

Summary
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative disorder. Mutations in presenilins 1 and 2 (PS1 and PS2) account for ∼40% of familial AD (FAD) cases. FAD mutations and genetic deletions of presenilins have been associated with calcium (Ca2+) signaling abnormalities. We demonstrate that wild-type presenilins, but not PS1-M146V and PS2-N141I FAD mutants, can form low-conductance divalent-cation-permeable ion channels in planar lipid bilayers. In experiments with PS1/2 double knockout (DKO) mouse embryonic fibroblasts (MEFs), we find that presenilins account for ∼80% of passive Ca2+ leak from the endoplasmic reticulum. Deficient Ca2+ signaling in DKO MEFs can be rescued by expression of wild-type PS1 or PS2 but not by expression of PS1-M146V or PS2-N141I mutants. The ER Ca2+ leak function of presenilins is independent of their γ-secretase activity. Our data suggest a Ca2+ signaling function for presenilins and provide support for the “Ca2+ hypothesis of AD.”
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that currently affects nearly 2% of the population in industrialized countries. Most cases of AD are idiopathic and are characterized by late onset (>60 years of age). A small fraction of AD cases (familial AD, FAD) are characterized by an earlier onset and genetic inheritance. Mutations in presenilin 1 (PS1) and presenilin 2 (PS2) account for about 40% of all known FAD cases (Tandon and Fraser, 2002). Presenilins are 50 kDa proteins that contain nine transmembrane domains (Laudon et al., 2005) and reside in the endoplasmic reticulum (ER) membrane (Annaert et al., 1999). The complex of presenilins with nicastrin, aph-1, and pen-2 subunits functions as γ-secretase, which cleaves the amyloid precursor protein (APP) and releases the amyloid β-peptide (Aβ), the principal constituent of the amyloid plaques in the brains of AD patients. Consistent with the role of presenilins as catalytic subunits of γ-secretase (De Strooper et al., 1998; Wolfe et al., 1999), FAD mutations in presenilins affect APP processing.
In addition to changes in APP processing, FAD mutations in presenilins result in deranged calcium (Ca2+) signaling (reviewed in Smith et al., 2005). What is a mechanistic explanation of these findings? Do presenilins play a direct role in Ca2+ signaling? Here we establish that presenilins function as passive ER Ca2+ leak channels and that the FAD mutations of presenilins affect their ability to conduct Ca2+ ions. Obtained results provide a new insight into normal physiological function of presenilins and strengthen the emerging connection between deranged neuronal Ca2+ signaling and AD (Khachaturian, 1989; LaFerla, 2002; Mattson et al., 2000; Smith et al., 2005).
Results
Recombinant Presenilins Form Cation-Permeable Channels in Planar Lipid Bilayers
The predicted structure of presenilins includes nine transmembrane domains (Figure 1A), consistent with potential ion-channel or transporter function. We proposed that presenilins may mediate Ca2+ transport in cells. To test this hypothesis, we expressed human PS1 protein and PS1-M146V and PS1-ΔE9 FAD mutants in Sf9 cells by baculoviral infection. We also investigated PS1-D257A (Figure 1A), a mutant that abolishes γ-secretase activity of presenilins (Wolfe et al., 1999). The efficient expression of PS1, PS1-M146V, PS1-D257A, and PS1-ΔE9 was confirmed by Western blotting (Figure 1B). The heterologously overexpressed presenilins are not incorporated into the γ-secretase complex and are mostly present as holoproteins (Figure 1B). The uncleaved form of endogenous presenilins is also present in untransfected mammalian cells, including neurons (Annaert et al., 1999).
Figure 1. Expression of Presenilins in Sf9 Cells.
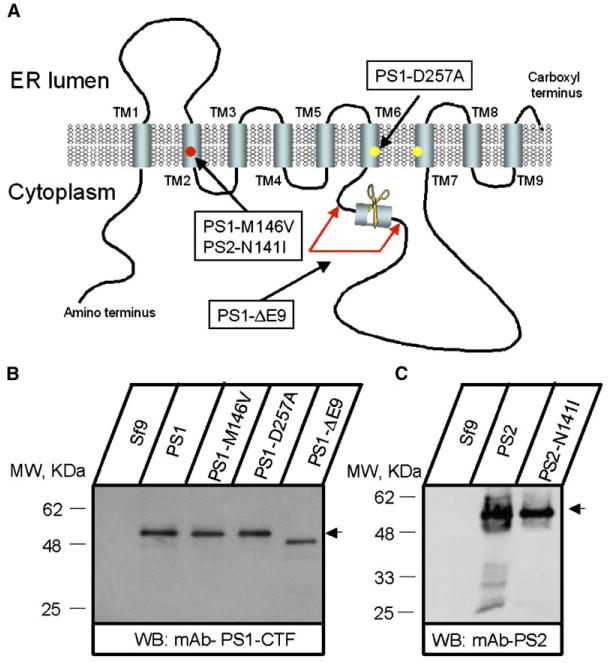
(A) Molecular model of presenilins (based on Laudon et al., 2005). The transmembrane domains (TM1–TM9), locations of aspartate residues critical for γ-secretase activity, and the site of endoproteolytic cleavage are indicated. Positions of PS1-M146V, PS1-ΔE9, PS1-D257A, and PS2-N141I mutations are shown.
