 and treated with/without ET-1 (1.0 nM) for 6 hours. (A) Survivin expression was determined by Western blotting, and the blot was stripped and re-probed for GAPDH. (B) Densitometric analysis. n = 3; * p < 0.01 vs. untreated, GFP-infected cells.
3.6. Survivin regulates ET-1 protection from apoptosis
Having shown that ET-1 increases Survivin expression through a mechanism dependent on Rho/ROCK, FAK and PI3K/AKT, and that each of these signaling intermediates is essential for ET-1 mediated resistance to apoptosis, we next sought to determine the role of Survivin in ET-1 mediated myofibroblast resistance to apoptosis. Normal lung fibroblasts were exposed to Fas/CHX in the presence or absence of ET-1 with/without a small molecule Survivin inhibitor (either YM155 or CAY10625; Figure 9) (Nakahara et al., 2007, Oikawa et al., 2010, Ryan et al., 2009). Each of these inhibitors significantly blocked ET-1 mediated protection from apoptosis. Neither of these Survivin inhibitors, when given alone, was sufficient to induce fibroblast apoptosis (10C, D and data not shown). Collectively, these studies support a mechanistic role for Survivin upregulation in ET-1 mediated myofibroblast resistance to apoptosis.
Figure 9. Survivin inhibition blocks ET-1-mediated protection from apoptosis.

(A–C) IMR-90 fibroblasts were subjected to the combination of Fas-activating antibody (Fas; CH11, 250 ng/ml) with CHX (500 ng/ml) with/without ET-1 (1.0 nM) and/or YM155, an inhibitor of Survivin (1.0, 10, or 100 nM) or with YM155 (1.0, 10 or 100 nM) alone for 16 hours. (A) Apoptosis was assessed by Western blotting for determination of cleaved PARP. Blots were stripped and probed for GAPDH. In separate experiments (not shown), no cleaved PARP was detected in cells treated with YM155 (1.0, 10 or 100 nM). (B) Densitometric analysis of cleaved PARP normalized to the expression in cells treated with Fas/CHX. * p < 0.05 vs. Fas/CHX. # p < 0.05 vs. Fas/CHX with ET-1. NS = non-significant difference; n =3 (C) Apoptosis in similarly treated cells was assessed by ELISA detection of histone associated DNA fragments.* p < 0.05 vs. Fas/CHX alone. # p < 0.05 vs. Fas/CHX with ET-1. NS = non-significant difference; n = 3. (D and E) IMR-90 fibroblasts were treated with Fas/CHX with/without ET-1 (1.0 nM) and/or CAY10625, a small molecule inhibitor of Survivin (2.5 or 7.5 μM) for 16 hours. Apoptosis was assessed by Western blotting for cleaved PARP. Blots were stripped and probed for GAPDH. (B) Densitometric analysis of cleaved PARP/GAPDH normalized to Fas/CHX-treated group. No cleaved PARP was detected in cells treated with CAY10625 alone or withET-1 and these groups are not shown in the densitometric analysis. * p < 0.001 vs. Fas/CHX-treated cells. # p < 0.001 vs. Fas/CHX with ET-1 or CAY10625 alone at any dose. NS = non-significant difference; n = 3.
4. DISCUSSION
Myofibroblasts are critical effectors of wound repair and their function must be temporally and spatially regulated to facilitate the restoration of tissue architecture and function following an injury. Myofibroblast apoptosis is an essential step in the normal resolution of wound repair, and fibrotic repair is characterized the accumulation of myofibroblasts with an apoptosis-resistant phenotype (Hinz et al., 2007). Because of the complex array of dynamic interactions between resident cells, recruited cells, the ECM, and soluble mediators present within the wound microenvironment, the mechanism(s) by which myofibroblasts acquire resistance to apoptosis are likely to be multifactorial. Studies have shown that soluble factors present in the wound microenvironment, such as TGF-β1 and plasminogen activator-inhibitor-1 (PAI-1) render normal lung myofibroblasts resistant to apoptotic stimuli (Chang et al., 2010, Horowitz et al., 2007, Horowitz et al., 2008). Additionally, we have previously reported that ET-1 promotes myofibroblast resistance to apoptosis through mechanisms that are independent of TGF-β1 (Kulasekaran et al., 2009). In this study, we enhance our insight into the mechanisms regulating myofibroblast susceptibility to apoptosis, showing that ET-1 activation of PI3K/AKT requires the Rho/ROCK-dependent activation of FAK. Moreover, we show that ROCK, FAK and PI3K/AKT are required for ET-1- induced resistance to apoptosis. Finally, we show that increased myofibroblast resistance to apoptosis can be induced by ET-1 mediated upregulation of Survivin (Figure 10).
