

Abstract
Sepsis is one of the leading causes of death in hospitals worldwide. Even with optimal therapy, severe sepsis results in 50% mortality, indicating variability in the response of individuals towards treatment. We hypothesize that the presence of pre-existing antibodies present in the blood before the onset of sepsis induced by cecal ligation and puncture (CLP) in mice, accounts for the differences in their survival. A Plasma Enhanced Killing (PEK) assay was performed to calculate the PEK capacity of plasma i.e. the ability of plasma to augment PMN killing of bacteria. PEK was calculated as PEK= (1/log (N)) × 100; where N= number of surviving bacteria; a higher PEK indicated better bacterial killing. A range of PEK in plasma collected from mice prior to CLP was observed, documenting individual differences in bacterial killing capacity. Mortality was predicted based on plasma IL-6 levels at 24 hr post CLP. Mice predicted to die (Die-P) had a lower PEK (<14) and higher peritoneal bacterial counts 24 hr post sepsis compared to those predicted to live (Live-P) with a PEK>16. Mice with PEK<14 were 3.1 times more likely to die compared to the PEK>16 group. To understand the mechanism of defense conferred by the pre-existing antibodies, binding of IgM or IgG to enteric bacteria was documented by flow cytometry. To determine the relative contribution of IgM or IgG, the immunoglobulins were specifically immuno-depleted from the naïve plasma samples and the PEK of the depleted plasma measured. Compared to naïve plasma, depletion of IgM had no effect on the PEK. However, depletion of IgG increased PEK suggesting that an inhibitory IgG binds to antigenic sites on bacteria preventing optimal opsonization of the bacteria. These data demonstrate that prior to CLP; circulating inhibitory IgG antibodies exist that prevent bacterial killing by PMNs in a CLP model of sepsis.
Introduction
In the United States, sepsis is the second-leading cause of death in non-coronary ICU patients, and the tenth-most common cause of death overall; according to data from the Centers for Disease Control and Prevention. Sepsis is common and also more dangerous in elderly, immunocompromised, and critically ill patients. It occurs in 1–2% of all hospitalizations and accounts for as much as 25% of intensive-care unit (ICU) bed utilization (1) (2). It is a major cause of death in ICUs worldwide, with mortality rates that range from 20% for sepsis to 40% for severe sepsis to >60% for septic shock (2–4). There are approximately 750,000 new sepsis cases each year, with at least 210,000 fatalities (5). The economic burden for the medical care has been estimated at 16.7 billion dollars annually (5) and is increasing. As medicine becomes more aggressive, with invasive procedures and immunosuppression; the incidence of sepsis is likely to continue to increase (3). The National Center for Health Statistics stated that from 2000 to 2007, the rate of hospitalization for septicemia or sepsis for persons aged 65–74 years increased 57%, from 6.5 per 1,000 to 10.2 indicating that there has not been much progress achieved in the treatment of sepsis (6).
Though Gram negative bacteria were initially thought to be the predominant organism responsible for sepsis (7), current studies increasingly indicate that Gram positive bacteria and fungal organisms are also common causes of sepsis (8). Acute respiratory failure, shock, acute renal failure, coagulopathy and multiple organ failure are some of the causes of mortality in septic patients (8, 9).
The cecal ligation and puncture (CLP) model of sepsis induces bacterial peritonitis thereby reproducing the complex immune response of sepsis similar to that with patients in ICU (10). In the murine model of sepsis, the first 5 days following CLP is defined as the acute phase while day 6–28 is defined as the chronic phase of sepsis in our laboratory (11). Mice which die during the early or acute phase of sepsis have an increase in body weight as well as increased plasma IL-6 levels indicating activation of the immune system (12). Both pro-inflammatory and anti-inflammatory cytokines have been shown to be elevated during this early or acute phase (13) and chronic sepsis is also characterized by an individualized inflammatory response (14). Neutrophil recruitment to the site of infection serves as a critical immune response for host survival (15).
It may be argued that septic patients die as a result of immunosuppression and failure to control bacterial growth rather than immunostimulation. This leads to the crucial question of understanding the mechanism for bacterial killing during the septic response. An inadequate host immune system that results in poor bacterial killing would represent a completely different mechanism of death compared to excessive inflammation. The present study attempts to decipher the mechanism by which some individuals die and others live while combating the same bacterial infection. We hypothesize that those mice that survive cecal ligation and puncture induced sepsis have pre-existing antibodies which opsonize and kill bacteria more efficiently.
Methods
Animals
Adult female ICR mice (from Harlan-Sprague Dawley, Inc., Indianapolis, IN) were used. Mice were acclimatized to our housing room for at least 5 days before surgery. This was a temperature controlled room with 12 hours light- 12 hours dark diurnal cycle. They were provided food and water ad libitum for the entire duration of the experiment. The experiments were approved by Boston University Animal Care and Use Committee.