(B and C) Expression of PS1, PS2, and mutants in Sf9 cells. Microsomes prepared from noninfected Sf9 cells (Sf9) and from Sf9 cells infected with PS1 and PS2 baculoviruses as indicated were analyzed by Western blotting with anti-PS1 (B) and anti-PS2 (C) monoclonal antibodies.
To investigate a potential ion-transport function of presenilins, we adapted a planar lipid bilayer (BLM) reconstitution technique (Tu et al., 2005a, 2005b). The ER microsomes from PS1-infected Sf9 cells were isolated and fused with BLM. Ba2+ ions (50 mM on the trans side) were used in these experiments as a current carrier. We did not detect ion currents across the BLM prior to fusion of ER microsomes (Figure 2A, first column, n = 59) or following fusion of microsomes from noninfected Sf9 cells (Figure 2A, second column, n = 11). In contrast, when microsomes from PS1-infected Sf9 cells were fused to the BLM, currents were observed in 7 out of 9 experiments (Figure 2A, third column). When compared to currents at 0 mV holding potential, the PS1-supported currents were increased at −10 mV holding potential and reduced at +10 mV holding potential (Figure 2A, third column), in agreement with the electrochemical gradient for Ba2+ ions. These data suggested that PS1 is able to facilitate transport of Ba2+ ions across the BLM, consistent with the Ca2+ channel function in vivo. In contrast to experiments with PS1 microsomes, much smaller currents were observed in experiments with PS1-M146V microsomes (Figure 2A, fourth column). The PS1-D257A mutant supported Ba2+ currents across the BLM similar to wild-type PS1 (Figure 2A, fifth column), and the PS1-ΔE9 mutant displayed enhanced channel function (Figure 2A, sixth column). By analogy with other known ion channels, the functional PS1 channels are likely to be multimers of several PS1 subunits. Can the PS1:PS1-M146V multimer function as an ion channel? To answer this question, we coinfected Sf9 cells with PS1 and PS1-M146V baculoviruses (in a 1:1 ratio), isolated ER microsomes, and fused them to planar lipid bilayers. We did not detect channel activity in these experiments (Figure 2A, seventh column), suggesting that the PS1-M146V mutant exerts a dominant-negative effect on ion-channel function of PS1.
Figure 2. PS1 and PS2 Form Small-Conductance Cation Channels in Planar Lipid Bilayers.
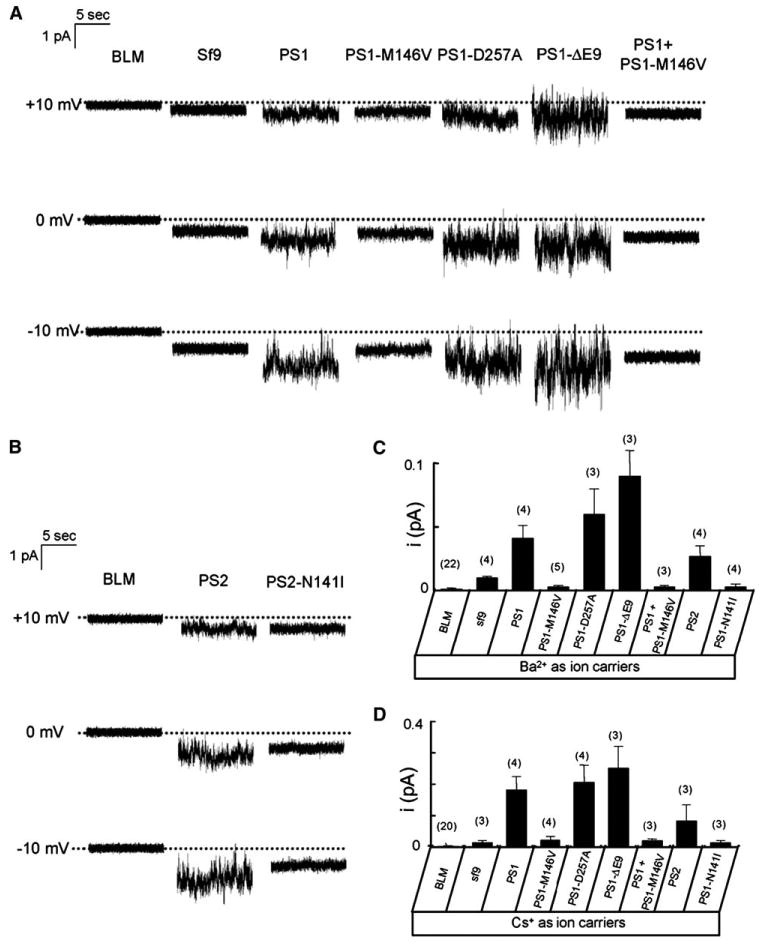
(A and B) Ba2+ currents recorded in planar lipid bilayer (BLM) experiments are shown for empty BLM (BLM), for microsomes from noninfected Sf9 cells (Sf9), and for microsomes from Sf9 cells infected with wild-type and mutant PS1 (A) and PS2 (B) baculoviruses as indicated. The dotted lines represent the zero level for the current traces. For each experiment, 10 s of continuous current recording is shown. Similar results were obtained in at least three experiments with each construct.
(C and D) The unitary current estimates are shown for experiments with 50 mM Ba2+ (C) or 100 mM Cs+ (D) in the trans compartment as mean ± SD (n = number of independent BLM experiments, shown above each bar).