Figure 10. Proposed mechanism of ET-1 regulation of myofibroblast susceptibility to apoptosis by upregulation of Survivin.
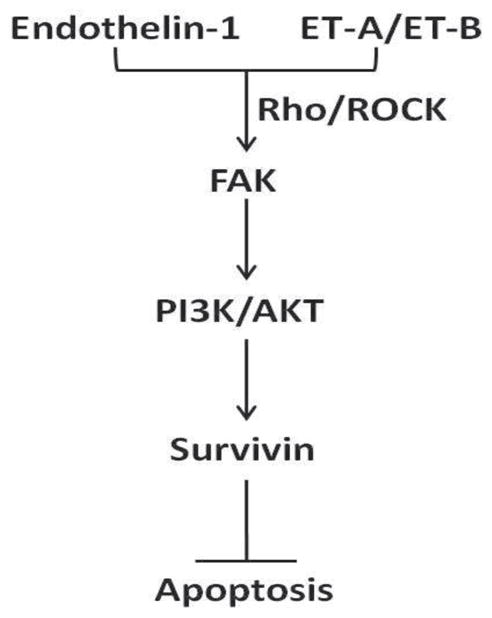
Endothelin-1 binding to its cognate receptors, ET-A and ET-B leads to the Rho-GTPase and ROCK-dependent activation of FAK. FAK activation then induces activation of PI3K and AKT, with the resultant increase in Survivin expression. Increased Survivin expression promotes myofibroblast resistance to apoptosis.
ET-1 and TGF-β1 are strongly associated with fibrogenesis through stimulation of myofibroblast differentiation, ECM synthesis, and resistance to apoptosis (Horowitz et al., 2004, Horowitz et al., 2007, Kulasekaran et al., 2009, Thannickal et al., 2003, Shi-Wen et al., 2004, Shi-wen et al., 2007a, Shi-Wen et al., 2007b, Shi-Wen et al., 2006). FAK is activated by TGF-β1 and contributes to myofibroblast survival and differentiation (Ding et al., 2008, Horowitz et al., 2007, Thannickal et al., 2003, Xia et al., 2004). In the current study, we show that ET-1, like TGF-β1, induces FAK activation in lung myofibroblasts. This finding is concordant with other studies linking ET-1 and FAK activation in mesenchymal cells (Cazaubon et al., 1997, Heidkamp et al., 2002, Kennedy et al., 2008, Rankin et al., 1994, Zachary et al., 1992). Indeed, one study found that FAK activation was required for myofibroblast secretion of ET-1 and that, in turn, ET-1 induced FAK activation, thereby establishing a profibrotic autocrine loop (Kennedy et al., 2008). The demonstration of a critical role for Rho/ROCK in FAK activation by ET-1 is consistent with studies implicating Rho family GTPases in FAK activation by both mechanical and soluble stimuli, including ET-1 (Eble et al., 2000, Koyama et al., 2003, Torsoni et al., 2005).
Although studies have linked ET-1 with FAK activation, there is little known about how this signaling pathway regulates mesenchymal cell behavior. One study found that FAK activation was required for ET-1 induced myocyte hypertrophy and another study reported that FAK activation by ET-1 in astrocytes stimulated cell cycle progression (Clemente et al., 2007, Koyama et al., 2004). In the current study we examined the role of FAK activation by ET-1 in the regulation of myofibroblast susceptibility to apoptosis, finding that the anti-apoptotic effects of ET-1 were reversed by inhibition of ROCK or FAK. Additionally, we show that PI3K/AKT activation by ET-1 requires FAK phosphorylation, suggesting that ET-1 mediated resistance to apoptosis requires PI3K/AKT activation downstream of FAK (Kulasekaran et al., 2009). These findings are consistent with other studies showing that FAK-mediated activation of AKT protects myofibroblasts from apoptosis (Huang et al., 2007, Xia et al., 2004). In contrast to our current findings with ET-1, we previously reported that TGF-β1 activation of PI3K/AKT, and the subsequent resistance to apoptosis, did not require FAK phosphorylation (Horowitz et al., 2007). A recent study showed that FAK was required as a scaffolding protein in TGF-β1 activation of PI3K/AKT, although phosphorylation of FAK was not necessary (Hong et al., 2011). Collectively, these studies indicate that PI3K/AKT and FAK interact to regulate myofibroblast apoptosis susceptibility, although the mechanism of interaction is contextual and may vary with different stimuli.