Sepsis Model
Cecal ligation and puncture which results in bacterial peritonitis was performed as first described (8) with some modifications (10). Briefly, under isoflurane anesthesia (5% induction followed by 3% for maintenance during surgery) a midline abdominal incision was performed through the skin and then through the linea alba. The cecum was exposed through the incision and ligated with 3-0 silk below the ileocecal valve; approximately two-thirds from the distal end of the cecum was ligated and then double punctured with a 16 gauge needle to induce sub-lethal sepsis. The double puncture involves piercing longitudinally from the distal tip of the cecum to the lateral wall of the cecum under the suture line followed by squeezing out some cecal content on both sides. The abdominal muscles were sutured with silk, and wound glue (Nexaband-Veterinary Products Laboratories, Phoenix, Az) was used to close the skin. Mice were resuscitated with 1 ml of warm (37× C) normal saline with buprenorphine (0.05 mg/kg) injected subcutaneously. Antibiotic treatment with 25 mg/kg imipenem (Merck, West Point, PA) was started 2 hours after surgery and continued every 12 hours for the first 5 days. Pain control was assured for the first 2 days post CLP with buprenorphine every 12 hours. The mice that were sacrificed at 24 hours received ketamine anesthesia and cervical dislocation was performed.
Sample collection
For the Plasma Enhanced Killing (PEK) assay, 50 μl of blood was collected twice from the facial vein with an interval of 3 days between the blood draws. The blood was diluted to 50 % using saline with heparin (100 μl heparin per 1 mL of saline), centrifuged for 5 minutes at 1000 × g at 4°C and the plasma from both collections were stored at −20°C till further analysis was done. At 6 and 24 hours post CLP, 20 μl of blood was collected from the facial vein followed by aspiration using a pipette and an EDTA (169 mM tripotassium salt) rinsed tip. The blood was diluted 1/10 in PBS with 1/50 EDTA, centrifuged for 5 min at 1000 × g at 4°C and the plasma IL-6 was measured by ELISA. For all endpoint sampling, blood was collected from the retro-orbital venous plexus. The peritoneal cavity was opened after sacrifice and washed with 1 ml of Hanks Balanced Salt Solution (HBSS) containing 50 mM EDTA and then a second time with 25 ml of the same solution. A portion of the first lavage fluid was used for microbiology cultures and the rest was centrifuged. For thioglycollate elicited neutrophils, the peritoneal cavity was washed with a total of 5 ml of HBSS.
Plasma Enhanced Killing (PEK) Assay
This assay was performed using thioglycollate elicited neutrophils, and enteric bacteria from a normal mouse, so that the cells and bacteria were consistent. The only assay variable was the source of the plasma, which came from individual mice. Thioglycollate was injected intra-peritoneally into a naïve mouse and cells recovered 4–6 hours later by peritoneal lavage using warm (37°C) HBSS. Red blood cells were lyzed with 3 ml of ACK lysis buffer (Invitrogen) and cells were resuspended in HBSS and counted using Beckman-Coulter particle counter model ZF (Coulter Electronics, Hialeah, FL). Bacteria were collected by excising and homogenizing the cecum (Brinkmann, Polytron PT 3000). The bacteria were resuspended in HBSS and quantified using the McFarland standard with measurement at 565 nm. The colony forming units (CFU) were verified by plating aliquots. To perform the plasma enhanced killing (PEK) assay, 50μl of plasma (previously diluted to 50%) from an individual mouse was incubated with an equal volume of bacteria (1.75 × 105 CFU) at 37°C with continuous shaking for 30 minutes to opsonize the bacteria. The opsonization process was stopped by placing this mixture on ice for 1 hour. The thioglycollate elicited neutrophils (1.75 × 104) were mixed with the opsonized bacteria and incubated in a 37°C shaker for 1 hour. The mixture was plated on blood agar plates (Fisher Scientific) in duplicates. Plates were incubated at 37° C in aerobic or anaerobic conditions for 24 hours and then read. The number of residual bacteria was determined. PEK was calculated by the following equation:
Where N = number of recovered bacteria. A lower number of recovered bacteria meant better bacterial killing which in turn lead to a higher PEK value. To state this more directly, a higher PEK number indicated better killing.
Depletion of IgG/IgM
Immuno 96 microwell plates (Nunc plates) were coated with goat anti-mouse IgM or goat anti-mouse IgG antibody (Bethyl Laboratories, diluted 1:400) and kept overnight at 4°C. The plates were blocked for 1 hour the next day in Casein blocking buffer followed by washing the plate in a Biotek (EL×405) microplate washer containing a wash buffer (PBS with 0.05% Tween-20) to remove the unbound antibodies. 50–100 μl of undiluted naïve plasma were added in each well and the plates were kept on a shaker at room temperature for 3 hrs. The supernatant containing IgG or IgM depleted plasma was collected and stored at −20°C for further use. Depletion was confirmed by ELISA of depleted plasma compared to that of the intact plasma.
Flow cytometry
Cecal bacteria corresponding to 1.0 McFarland Standard (3×108 CFU) were opsonized with 50% plasma as described above. The opsonized bacteria were washed twice with 0.5% BSA in HBSS and resuspended in 150 μl of FACS buffer (0.5% BSA in HBSS). The resulting bacteria were stained using 1.5 μl of phycoerythrin (PE) conjugated anti-mouse IgM antibody or 2μl of fluorescein isothiocyanate (FITC) conjugated anti-mouse IgG antibody (BD Pharmingen) and incubated at 4°C for 45 minutes in dark. PE conjugated Rat IgG2a,κ and FITC conjugated Rat IgG1,κ (BD Pharmingen) were used as isotype controls. Following incubation, the stained bacteria were washed twice and then fixed with 2% paraformaldehyde and analyzed by flow cytometry using a FACS Calibur (BD).