Monovalent cations carry large currents via most known Ca2+ channels in the absence of divalent cations. Thus, in the next series of experiments, we repeated BLM reconstitution experiments using 100 mM Cs+ on the trans side of the membrane. Cs+ ions were used in these experiments to minimize currents via endogenous potassium channels present in ER microsomes. The results of bilayer experiments obtained using Cs+ as a current carrier were consistent with results obtained using Ba2+, but the currents were ∼4 times larger in the amplitude (see Figure S1A in the Supplemental Data available with this article online).
PS1 and PS2 are two highly homologous mammalian presenilin isoforms. Does PS2 share ion-channel function with PS1? To answer this question, we generated baculovirus encoding human wild-type PS2 and the PS2-N141I FAD mutant (Figure 1A) and confirmed efficient expression of these proteins in Sf9 cells by Western blotting (Figure 1C). We found that recombinant PS2, but not recombinant PS2-N141I, supported Ba2+ and Cs+ currents in the BLM (Figure 2B; Figure S1B).
Channels Formed by Presenilins Have Very Low Conductance
We have not been able to resolve individual single-channel opening events in our experiments, and, instead, PS-mediated currents had a “noisy” appearance (Figure 2). These types of currents are typical for channels with very low conductance, such as, for example, ICRAC Ca2+ currents (Zweifach and Lewis, 1993). To estimate the size of the unitary current via PS-supported channels, we applied a modification of the stationary noise analysis technique (see the Supplemental Data for details). By applying this method, we estimated that, at 0 mV, the size of the unitary Ba2+ currents supported by PS1 was equal to 0.04 ± 0.01 pA (n = 4); that is, ∼50 times lower than the size of the unitary current via InsP3R1 (2 pA) observed in identical recording conditions (Tu et al., 2005b). In the same conditions, the size of the unitary current was estimated to be equal to 0.06 ± 0.02 pA (n = 3) for PS1-D257A, 0.09 ± 0.02 pA (n = 3) for PS1-DE9, and 0.03 ± 0.01 pA (n = 4) for PS2 (Figure 2C). The size of the unitary Cs+ currents at 0 mV was estimated to be equal to 0.18 ± 0.04 pA (n = 4) for PS1, 0.21 ± 0.05 pA (n = 4) for PS1-D257A, 0.25 ± 0.07 pA (n = 3) for PS1-DE9, and 0.09 ± 0.05 pA (n = 3) for PS2 (Figure 2D). Thus, the size of the Cs+ unitary current was 3- to 5-fold larger than the size of the Ba2+ unitary current for the wild-type PS1, wild-type PS2, and the mutants that have been tested. In additional experiments, we performed recordings of PS1-mediated channel activity using 200 mM Cs+ as a current carrier. We found that the doubling of trans Cs+ concentration led to a significant increase in the size of the observed currents but that the unitary channel openings still could not be resolved (Figure S2). By noise analysis, we estimated the size of the unitary currents supported by PS1 in experiments with 200 mM Cs+ to be equal to 0.51 ± 0.09 pA (n = 3).