Accumulating evidence from animal models supports the relevance of FAK and AKT activation in the pathobiology of fibrosis, although the mechanisms involved are not clear. Using the murine model of bleomycin-induced pulmonary fibrosis, we found that FAK and AKT activation were markedly increased in fibrotic lungs and that lung fibrosis was significantly reduced by pharmacologic inhibition of these kinases (Vittal et al., 2005). Subsequent reports have shown that inhibition of the PI3K/AKT pathway blocks lung fibrosis in murine models induced by bleomycin, TGF-β1 overexpression and TGF-α overexpression (Kang et al., 2007, Korfhagen et al., 2009, Wei et al., 2010). Consistently, studies show an association between decreased PTEN (phosphatase and tensin homolog, a negative regulator of PI3K/AKT), increased myofibroblast resistance to apoptosis, and lung fibrogenesis in murine models (Nho et al., 2006, White et al., 2006). Outside of the lung, a recent study showed that FAK and AKT activation in myofibroblasts were essential for hypertrophic scar formation in a murine skin wound model (Paterno et al., 2011). Similar reports have associated mesenchymal cell activation of FAK and AKT with fibrosis in the liver (Jiang et al., 2004, Parsons et al., 2007) and kidney (Hayashida et al., 2007, Kato et al., 2009), with myocardial hypertrophy (Clemente et al., 2007), and with wound-healing after myocardial infarction (Shimazaki et al., 2008). Taken together, these studies indicate that FAK and AKT may function as convergence points of fibrogenesis, regardless of the organ involved or the inciting stimulus.
The downstream mechanism by which ET-1 activation of AKT promotes myofibroblast resistance to apoptosis has not been previously reported. Survivin is a member of IAP family of cytoprotective proteins, which also includes cIAP1, cIAP2 and X-linked inhibitor of apoptosis (XIAP). Expression of the IAP family proteins allows cells to circumvent apoptosis through a variety of mechanisms including direct inhibition of caspase activation (Altieri, 2010). While some studies show that Survivin can directly block caspase activation, it has also been shown to promote cell survival indirectly by preventing Smac/Diablo from inhibiting XIAP, facilitating direct inhibition of caspase activation by XIAP (Altieri, 2010, Vucic and Fairbrother, 2007). In the current study, we found that ET-1 increases Survivin expression in normal lung myofibroblasts through a mechanism mediated by ROCK, FAK, and PI3K/AKT. Substantiating a role for Survivin in the regulation of myofibroblast susceptibility to apoptosis, inhibition of Survivin completely restored susceptibility to myofibroblasts treated with ET-1. Inhibition of Survivin in the absence of Fas/CHX was not, however, sufficient to induce apoptosis, demonstrating that while increased Survivin expression can suppress apoptosis, Survivin expression is not required for cell survival in the absence of an apoptotic stimulus.
The increased expression of Survivin following a single exposure to ET-1 was transient, and Survivin expression returned to baseline levels at 24 hours. This finding may explain, in part, the extensive cell death observed at 48 hours following exposure to Fas/CHX despite treatment with ET-1. Moreover, this transient increase suggests that recurrent or continuous exposure to ET-1 may be required to promote sustained increases in Survivin and continued resistance to apoptosis. Conceptually, this finding is congruent with the hypothesis that IPF results from a chronic or recurrent stimulus of lung injury.