Peritoneal bacterial cultures after cecal ligation and puncture
The peritoneal lavage fluid collected 24 hours post CLP was cultured on 5% sheep blood agar plates. Plates were incubated at 37° C in aerobic or anaerobic conditions for 16 hours and then they were read and the number of CFU was determined.
ELISA
Plasma levels of IL-6 were determined according to a protocol already published by our laboratory (16). IgM and IgG ELISA were performed to look for total levels of IgM and IgG before and after depletion of the naïve plasma.
Statistical analysis
Statistics were performed using the computer program Prism 5 (GraphPad Software, San Diego, CA). For survival, the log rank survival was performed with a 95% confidence interval. The chi-square test was used to calculate the odds ratio. One-way ANOVA followed by Dunnett’s Multiple Comparison Test or Newman-Keuls was used to compare multiple groups. When two groups were considered, an unpaired Student t test was used. Paired Student t test was performed to evaluate the change in PEK value before and after IgM/IgG depletion. P value <0.05 was considered statistically significant.
RESULTS
Differential mortality rate following CLP in mice
All mice undergoing CLP were age and sex matched, had the same surgical procedure performed and identical antibiotic and fluid therapy for post surgery monitoring. Even with this consistent CLP protocol not all mice died indicating variability in the septic response. Similar to previous reports, 65% mortality was observed during the acute phase of sepsis (Fig. 1A). Our laboratory and others have established plasma IL-6 as a biomarker of sepsis mortality in mice undergoing CLP (17) (18, 19). At 24 hr post CLP, 20 μl of blood was drawn from the facial vein and plasma IL-6 measured. Facial vein bleeding does not affect the overall health of the animal (13). We verified that plasma IL-6 will predict mortality by following the mice for 7 days post CLP. Since day 5 post CLP marks the end of the acute phase, the plasma IL-6 levels were matched to the actual status (alive or dead) of the animal on day 5 (Fig. 1B). Two discrete IL-6 levels were determined in order to optimize sensitivity and specificity. A Receiver Operator Characteristic (ROC) curve at 24 hours indicated that plasma IL-6 levels < 1.7 ng/ml provided 100% sensitivity for the mice which were alive at day 5 post CLP and levels > 12.6 ng/ml provided 100% specificity (Fig. 1C) similar to our previously published results (20). A Kaplan-Meier survival curve dividing mice into 2 groups based on these discrimination values showed approximately 85% survival with IL-6 < 1.7 ng/ml and less than 20% survival with IL-6 >12.6 ng/ml (Fig. 1D). For several experiments where the mice needed to be sacrificed, the outcome was predicted based on the plasma IL-6 at 24 hour post CLP. Mice having IL-6 < 1.6 ng/ml and those having IL-6 >12.6 ng/ml were categorized as Live-Predicted (Live-P) and Die-Predicted (Die-P) respectively. As shown in figure 1, individual mice responded differently to CLP mediated sepsis. Plasma IL-6 accurately predicts mortality only for the first 5 days following CLP; therefore survival was followed for the acute phase instead of 28 days.
Figure 1. Plasma IL-6 predicts mortality.

(A) The CLP protocol results in 65% mortality during the first 5 days. None of the mice died during the first 24 hours, such that plasma samples were available for each mouse. n= 22. (B) Plasma IL-6 levels 24 hour post CLP were significantly higher in those mice that died. n = 22–25 for each group, data were combined from 3 independent experiments. Results are shown as mean ± SEM. (C) Receiver Operator Characteristic (ROC) curve to test for specificity and sensitivity of 24 hour plasma IL-6 to predict outcome. The area under the curve (AUC) has 100 % specificity (12.6 ng/ml) and 100% sensitivity (1.7 ng/ml) with AUC close to 1 making it an excellent biomarker. (D) Survival curve for mice during the acute phase of sepsis based on their plasma IL-6 cut offs. p = 0.0025 from Log-rank (Mantel-Cox) Test. n= 11 for IL-6 <1.6 ng/ml and n=17 IL-6>12.7 ng/ml.
Plasma antibody binds to enteric bacteria and augments phagocytic killing
The CLP model mimics the clinical scenario where even with optimal treatment some patients expire while others survive (21). The murine studies have a relatively homogeneous cohort where the mice are all the same age, weight, gender and have the same fluid and antibiotic regime post surgery. Our hypothesis for the variation in the response to a uniform septic challenge may be the presence of a pre-existing “factor” present in the plasma of mice which functions differently between different mice.