To determine the single-channel conductance of PS1-formed channels, we used noise analysis to estimate the size of microscopic unitary currents at −20 mV, −10 mV, 0 mV, and +10 mV transmembrane voltages. From these results, we determined that the average microscopic conductance with Ba2+ as a current carrier (γBa) is equal to 1.2 pS for PS1, 0.002 pS for PS1-M146V, and 2.6 pS for PS1-DE9 (Figure S3A). With Cs+ as a current carrier, the average microscopic conductance (γCs) is equal to 5.2 pS for PS1, 0.7 pS for PS1-M146V, and 8.3 pS for PS1-DE9 (Figure S3B).
Purified PS1 Forms Cation Channels in the Bilayers
Is the presenilin protein itself or some additional protein or proteins present in the Sf9 cell microsomal preparation responsible for the channel activity observed in the BLM (Figure 2)? To answer this question, we generated baculoviruses encoding amino-terminal His-tagged PS1 and PS1-M146V proteins. The microsomes from Sf9 cells infected with His-PS1 and His-PS1-M146V baculoviruses were solubilized in 1% CHAPS and used for purification of His-PS1 and His-PS1-M146V proteins on an Ni-NTA agarose column as previously described (Shah et al., 2005). The purified His-PS1 and His-PS1-M146V proteins were reconstituted into liposomes composed of phospholipids, ergosterol, and nystatin. The resulting proteoliposomes were highly enriched for His-PS1 and His-PS1-M146V proteins (Figures 3A and 3B). Obtained liposomes were used for planar lipid bilayer reconstitution experiments performed in the presence of NaCl on both sides of the membrane. Consistent with the previous report (Woodbury and Miller, 1990), fusion of ergosterol/nystatin-containing liposomes with the bilayer resulted in the appearance of large transient currents (Figure 3C). Perfusion of the cis chamber terminated fusion of liposomes with the bilayer and terminated nystatin-mediated channel activity due to reduction in local ergosterol concentration (Figure 3C). When the remaining currents were analyzed, we observed Na+ currents across the bilayer in experiments with His-PS1 liposomes in 5 out of 7 experiments (Figure 3D, third column). In contrast, no significant current activity was observed in experiments with protein-free liposomes (Figure 3D, second column, n = 5) and with His-PS1-M146V liposomes (Figure 3D, fourth column, n = 4). The noise analysis performed as described above yielded an average microscopic conductance (γNa) equal to 5 pS for His-PS1 liposomes and 0.003 pS for His-PS1-M146V liposomes (Figure S4).
Figure 3. Purified His-PS1 Forms Na+ Channels in Planar Lipid Bilayers.
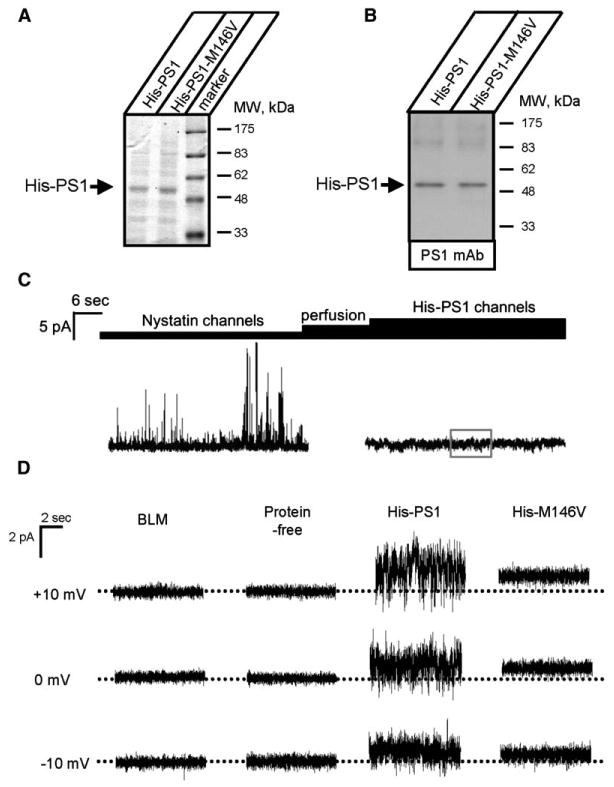
(A and B) Proteoliposomes containing purified His-PS1 and His-PS1-M146V proteins were analyzed by SDS gel electrophoresis followed by Coomassie staining (A) or Western blotting (B).