As with other IAPs, Survivin has been best studied for its role in neoplastic disease (Altieri, 2010). It is now recognized that fibrosis shares many biologic features with neoplastic processes, including aberrant cell accumulation due to evasion of apoptosis mediated by AKT (Vancheri et al., 2010). Consistent with our findings, one recent study reported that overexpression of active PI3K increased Survivin expression in mesenchymal cells (Zhao et al., 2010). Moreover, we recently showed that PGE2-sensitized fibroblasts to apoptosis while decreasing basal levels of Survivin expression (Huang et al., 2009). Supporting a critical role for PI3K/AKT in the regulation of Survivin expression, PGE2 increases PTEN activity and decreases PI3K/AKT activity in lung fibroblasts (Maher et al., 2010, White et al., 2005).
The findings presented here, taken in context with prior reports from our laboratory and others, indicate that Survivin may represent a common downstream mechanism by which PI3K/AKT regulates myofibroblast survival and, therefore, fibrogenesis. In this study, we demonstrate for the first time that ET-1 mediated upregulation of an IAP leads to myofibroblast resistance to an apoptotic stimulus. In contrast, PGE2 was shown to sensitize fibroblasts to apoptosis while decreasing basal expression of both Survivin and XIAP (Huang et al., 2009, Maher et al., 2010). While each of these studies demonstrated the effect of PGE2 on a different IAP, the findings are complementary, as Survivin functions to stabilize XIAP through inhibition of Smac/Diablo (Altieri, 2010). Combined with our current report, these studies strongly support a role for the PI3K/AKT-regulated induction or suppression of IAPs in the modulation of mesenchymal cell susceptibility to apoptosis.
Although no reports have specifically examined the role of IAPs in fibrogenesis in-vivo, a murine model of reversible liver fibrosis showed a significant increase in Survivin expression which correlated with collagen synthesis during the fibrotic phase; moreover, Survivin expression dramatically decreased during the resolution phase, which coincides with stellate cell (myofibroblast) apoptosis (Marsillach et al., 2008). Other studies reported that myofibroblasts from keloid scars had increased expression of cIAP-1 and that XIAP expression was increased in a model of hypertension-induced cardiac fibrosis.(Messadi et al., 2004, Ammarguellat et al., 2002). In the latter study, blockade of the ET-A receptor led to the decreased XIAP expression, implicating ET-1 in the upregulation of XIAP in this model (Ammarguellat et al., 2002).
Tissue fibrosis is implicated in up to 45% of chronic diseases in humans, but specific anti-fibrotic interventions are lacking (Wynn, 2008). The mechanisms regulating myofibroblast apoptosis in normal and fibrotic repair processes remain poorly understood, although it is increasingly appreciated that enhancement of myofibroblast susceptibility to apoptosis holds promise as a novel anti-fibrotic strategy (Hernandez-Gea and Friedman, 2011, Hinz and Gabbiani, 2010). This study shows, for the first time, that ET-1 modulates myofibroblasts susceptibility to apoptosis by increasing Survivin expression through activation of FAK and PI3K/AKT. These findings suggest that Survivin, and other IAPs, may contribute to fibrosis by promoting myofibroblast survival and that IAPs may represent novel targets for intervention in fibrotic disease.