We sought to determine whether plasma samples obtained prior to CLP would have a difference in the capacity to enhance neutrophil killing of enteric bacteria. To explore this further a Plasma Enhanced Killing (PEK) assay was developed as described in the methods. This assay quantifies the killing capacity of the naïve plasma. Plasma without bacteria did not show any bacterial growth, indicating that our blood collection technique did not result in contaminated plasma (Fig 2A). The initial inoculum of bacteria at the start of the assay was same for all the samples tested, i.e. 1×105 CFU and was called CFU (initial). Bacteria are known to have a doubling time of about 20 minutes (22), the bacterial count obtained at the end of the assay should be approximately 1×107 CFU which was verified in the assay (Fig 2A). We called this bacterial count as CFU (final). The assay should be able to test whether plasma alone can kill bacteria or the presence of neutrophils augments bacterial killing by plasma. Plasma alone plus bacteria did slightly reduce the number of CFUs, but neutrophils alone plus bacteria had essentially no impact on bacterial growth (Fig. 2B). The complete assay for bacterial killing includes neutrophils, bacteria and plasma and the individual plasma samples showed a wide range in the ability of the plasma to enhance killing of the bacteria by neutrophils. Once the assay was established we calculated an assay number to clearly communicate the results. The PEK assay number was calculated as PEK= (1/log (N)) × 100, where N = number of recovered bacteria. With this assay, a higher PEK number means better bacterial killing by the plasma. As observed in figure 2C, plasma from individual mice showed a range enhancement of bacterial killing. The next experiments examined whether the ability of plasma to kill bacteria, as determined by the PEK numbers, served as a mechanism for better sepsis survival after CLP.
Figure 2. Plasma Enhanced Killing (PEK) assay using naïve plasma.

(A) Assay controls. The initial inoculum of bacteria was the number of bacteria combined with the plasma (CFU initial). The actual CFUs were verified by culture. Plasma only shows that no bacteria were present when plasma alone was cultured. The number of bacteria at the end of the 2 hour assay (CFU final) showed a significant increase from the initial inoculum, as expected. bdl = below detection level. (B) Plasma alone mixed with bacteria showed a slight decrease in the number of CFUs compared to the CFU final. Neutrophils plus bacteria shows that neutrophils alone had limited ability to kill bacteria. However, plasma + bacteria + PMNs showed a wide range in the capacity of the plasma from an individual mouse to enhanced bacterial killing. (C) Data from panel B was re-graphed using the equation described in methods. Each symbol is the result from an individual mouse. A higher PEK number indicates greater capacity to kill bacteria. Data cumulative of 3 independent experiments. n= 22 for naïve plasma.
Plasma enhanced killing predicts survival in CLP model of sepsis
Plasma was obtained from the facial vein 3–4 days prior to CLP to use for the PEK assay. It is important to note that the PEK value was determined using plasma samples obtained prior to CLP, i.e. before the onset of sepsis. The first study tested whether CLP survivors had higher PEK numbers compared to non-survivors. We observed that mice that were alive at day 5 post CLP have a significantly higher PEK capacity of their naïve plasma compared to those mice which were dead by day 5 post CLP (Fig. 3A). To rigorously test whether PEK predicts survival, a ROC curve analysis was established (Fig. 3B). The Area Under the Curve (AUC) is a measure of the robustness of the predictive value of a test. The AUC in figure 3B is 0.96 making it an excellent predictor of CLP induced sepsis mortality. The pre-CLP plasma showed PEK levels >16.5 provided 100% sensitivity for the mice which were alive at day 5 post CLP and levels < 14 provided 100% specificity (Fig. 3C). A Kaplan-Meier survival curve with the PEK cutoffs showed that mice with a PEK <14 of their naïve plasma had a significantly higher mortality rate during acute phase of sepsis (Fig. 3C, p<0.003 high vs. low PEK by log-rank test). Further confirmation of the ability of the PEK assay to predict outcome was assessed by calculating the odds ratios. Mice having PEK < 14 had a relative risk of sepsis mortality that was 3.1 times greater than mice with a PEK > 16.5 (95% confidence interval: 1.3 to 7.0, data not shown). Though PEK <14 and PEK>16.5 appear to have a narrow range, upon looking at the CFU’s of the recovered bacteria from the PEK assay, we observed that PEK < 14 had CFUs in the order of 107 or higher while PEK >16.5 were in the order of 105 or lower thereby indicating a minimum of 2 log difference between the survivors and non-survivors. A subset of mice undergoing CLP were sacrificed at 24 hours and their predicted mortality calculated based on plasma IL-6 levels. Those mice predicted to live had significantly higher PEK values. (Fig. 3D)
Figure 3. Comparison of plasma enhanced killing sepsis mortality.

(A) Mice alive after 5 days of CLP-induced sepsis had a much higher PEK capacity in plasma samples obtained prior to the onset of sepsis. n= 18 for Live and 13 for Die group. p<0.05 when compared to Live. Results are shown as mean ± SEM and each symbol is the value for an individual mouse. Data are representative of 3 independent experiments. (B) Receiver Operator Characteristic (ROC) curve to test for specificity and sensitivity of PEK capacity of pre-procedure plasma. Area under the curve (AUC) represents 100 % specificity (PEK<14) and 100% sensitivity (PEK >16). (C) Survival curve for mice during the acute phase of sepsis based on their pre-CLP plasma PEK capacity discrimination values. p = 0.0025 from Log-rank (Mantel-Cox) test. n= 18 for PEK > 16 and n=13 PEK <14. (D) Mice predicted to die based on plasma IL-6 levels had lower PEK values compared to mice predicted to live. n= 9 and 7 for Live-P and Die-P respectively. Results are shown as box and whiskers with 10–90 percentile range ± SEM. p<0.05 when compared to Live-P.