(C) Nystatin/ergosterol-mediated fusion of His-PS1 proteoliposomes with BLM. Large nystatin channels are observed upon fusion of proteoliposomes with the bilayer. The activity of nystatin channels is terminated following perfusion of the cis chamber.
(D) Na+ currents are shown for empty BLM (BLM) and for experiments with protein-free liposomes and proteoliposomes containing purified His-PS1 and His-PS1-M146V. The dotted lines represent the zero level for the current traces. The direction of the current is consistent with Na+ as a current carrier (600 mM NaCl cis and 150 mM NaCl trans). For each experiment, 10 s of continuous current recording is shown. Similar results were obtained in at least three experiments with each batch of liposomes.
Ca2+ Signaling Defects in Presenilin Double-Knockout (DKO) Mouse Embryonic Fibroblasts
To determine the physiological role of Ca2+ channels formed by presenilins, we initiated a series of Ca2+ imaging experiments with fibroblasts derived from presenilin double-knockout mice (Herreman et al., 2000) (DKO cells). Control experiments were performed with fibroblasts derived from wild-type mice (MEF cells). In our experiments, we loaded MEF and DKO cells with Fura-2 Ca2+ imaging dye and triggered InsP3R-mediated Ca2+ release from the ER by addition of 300 nM bradykinin (BK), an agonist of PLC-coupled BK receptor. The cytosolic Ca2+ concentration in these experiments is calculated from the ratio of Fura-2 signals at 340 nm and 380 nm excitation wavelengths shown by the pseudocolor images (Figure 4A). Prior to stimulation with BK, cytosolic Ca2+ was equal to 187 ± 41 nM (n = 28) in MEF cells and 135 ± 32 nM (n = 23) in DKO cells, significantly (p < 0.05) lower. Application of 300 nM BK induced a much larger Ca2+ response in DKO cells than in MEF cells (Figures 4A and 4B). On average, the difference between the peak and the basal Ca2+ levels (Δ[Ca2+]) was equal to 225 ± 103 nM (n = 28) for MEF cells and 787 nM ± 76 (n = 23) for DKO cells (Figure 4C). Thus, the amplitude of BK-induced Ca2+ responses was 3.5-fold larger in DKO fibroblasts than in wild-type MEFs.
Figure 4. Ca2+ Signaling in Wild-Type and Presenilin DKO Fibroblasts.
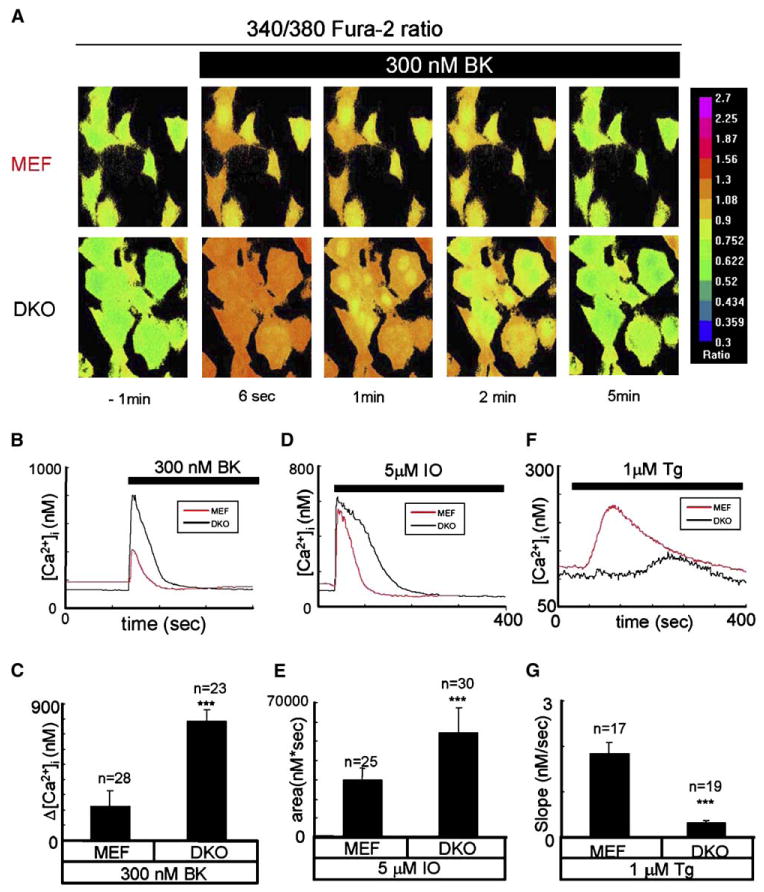
(A) Representative Fura-2 images of bradykinin (BK) induced Ca2+ responses in MEF (top) and DKO (bottom) cells. 340/380 Fura-2 ratios are shown at time points indicated. The pseudocolor calibration scale for 340/380 ratios is shown on the right. Three hundred nanomolar BK was added at t = 0.