Supplementary Material
Acknowledgments
These studies were supported by National Institutes of Health (NIH) grants K08 HL081059 (J.C.H.) and R01 HL105489 (J.C.H.) and funding from Atlantic Philanthropies Inc. (USA), American College of Chest Physicians/The Chest Foundation, the John A. Hartford Foundation, and the Association of Specialty Professors (J.C.H.), NIH R01 HL078871 (T.H.S.), NIH R01 HL 085083 (E.S.W.), and NIH K08 HL094657 (S.K.H.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J. 2010;430:199–205. doi: 10.1042/BJ20100814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammarguellat FZ, Gannon PO, Amiri F, Schiffrin EL. Fibrosis, matrix metalloproteinases, and inflammation in the heart of DOCA-salt hypertensive rats: role of ET(A) receptors. Hypertension. 2002;39:679–684. doi: 10.1161/hy0202.103481. [DOI] [PubMed] [Google Scholar]
- Buhling F, Wille A, Rocken C, Wiesner O, Baier A, Meinecke I, Welte T, Pap T. Altered expression of membrane-bound and soluble CD95/Fas contributes to the resistance of fibrotic lung fibroblasts to FasL induced apoptosis. Respir Res. 2005;6:37. doi: 10.1186/1465-9921-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazaubon S, Chaverot N, Romero IA, Girault JA, Adamson P, Strosberg AD, Couraud PO. Growth factor activity of endothelin-1 in primary astrocytes mediated by adhesion-dependent and-independent pathways. J Neurosci. 1997;17:6203–6212. doi: 10.1523/JNEUROSCI.17-16-06203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Wei K, Jacobs SS, Upadhyay D, Weill D, Rosen GD. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J Biol Chem. 2010;285:8196–8206. doi: 10.1074/jbc.M109.025684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente CF, Tornatore TF, Theizen TH, Deckmann AC, Pereira TC, Lopes-Cendes I, Souza JR, Franchini KG. Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ Res. 2007;101:1339–1348. doi: 10.1161/CIRCRESAHA.107.160978. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008;283:35622–35629. doi: 10.1074/jbc.M804036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Gladson CL, Wu H, Hayasaka H, Olman MA. Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J Biol Chem. 2008;283:26839–26849. doi: 10.1074/jbc.M803645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eble DM, Strait JB, Govindarajan G, Lou J, Byron KL, Samarel AM. Endothelin-induced cardiac myocyte hypertrophy: role for focal adhesion kinase. Am J Physiol Heart Circ Physiol. 2000;278:H1695–1707. doi: 10.1152/ajpheart.2000.278.5.H1695. [DOI] [PubMed] [Google Scholar]
- Evans NJ, Walker JW. Endothelin receptor dimers evaluated by FRET, ligand binding, and calcium mobilization. Biophys J. 2008;95:483–492. doi: 10.1529/biophysj.107.119206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca C, Abraham D, Renzoni EA. Endothelin in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;44:1–10. doi: 10.1165/rcmb.2009-0388TR. [DOI] [PubMed] [Google Scholar]
- Frankel SK, Cosgrove GP, Cha SI, Cool CD, Wynes MW, Edelman BL, Brown KK, Riches DW. TNF-alpha sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apoptosis. Am J Respir Cell Mol Biol. 2006;34:293–304. doi: 10.1165/rcmb.2005-0155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, Cance WG. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51:7405–7416. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregan B, Jurgensen J, Papsdorf G, Furkert J, Schaefer M, Beyermann M, Rosenthal W, Oksche A. Ligand-dependent differences in the internalization of endothelin A and endothelin B receptor heterodimers. J Biol Chem. 2004;279:27679–27687. doi: 10.1074/jbc.M403601200. [DOI] [PubMed] [Google Scholar]
- Hafizi S, Wharton J, Chester AH, Yacoub MH. Profibrotic effects of endothelin-1 via the ETA receptor in cultured human cardiac fibroblasts. Cell Physiol Biochem. 2004;14:285–292. doi: 10.1159/000080338. [DOI] [PubMed] [Google Scholar]
- Hayashida T, Wu MH, Pierce A, Poncelet AC, Varga J, Schnaper HW. MAP-kinase activity necessary for TGFbeta1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J Cell Sci. 2007;120:4230–4240. doi: 10.1242/jcs.03492. [DOI] [PubMed] [Google Scholar]
- Heidkamp MC, Bayer AL, Kalina JA, Eble DM, Samarel AM. GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circ Res. 2002;90:1282–1289. doi: 10.1161/01.res.0000023201.41774.ea. [DOI] [PubMed] [Google Scholar]
- Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- Hinz B, Gabbiani G. Fibrosis: recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol Rep. 2010;2:78. doi: 10.3410/B2-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Wilkes MC, Penheiter SG, Gupta SK, Edens M, Leof EB. Non-Smad transforming growth factor-beta signaling regulated by focal adhesion kinase binding the p85 subunit of phosphatidylinositol 3-kinase. J Biol Chem. 2011;286:17841–17850. doi: 10.1074/jbc.M111.233676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, Cui Z, Thannickal VJ. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–1367. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JC, Rogers DS, Sharma V, Vittal R, White ES, Cui Z, Thannickal VJ. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761–771. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JC, Rogers DS, Simon RH, Sisson TH, Thannickal VJ. Plasminogen activation induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am J Respir Cell Mol Biol. 2008;38:78–87. doi: 10.1165/rcmb.2007-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JC, Thannickal VJ. Idiopathic pulmonary fibrosis: new concepts in pathogenesis and implications for drug therapy. Treat Respir Med. 2006;5:325–342. doi: 10.2165/00151829-200605050-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Khoe M, Befekadu M, Chung S, Takata Y, Ilic D, Bryer-Ash M. Focal adhesion kinase mediates cell survival via NF-kappaB and ERK signaling pathways. Am J Physiol Cell Physiol. 2007;292:C1339–1352. doi: 10.1152/ajpcell.00144.2006. [DOI] [PubMed] [Google Scholar]
- Huang SK, White ES, Wettlaufer SH, Grifka H, Hogaboam CM, Thannickal VJ, Horowitz JC, Peters-Golden M. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 2009;23:4317–4326. doi: 10.1096/fj.08-128801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HQ, Zhang XL, Liu L, Yang CC. Relationship between focal adhesion kinase and hepatic stellate cell proliferation during rat hepatic fibrogenesis. World J Gastroenterol. 2004;10:3001–3005. doi: 10.3748/wjg.v10.i20.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–1093. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada N, Seki S, Kuroki T, Kaneda K. ROCK inhibitor Y-27632 attenuates stellate cell contraction and portal pressure increase induced by endothelin-1. Biochem Biophys Res Commun. 1999;266:296–300. doi: 10.1006/bbrc.1999.1823. [DOI] [PubMed] [Google Scholar]
- Kennedy L, Shi-Wen X, Carter DE, Abraham DJ, Leask A. Fibroblast adhesion results in the induction of a matrix remodeling gene expression program. Matrix Biol. 2008;27:274–281. doi: 10.1016/j.matbio.2008.01.004. [DOI] [PubMed] [Google Scholar]
- King TE, Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA, Jr, Flint A, Thurlbeck W, Cherniack RM. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001;164:1025–1032. doi: 10.1164/ajrccm.164.6.2001056. [DOI] [PubMed] [Google Scholar]
- Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, Weaver TE, Guenther A. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korfhagen TR, Le Cras TD, Davidson CR, Schmidt SM, Ikegami M, Whitsett JA, Hardie WD. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2009;41:562–572. doi: 10.1165/rcmb.2008-0377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama Y, Yoshioka Y, Matsuda T, Baba A. Focal adhesion kinase is required for endothelin-induced cell cycle progression of cultured astrocytes. Glia. 2003;43:185–189. doi: 10.1002/glia.10240. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Yoshioka Y, Shinde M, Matsuda T, Baba A. Focal adhesion kinase mediates endothelin-induced cyclin D3 expression in rat cultured astrocytes. J Neurochem. 2004;90:904–912. doi: 10.1111/j.1471-4159.2004.02546.x. [DOI] [PubMed] [Google Scholar]
- Kulasekaran P, Scavone CA, Rogers DS, Arenberg DA, Thannickal VJ, Horowitz JC. Endothelin-1 and transforming growth factor-beta1 independently induce fibroblast resistance to apoptosis via AKT activation. Am J Respir Cell Mol Biol. 2009;41:484–493. doi: 10.1165/rcmb.2008-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepparanta O, Pulkkinen V, Koli K, Vahatalo R, Salmenkivi K, Kinnula VL, Heikinheimo M, Myllarniemi M. Transcription factor GATA-6 is expressed in quiescent myofibroblasts in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;42:626–632. doi: 10.1165/rcmb.2009-0021OC. [DOI] [PubMed] [Google Scholar]