The mice sacrificed 24 hours after CLP had the number of bacteria in the peritoneal lavage fluid determined. Figure 4 demonstrates that mice with PEK >16.5 had a significantly lower peritoneal CFU count post sepsis compared to those mice with PEK <14. Additionally, those mice with a high PEK and increased numbers of recovered peritoneal bacteria were predicted to die based on plasma IL-6 levels. Our CLP procedure involves giving antibiotics (25 mg/kg Imipenem) for first 5 days following CLP (23). As a result of this dose and duration of antibiotic therapy no blood bacteria are observed at 24 hr post CLP (23) (24). However, this dose of antibiotics is not sufficient to eradicate bacteria in the peritoneal cavity and hence we observe bacterial CFUs in the peritoneal lavage (15, 25). Thus, our data showed Live-P mice had greater plasma enhanced killing of bacteria in vitro (i.e. high PEK) and post CLP in vivo (i.e. low peritoneal CFU). A significant correlation (data not shown) was observed between the PEK values and the peritoneal CFU.
Figure 4. Peritoneal bacteria in mice after sepsis.
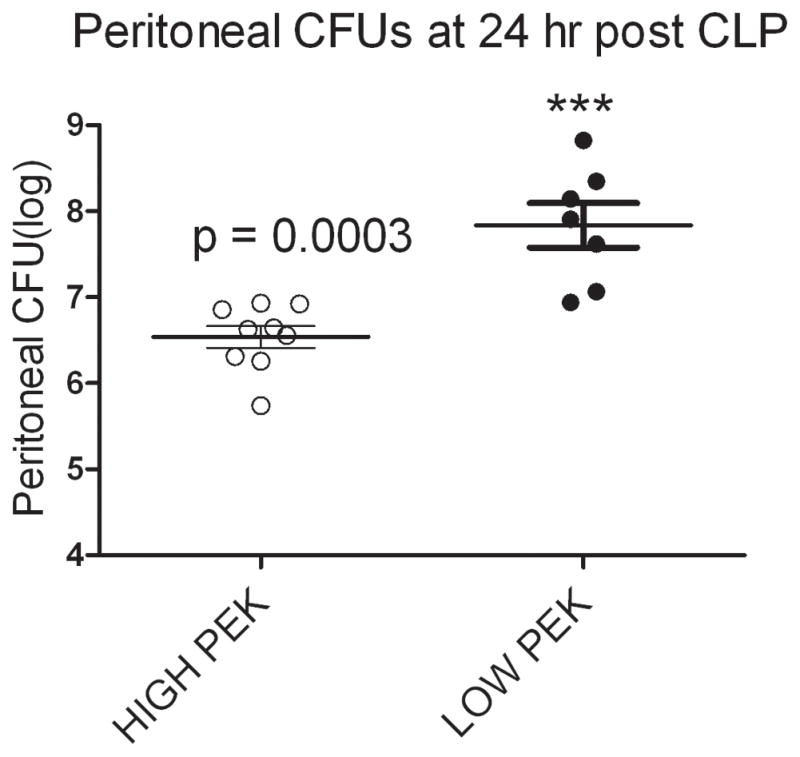
Mice were stratified into High PEK or Low PEK based on the PEK assay of the naïve plasma. CLP were performed on these mice and the peritoneal lavage at 24 hour post CLP was harvested. Mice with low PEK (PEK <14) value had greater bacterial counts compared to those with high PEK (PEK >16). n= 9 and 7 for High PEK and Low PEK respectively and are the same mice used in figure 3D. p<0.05 when compared to High PEK.
Plasma IgG accounts for the differences in PEK values
Several reports have demonstrated the presence of natural antibodies in regulating inflammation and in different diseases including cancer (26). Natural Immunoglobulin M (IgM) has been known to provide immediate defense against systemic bacterial infection (27). Our PEK assay results established the presence of a factor in the plasma which augments bacterial killing in the presence of PMNs. The next experiments sought to further characterize this factor. We first wanted to test whether pre-existing antibodies in the plasma i.e. IgM and IgG are able to bind to enteric bacteria. Plasma was used to opsonize bacteria and the opsonized bacteria were stained for bacterial bound mouse IgM or mouse IgG by flow cytometry. The Mean Fluorescence Intensity (MFI) of the individual plasma samples was higher than that of the isotype control indicating the presence of plasma IgM or IgG which is able to bind to the enteric bacteria (Fig. 5A and B). Further, we observed that the samples on an average had a three-fold increase in plasma IgG binding to enteric bacteria compared to the isotype control.
Figure 5. Plasma IgM and IgG binds to enteric bacteria.