(B) The time course of BK-induced [Ca2+] changes in MEF (red) and DKO (black) representative cells is shown.
(C) The average amplitude of BK-induced Ca2+ release from MEF and DKO cells is shown as mean ± SD (n = number of cells).
(D) The time course of ionomycin (IO) induced [Ca2+] changes in MEF (red) and DKO (black) representative cells is shown.
(E) The average size of the ionomycin-releasable Ca2+ pool is shown for MEF and DKO cells as mean ± SD (n = number of cells).
(F) The time course of thapsigargin (Tg) induced [Ca2+] changes in MEF (red) and DKO (black) representative cells is shown.
(G) The average slope of thapsigargin-induced Ca2+ elevation is shown for MEF and DKO cells as mean ± SD (n = number of cells). The data from at least three independent experiments with BK, IO, and Tg were combined for analysis. The average amplitude of BK-induced Ca2+ response, the average size of the IO-releasable Ca2+ pool, and the average rate of Tg-induced Ca2+ leak are significantly (***p < 0.05) different in DKO cells compared to MEF cells.
What is an explanation for potentiated BK-induced Ca2+ release in DKO cells? No significant changes in InsP3R1 expression levels were detected in DKO fibroblasts when compared to MEFs by Western blotting (Figure S5). DKO and MEF cells did not respond to caffeine (data not shown), ruling out an involvement of the ryanodine receptors. Thus, we reasoned that the DKO and MEF cells may differ in the filling state of ER Ca2+ stores. To compare the levels of Ca2+ stored in the ER, in the next series of experiments, we evaluated Ca2+ signals induced in MEF and DKO cells by application of 5 mM ionomycin. Ionomycin is an ionophore that induces formation of Ca2+-permeable pores in cellular membranes, leading to complete emptying of ER Ca2+ stores independently from the InsP3R activation. We found that application of ionomycin resulted in more massive and longer-lasting Ca2+ signals in DKO cells than in MEF cells (Figure 4D). To estimate total ER Ca2+ content from these data, we integrated the area under the ionomycin-induced cytosolic Ca2+ curve for both types of cells. On average, the area under the curve was equal to 30 ± 6 mM3s (n = 25) for MEF cells and 55 ± 13 mM3s (n = 30) for DKO cells (Figure 4E). Thus, the size of the ionomycin-sensitive Ca2+ pool is 1.8-fold larger in DKO cells than in MEF cells, consistent with overfilled ER Ca2+ stores in DKO cells.
These results led us to propose that presenilins act as ER Ca2+ leak channels that facilitate passive Ca2+ leak across the ER membrane. To test this hypothesis more directly, we evaluated Ca2+ signals induced in MEF and DKO cells by thapsigargin. Application of thapsigargin blocks SERCA Ca2+ pump activity, leading to passive leak of Ca2+ from the ER to the cytosol via the endogenous ER Ca2+ leak pathway. We found that application of 1 μM thapsigargin caused rapid and large cytosolic Ca2+ elevation in MEF cells, but much smaller and delayed cytosolic Ca2+ elevation in DKO cells (Figure 4F). On average, we found that cytosolic Ca2+ elevation could be detected at 16 ± 4 s (n = 17) after application of thapsigargin to MEF cells and at 157 ± 14 s (n = 19) after application of thapsigargin to DKO cells. To estimate the rate of endogenous Ca2+ leak in these experiments, we measured a slope of cytosolic Ca2+ increase in thapsigargin-exposed cells (release rate). We found that, on average, the release rate was equal to 1.85 ± 0.24 nM/s (n = 17) in MEF cells and 0.33 ± 0.05 nM/s (n = 19) in DKO cells (Figure 4G). Thus, genetic deletion of presenilins resulted in a 5.6-fold reduction in the rate of Ca2+ leak across the ER membrane, suggesting that in MEF fibroblasts, presenilins account for ∼80% of the total endogenous ER Ca2+ leak activity.