- Maher TM, Evans IC, Bottoms SE, Mercer PF, Thorley AJ, Nicholson AG, Laurent GJ, Tetley TD, Chambers RC, McAnulty RJ. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:73–82. doi: 10.1164/rccm.200905-0674OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsillach J, Ferre N, Camps J, Rull A, Beltran R, Joven J. Changes in the expression of genes related to apoptosis and fibrosis pathways in CCl4-treated rats. Mol Cell Biochem. 2008;308:101–109. doi: 10.1007/s11010-007-9617-0. [DOI] [PubMed] [Google Scholar]
- Masamune A, Satoh M, Kikuta K, Suzuki N, Shimosegawa T. Endothelin-1 stimulates contraction and migration of rat pancreatic stellate cells. World J Gastroenterol. 2005;11:6144–6151. doi: 10.3748/wjg.v11.i39.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messadi DV, Doung HS, Zhang Q, Kelly AP, Tuan TL, Reichenberger E, Le AD. Activation of NFkappaB signal pathways in keloid fibroblasts. Arch Dermatol Res. 2004;296:125–133. doi: 10.1007/s00403-004-0487-y. [DOI] [PubMed] [Google Scholar]
- Moodley YP, Caterina P, Scaffidi AK, Misso NL, Papadimitriou JM, McAnulty RJ, Laurent GJ, Thompson PJ, Knight DA. Comparison of the morphological and biochemical changes in normal human lung fibroblasts and fibroblasts derived from lungs of patients with idiopathic pulmonary fibrosis during FasL-induced apoptosis. J Pathol. 2004;202:486–495. doi: 10.1002/path.1531. [DOI] [PubMed] [Google Scholar]
- Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, Kita A, Tominaga F, Yamanaka K, Kudoh M, Sasamata M. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- Nho RS, Xia H, Diebold D, Kahm J, Kleidon J, White E, Henke CA. PTEN regulates fibroblast elimination during collagen matrix contraction. J Biol Chem. 2006;281:33291–33301. doi: 10.1074/jbc.M606450200. [DOI] [PubMed] [Google Scholar]
- Oikawa T, Unno Y, Matsuno K, Sawada J, Ogo N, Tanaka K, Asai A. Identification of a small-molecule inhibitor of the interaction between Survivin and Smac/DIABLO. Biochem Biophys Res Commun. 2010;393:253–258. doi: 10.1016/j.bbrc.2010.01.113. [DOI] [PubMed] [Google Scholar]
- Parsons CJ, Takashima M, Rippe RA. Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S79–84. doi: 10.1111/j.1440-1746.2006.04659.x. [DOI] [PubMed] [Google Scholar]
- Paterno J, Vial IN, Wong VW, Rustad KC, Sorkin M, Shi Y, Bhatt KA, Thangarajah H, Glotzbach JP, Gurtner GC. Akt-mediated mechanotransduction in murine fibroblasts during hypertrophic scar formation. Wound Repair Regen. 2011;19:49–58. doi: 10.1111/j.1524-475X.2010.00643.x. [DOI] [PubMed] [Google Scholar]
- Rankin S, Morii N, Narumiya S, Rozengurt E. Botulinum C3 exoenzyme blocks the tyrosine phosphorylation of p125FAK and paxillin induced by bombesin and endothelin. FEBS Lett. 1994;354:315–319. doi: 10.1016/0014-5793(94)01148-6. [DOI] [PubMed] [Google Scholar]
- Ryan BM, O’Donovan N, Duffy MJ. Survivin: a new target for anti-cancer therapy. Cancer Treat Rev. 2009;35:553–562. doi: 10.1016/j.ctrv.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Shi-Wen X, Chen Y, Denton CP, Eastwood M, Renzoni EA, Bou-Gharios G, Pearson JD, Dashwood M, du Bois RM, Black CM, Leask A, Abraham DJ. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell. 2004;15:2707–2719. doi: 10.1091/mbc.E03-12-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-Wen X, Denton CP, Dashwood MR, Holmes AM, Bou-Gharios G, Pearson JD, Black CM, Abraham DJ. Fibroblast matrix gene expression and connective tissue remodeling: role of endothelin-1. J Invest Dermatol. 2001;116:417–425. doi: 10.1046/j.1523-1747.2001.01256.x. [DOI] [PubMed] [Google Scholar]
- Shi-wen X, Kennedy L, Renzoni EA, Bou-Gharios G, du Bois RM, Black CM, Denton CP, Abraham DJ, Leask A. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum. 2007a;56:4189–4194. doi: 10.1002/art.23134. [DOI] [PubMed] [Google Scholar]