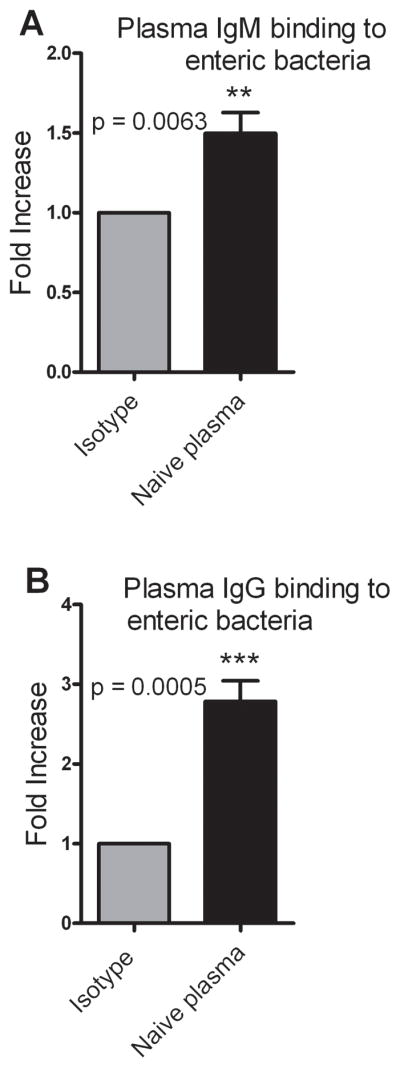
Plasma was used to opsonize enteric bacteria and the opsonized bacteria were stained with either PE anti–mouse IgM or FITC anti–mouse IgG. Isotype controls for IgM and IgG were PE Rat IgG2a, κ and FITC Rat IgG1, κ respectively. (A) Plasma IgM binding to enteric bacteria represented as a fold increase over isotype control. n = 8. (B) Plasma IgG binding of enteric bacteria represented as a fold increase over isotype control. n = 7. Data are the combined results of 3 independent experiments. p < 0.05 when compared to isotype control.
Since, plasma IgM was able to bind to enteric bacteria, we wanted to explore if IgM was responsible for differential PEK. PEK was measured for naïve, IgM depleted and IgG depleted plasma on an individual basis. Upon IgM depletion there was a slight (1.5%) decrease in the PEK (Fig 6A). Depletion of IgG however, improved the PEK significantly (Fig. 6B) indicating that when IgG was depleted, there was better bacterial killing. For all the plasma tested for PEK before and after IgG depletion, we observed that depletion of IgG lead to an increase in PEK for every sample. A paired t-test confirmed that depletion of IgG resulted in a statistically significant increase in the PEK value. On an average, depletion of plasma IgM had no effect on PEK but there was a significant increase in the PEK of IgG depleted plasma compared to naïve or IgM depleted plasma (Fig. 6C). Depletion of IgM and IgG was confirmed by ELISA (Fig. 6D and 6E respectively). We were further able to show that depletion of IgM had no effect on the total IgG levels and vice-versa (data not shown).
Figure 6. Critical role of plasma IgG in determining PEK capacity.

(A) PEK of IgM depleted plasma when compared to the naïve pre-CLP plasma. Each symbol is an individual mouse comparing naïve and IgM depleted (IgM−) PEK values. (B) Change in PEK of IgG depleted plasma when compared to the naïve pre-CLP plasma. Each symbol is an individual mouse comparing naïve and IgG depleted (IgG−) PEK values. In every sample, depletion of IgG increases the PEK numbers. (C) Depletion of IgM does not change the PEK value, while depletion of IgG results in a significant increase. n=7 both groups. p < 0.05 when compared to naive and IgG depleted plasma. (D) Plasma IgM levels before and after depletion. (E) Plasma IgG levels before and after depletion.
DISCUSSION
Our results indicate the presence of an inhibitory IgG antibody in the plasma with preferential binding affinity to those antigenic sites of the bacteria which would otherwise be recognized by other plasma factors leading to its opsonization and killing. In mice which die, the inhibitory IgG might prevent killing of bacteria leading to a low PEK. For the same mouse, post CLP there is less clearance of bacteria and hence they have a higher peritoneal CFU post sepsis. Our data suggest that mice which live post sepsis probably have lower levels of the inhibitory IgG in their plasma. In classical scenario mice which live post CLP might have decreased levels of the inhibitory IgG, in addition to other antibodies which kill the bacteria. Mice which die would have an excess of blocking IgG which prevents the killing the bacteria. Upon IgG depletion, the inhibitory IgG are removed rendering an increase in bacterial killing thereby increasing the PEK. Our experiments suggest that naturally occurring IgG and IgM play a critical role in mediating protection against bacterial infection even before the onset of sepsis. This is consistent with previous findings showing that mice which lack secreted IgM were more susceptible to CLP induced lethality (28). Naturally occurring IgM antibodies have also been reported to be responsible for increased ischemia/reperfusion injury (29) and bacterial clearance (30). Based on our results, it is probable that enhanced opsonization of bacteria by the plasma would lead to bacterial clearance. IVIG therapy has been shown to have an anti-inflammatory effect (31) and improves lung injury in CLP mediated sepsis in rats (32). However, in all these studies conducted so far the actual mechanism of improved survival has not been addressed. Based on our hypothesis, it might be possible that antibodies used in these trials would overcome the blocking IgG thereby increasing bacterial clearance and survival chances in sepsis.