Rescue of Ca2+ Signaling Defects in DKO Fibroblasts
The defects in Ca2+ signaling observed in DKO fibroblasts (Figure 4) provided an opportunity for a series of rescue experiments. In these experiments, DKO fibroblasts were transfected with EGFP plasmid alone (EGFP control) or EGFP plasmid together with the presenilin expression constructs and analyzed by Fura-2 Ca2+ imaging. In the first series of experiments, DKO fibroblasts were transfected with EGFP, EGFP + PS1, and EGFP+PS1-M146V constructs (Figure 5A). Under resting conditions, the average basal Ca2+ levels were equal to 183 ± 24 nM (n = 7) for EGFP-transfected cells, 296 ± 28 nM (n = 12) for EGFP + PS1-transfected cells, and 194 ± 42 nM (n = 17) for EGFP+PS1-M146V-transfected cells. The statistical analysis revealed that resting Ca2+ levels were significantly (p < 0.05) higher in EGFP + PS1-transfected cells than in cells transfected with EGFP or EGFP + PS1-M146V. Application of 300 nM BK induced large and transient Ca2+ signals in EGFP-transfected and EGFP + PS1-M146V-transfected cells but smaller and longer-lasting Ca2+ signals in EGFP + PS1-transfected cells (Figures 5A and 5B). On average, the difference between the peak and basal Ca2+ levels (Δ[Ca2+]) in BK-stimulated cells was equal to 702 ± 82 nM (n = 7) for EGFP-transfected cells, 258 ± 92 nM (n = 12) for EGFP + PS1-transfected cells, and 633 ± 125 nM (n = 17) for EGFP + PS1-M146V-transfected cells (Figure 5C).
Figure 5. Rescue of Ca2+ Signaling Defects in DKO Fibroblasts.
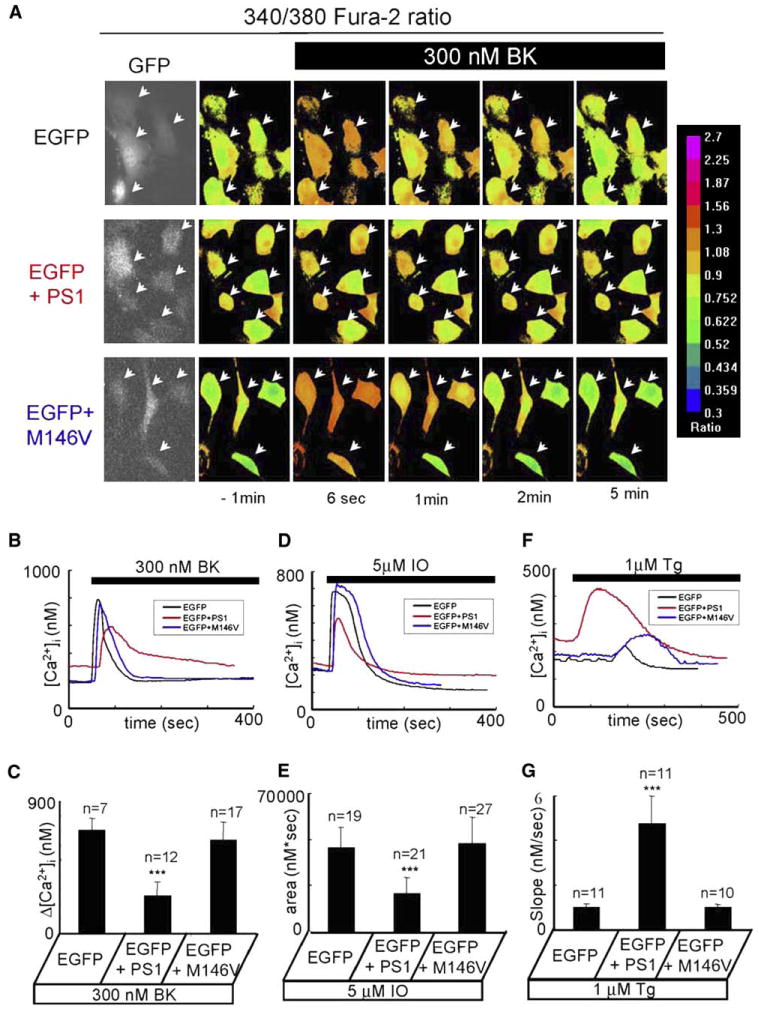
(A) Representative Fura-2 images of BK-induced Ca2+ responses are shown for DKO fibroblasts transfected with EGFP (first row), EGFP + PS1 (second row), and EGFP + PS1-M146V (third row) expression plasmids. The transfected cells were identified by GFP imaging (first column). The data are presented and analyzed as explained in the Figure 4 legend.