- Shi-Wen X, Renzoni EA, Kennedy L, Howat S, Chen Y, Pearson JD, Bou-Gharios G, Dashwood MR, du Bois RM, Black CM, Denton CP, Abraham DJ, Leask A. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol. 2007b;26:625–632. doi: 10.1016/j.matbio.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Shi-Wen X, Rodriguez-Pascual F, Lamas S, Holmes A, Howat S, Pearson JD, Dashwood MR, du Bois RM, Denton CP, Black CM, Abraham DJ, Leask A. Constitutive ALK5-independent c-Jun N-terminal kinase activation contributes to endothelin-1 overexpression in pulmonary fibrosis: evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Mol Cell Biol. 2006;26:5518–5527. doi: 10.1128/MCB.00625-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson MS, Ismail-Beigi F. Endothelin-1 increases collagen accumulation in renal mesangial cells by stimulating a chemokine and cytokine autocrine signaling loop. J Biol Chem. 2011;286:11003–11008. doi: 10.1074/jbc.M110.190793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, Parsons JT. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–14852. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K, Hara N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur Respir J. 2002;20:359–368. doi: 10.1183/09031936.02.00252602. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3:350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Torsoni AS, Marin TM, Velloso LA, Franchini KG. RhoA/ROCK signaling is critical to FAK activation by cyclic stretch in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2005;289:H1488–1496. doi: 10.1152/ajpheart.00692.2004. [DOI] [PubMed] [Google Scholar]
- Tostes RC, Touyz RM, He G, Ammarguellat F, Schiffrin EL. Endothelin A receptor blockade decreases expression of growth factors and collagen and improves matrix metalloproteinase-2 activity in kidneys from stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2002;39:892–900. doi: 10.1097/00005344-200206000-00015. [DOI] [PubMed] [Google Scholar]
- Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35:496–504. doi: 10.1183/09031936.00077309. [DOI] [PubMed] [Google Scholar]
- Vittal R, Horowitz JC, Moore BB, Zhang H, Martinez FJ, Toews GB, Standiford TJ, Thannickal VJ. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166:367–375. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic D, Fairbrother WJ. The inhibitor of apoptosis proteins as therapeutic targets in cancer. Clin Cancer Res. 2007;13:5995–6000. doi: 10.1158/1078-0432.CCR-07-0729. [DOI] [PubMed] [Google Scholar]
- Wei X, Han J, Chen ZZ, Qi BW, Wang GC, Ma YH, Zheng H, Luo YF, Wei YQ, Chen LJ. A phosphoinositide 3-kinase-gamma inhibitor, AS605240 prevents bleomycin-induced pulmonary fibrosis in rats. Biochem Biophys Res Commun. 2010;397:311–317. doi: 10.1016/j.bbrc.2010.05.109. [DOI] [PubMed] [Google Scholar]
- Wendel M, Petzold A, Koslowski R, Kasper M, Augstein A, Knels L, Bleyl JU, Koch T. Localization of endothelin receptors in bleomycin-induced pulmonary fibrosis in the rat. Histochem Cell Biol. 2004;122:507–517. doi: 10.1007/s00418-004-0708-7. [DOI] [PubMed] [Google Scholar]
- White ES, Atrasz RG, Dickie EG, Aronoff DM, Stambolic V, Mak TW, Moore BB, Peters-Golden M. Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am J Respir Cell Mol Biol. 2005;32:135–141. doi: 10.1165/rcmb.2004-0126OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, Hogaboam CM, Flaherty KR, Martinez FJ, Kontos CD, Toews GB. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10) Am J Respir Crit Care Med. 2006;173:112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Li YS, Haga JH, Kaunas R, Chiu JJ, Su FC, Usami S, Chien S. Directional shear flow and Rho activation prevent the endothelial cell apoptosis induced by micropatterned anisotropic geometry. Proc Natl Acad Sci U S A. 2007;104:1254–1259. doi: 10.1073/pnas.0609806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. 2004;279:33024–33034. doi: 10.1074/jbc.M313265200. [DOI] [PubMed] [Google Scholar]
- Zachary I, Sinnett-Smith J, Rozengurt E. Bombesin, vasopressin, and endothelin stimulation of tyrosine phosphorylation in Swiss 3T3 cells. Identification of a novel tyrosine kinase as a major substrate. J Biol Chem. 1992;267:19031–19034. [PubMed] [Google Scholar]
- Zhao P, Meng Q, Liu LZ, You YP, Liu N, Jiang BH. Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem Biophys Res Commun. 2010;395:219–224. doi: 10.1016/j.bbrc.2010.03.165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.