A recent study illustrated the presence of blocking IgG that prevented killing of group B meningococci by human sera (33), further strengthening the hypothesis that blocking immunoglobulins plays a role in many other diseases including meningitis. This study observed that the blocking IgG was directed against a meningococcal antigen H.8. They were able to show that the Fc region of the IgG was required for its blocking activity, the blocking procedure required glycosylation and that this blocking resulted from inhibition of the classical complement pathway. These results extend back to the first of its kind of experiment in 1894 when the phenomenon of blocking of bacterial killing was first observed in animals given excess of immune serum.
To our knowledge, these data are the first to emphasize the presence of a strong repertoire of pre-existing antibodies which have bactericidal killing capacity even before the onset of bacterial peritonitis. Mice which survive are able to clear bacteria better as evidenced by the PEK values in their naïve pre-CLP plasma as well as reduced peritoneal bacterial counts after developing sepsis. We have identified naturally occurring blocking IgGs that prevent bacterial killing which may be a possible mechanism of increased mortality in mice which are susceptible to CLP. Our studies on depletion of IgM/IgG and its consequent PEK were in vitro experiments. Total depletion of antibodies in vivo would lead to an immune deficiency condition called agammaglobulinemia and would actually pre-dispose the host to severe infections (34). Thus, this study were restricted to in vitro experiments.
This paves the way for clinical studies in future whereby the PEK assay could serve as a potential biomarker for predicting the outcome of normal healthy individuals upon sepsis infection. Inhibiting these pre-existing antibodies which block bacterial opsonization and killing can be exploited in future IVIG therapies.
Acknowledgments
Supported by NIH grant: RO1 GM082962-01A2
We thank Elizabeth Duffy for her expert laboratory management which allowed the experiments to progress quickly.
Abbreviations used in this paper
- CLP
cecal ligation and puncture
- PEK
plasma enhanced killing
- Live-P
predicted to live past day 5 post CLP
- Die-P
predicted to die in the first 5-days post CLP
- CFU
colony forming units
- IgM/IgG
Immunoglobulin M/G
- ELISA
enzyme linked immunosorbent assay
Footnotes
Manuscript was submitted as one of the New Investigator Nominees at the 34th Annual Conference of the Shock Society held at Norfolk, VA 11th-14th June, 2011.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Angus DC, Pereira CA, Silva E. Epidemiology of Severe Sepsis around the World. Endocr Metab Immune Disord Drug Targets. 2006;6(2):207–12. doi: 10.2174/187153006777442332. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of Severe Sepsis in the United States: Analysis of Incidence, Outcome, and Associated Costs of Care. Crit Care Med. 2001;29(7):1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Angus DC, Wax RS. Epidemiology of Sepsis: An Update. Crit Care Med. 2001;29(7 Suppl):S109–16. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 4.Quartin AA, Schein RM, Kett DH, Peduzzi PN. Magnitude and Duration of the Effect of Sepsis on Survival. Department of Veterans Affairs Systemic Sepsis Cooperative Studies Group. Jama. 1997;277(13):1058–63. [PubMed] [Google Scholar]
- 5.Martin GS, Mannino DM, Eaton S, Moss M. The Epidemiology of Sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–54. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 6.Russell JA. Management of Sepsis. N Engl J Med. 2006;355(16):1699–713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 7.Perl TM. Long-Term Survival and Function after Suspected Gram-Negative Sepsis. Jama. 1995;274(4):338–45. [PubMed] [Google Scholar]
- 8.Wichterman KA, Baue AE, Chaudry IH. Sepsis and Septic Shock--a Review of Laboratory Models and a Proposal. J Surg Res. 1980;29(2):189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 9.Remick DG. Pathophysiology of Sepsis. Am J Pathol. 2007;170(5):1435–44. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebong SJ, Call DR, Bolgos G, Newcomb DE, Granger JI, O’Reilly M, et al. Immunopathologic Responses to Non-Lethal Sepsis. Shock. 1999;12(2):118–26. doi: 10.1097/00024382-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Xiao H, Siddiqui J, Remick DG. Mechanisms of Mortality in Early and Late Sepsis. Infect Immun. 2006;74(9):5227–35. doi: 10.1128/IAI.01220-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Remick DG, Bolgos G, Copeland S, Siddiqui J. Role of Interleukin-6 in Mortality from and Physiologic Response to Sepsis. Infect Immun. 2005;73(5):2751–7. doi: 10.1128/IAI.73.5.2751-2757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osuchowski MF, Craciun FL, Schuller E, Sima C, Gyurko R, Remick DG. Untreated Type 1 Diabetes Increases Sepsis-Induced Mortality without Inducing a Prelethal Cytokine Response. Shock. 2010;34(4):369–76. doi: 10.1097/SHK.0b013e3181dc40a8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osuchowski MF, Welch K, Yang H, Siddiqui J, Remick DG. Chronic Sepsis Mortality Characterized by an Individualized Inflammatory Response. J Immunol. 2007;179(1):623–30. doi: 10.4049/jimmunol.179.1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craciun FL, Schuller ER, Remick DG. Early Enhanced Local Neutrophil Recruitment in Peritonitis-Induced Sepsis Improves Bacterial Clearance and Survival. J Immunol. 2010;185(11):6930–8. doi: 10.4049/jimmunol.1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nemzek JA, Siddiqui J, Remick DG. Development and Optimization of Cytokine Elisas Using Commercial Antibody Pairs. J Immunol Methods. 2001;255(1–2):149–57. doi: 10.1016/s0022-1759(01)00419-7. [DOI] [PubMed] [Google Scholar]
- 17.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at Six: Interleukin-6 Measured 6 H after the Initiation of Sepsis Predicts Mortality over 3 Days. Shock. 2002;17(6):463–7. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 18.van Griensven M, Probst C, Muller K, Hoevel P, Pape HC. Leukocyte-EndothelialInteractions Via Icam-1 Are Detrimental in Polymicrobial Sepsis. Shock. 2006;25(3):254–9. doi: 10.1097/01.shk.0000196497.49683.13. [DOI] [PubMed] [Google Scholar]
- 19.Tschop J, Nogueiras R, Haas-Lockie S, Kasten KR, Castaneda TR, Huber N, et al. Cns Leptin Action Modulates Immune Response and Survival in Sepsis. J Neurosci. 2010;30(17):6036–47. doi: 10.1523/JNEUROSCI.4875-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Craciun FL, Schuller ER, Remick DG. Early Enhanced Local Neutrophil Recruitment in Peritonitis-Induced Sepsis Improves Bacterial Clearance and Survival. J Immunol. 185(11):6930–8. doi: 10.4049/jimmunol.1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Backer D, Biston P, Devriendt J, Madl C, Chochrad D, Aldecoa C, et al. Comparison of Dopamine and Norepinephrine in the Treatment of Shock. N Engl J Med. 2010;362(9):779–89. doi: 10.1056/NEJMoa0907118. [DOI] [PubMed] [Google Scholar]
- 22.Irwin PL, Nguyen LH, Paoli GC, Chen CY. Evidence for a Bimodal Distribution of Escherichia Coli Doubling Times Below a Threshold Initial Cell Concentration. BMC Microbiol. 2010;10:207. doi: 10.1186/1471-2180-10-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newcomb D, Bolgos G, Green L, Remick DG. Antibiotic Treatment Influences Outcome in Murine Sepsis: Mediators of Increased Morbidity. Shock. 1998;10(2):110–7. doi: 10.1097/00024382-199808000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Belikoff B, Hatfield S, Sitkovsky M, Remick DG. Adenosine Negative Feedback on A2a Adenosine Receptors Mediates Hyporesponsiveness in Chronically Septic Mice. Shock. 2011;35(4):382–7. doi: 10.1097/SHK.0b013e3182085f12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vianna RC, Gomes RN, Bozza FA, Amancio RT, Bozza PT, David CM, et al. Antibiotic Treatment in a Murine Model of Sepsis: Impact on Cytokines and Endotoxin Release. Shock. 2004;21(2):115–20. doi: 10.1097/01.shk.0000111828.07309.21. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz-Albiez R, Monteiro RC, Rodriguez M, Binder CJ, Shoenfeld Y. Natural Antibodies, Intravenous Immunoglobulin and Their Role in Autoimmunity, Cancer and Inflammation. Clin Exp Immunol. 2009;158 (Suppl 1):43–50. doi: 10.1111/j.1365-2249.2009.04026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boes M. Role of Natural and Immune Igm Antibodies in Immune Responses. Mol Immunol. 2000;37(18):1141–9. doi: 10.1016/s0161-5890(01)00025-6. [DOI] [PubMed] [Google Scholar]
- 28.Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A Critical Role of Natural Immunoglobulin M in Immediate Defense against Systemic Bacterial Infection. J Exp Med. 1998;188(12):2381–6. doi: 10.1084/jem.188.12.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang M, Michael LH, Grosjean SA, Kelly RA, Carroll MC, Entman ML. The Role of Natural Igm in Myocardial Ischemia-Reperfusion Injury. J Mol Cell Cardiol. 2006;41(1):62–7. doi: 10.1016/j.yjmcc.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H, et al. Control of Early Viral and Bacterial Distribution and Disease by Natural Antibodies. Science. 1999;286(5447):2156–9. doi: 10.1126/science.286.5447.2156. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Ward MF, Sama AE. Novel Hmgb1-Inhibiting Therapeutic Agents for Experimental Sepsis. Shock. 2009;32(4):348–57. doi: 10.1097/SHK.0b013e3181a551bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagiwara S, Iwasaka H, Hasegawa A, Asai N, Noguchi T. High-Dose Intravenous Immunoglobulin G Improves Systemic Inflammation in a Rat Model of Clp-Induced Sepsis. Intensive Care Med. 2008;34(10):1812–9. doi: 10.1007/s00134-008-1161-1. [DOI] [PubMed] [Google Scholar]
- 33.Ray TD, Lewis LA, Gulati S, Rice PA, Ram S. Novel Blocking Human Igg Directed against the Pentapeptide Repeat Motifs of Neisseria Meningitidis Lip/H.8 and Laz Lipoproteins. J Immunol. 2011;186(8):4881–94. doi: 10.4049/jimmunol.1003623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taccone FS, Stordeur P, De Backer D, Creteur J, Vincent JL. Gamma-Globulin Levels in Patients with Community-Acquired Septic Shock. Shock. 2009;32(4):379–85. doi: 10.1097/SHK.0b013e3181a2c0b2. [DOI] [PubMed] [Google Scholar]