(B) The time course of BK-induced [Ca2+] changes in representative DKO cells transfected with EGFP (black), EGFP + PS1 (red), and EGFP + PS1-M146V (blue) constructs is shown.
(C) The average amplitude of BK-induced Ca2+ release from DKO cells transfected with EGFP, EGFP + PS1, and EGFP + PS1-M146V constructs is shown as mean ± SD (n = number of cells).
(D) The time course of IO-induced [Ca2+] changes in representative DKO cells transfected with EGFP (black), EGFP + PS1 (red), and EGFP + PS1-M146V (blue) constructs is shown.
(E) The average size of the IO-sensitive Ca2+ pool is shown for DKO cells transfected with EGFP, EGFP + PS1, and EGFP + PS1-M146V constructs as mean ± SD (n = number of cells).
(F) The time course of Tg-induced [Ca2+] changes in representative DKO cells transfected with EGFP (black), EGFP + PS1 (red), and EGFP + PS1-M146V (blue) constructs is shown.
(G) The average slope of Tg-induced Ca2+ elevation is shown for DKO cells transfected with EGFP, EGFP + PS1, and EGFP + PS1-M146V constructs as mean ± SD (n = number of cells).
The data from at least three independent experiments for each experimental condition were combined for analysis. The average amplitude of BK-induced Ca2+ response, the average size of the IO-releasable Ca2+ pool, and the average rate of Tg-induced Ca2+ leak are significantly (***p < 0.05) different in DKO cells transfected with EGFP + PS1 compared to DKO cells transfected with EGFP or EGFP + PS1-M146V.
To interpret these results, we reasoned that expression of wild-type PS1 rescues the endogenous Ca2+ leak pathway in DKO cells and reduces intraluminal Ca2+ levels and the amount of released Ca2+. The increase in resting Ca2+ levels observed in PS1-transfected cells (Figures 5A and 5B) can also be explained by increased “leakiness” of ER Ca2+ stores under resting conditions. The longer duration of the BK-induced Ca2+ transient in PS1-transfected cells (Figures 5A and 5B) most likely reflects the fact that the SERCA Ca2+ pump requires more time to pump released Ca2+ back into the “leaky” stores. Our BLM reconstitution experiments demonstrated that Ca2+ channel function is impaired in the PS1-M146V FAD mutant (Figure 2A). Consistent with these observations, expression of the PS1-M146V mutant in DKO cells had no significant effect on resting Ca2+ levels or the amplitude and duration of BK-induced Ca2+ response (Figures 5A–5C), presumably because the PS1-M146V mutant was not able to facilitate Ca2+ leak from the ER.
To test these ideas more directly, we evaluated the size of the ionomycin-sensitive Ca2+ pool in transfected DKO cells. We found that addition of 5 μM ionomycin induced a large and long-lasting elevation of cytosolic Ca2+ levels in EGFP-transfected and EGFP + PS1-M146V-transfected cells, but a smaller in amplitude and shorter in duration Ca2+ elevation in EGFP + PS1-transfected cells (Figure 5D). On average, the area under the Ca2+ curve was equal to 46 ± 9 μM×s (n = 19) for EGFP-transfected cells, 26 ± 7 μM×s (n = 21) for cells transfected with EGFP + PS1, and 48 ± 11 μM×s (n = 27) for cells transfected with EGFP + PS1-M146V (Figure 5E). Thus, expression of wild-type PS1 resulted in a 1.8-fold reduction in ER Ca2+ content in DKO cells, whereas expression of the PS1-M146V mutant had no significant effect.
As an additional test of our hypothesis, we evaluated thapsigargin-induced Ca2+ signals in transfected DKO cells. We found that application of 1 mM thapsigargin resulted in rapid and massive cytosolic Ca2+ elevation in EGFP + PS1-transfected cells but delayed and much smaller Ca2+ elevation in EGFP-transfected and EGFP + PS1-M146V-transfected cells (Figure 5F). On average, we found that cytosolic Ca2+ elevation could be detected at 17 ± 3 s (n = 11) after application of thapsigargin to DKO cells transfected with EGFP + PS1, at 133 ± 17 s (n = 11) for DKO cells transfected with EGFP, and at 148 ± 18 s (n = 10) for DKO cells transfected with EGFP + PS1-M146V. Further analysis revealed that the average rate of thapsigargin-induced Ca2+ leak was equal to 1.0 ± 0.2 nM/s (n = 11) for EGFP-transfected DKO cells, 5 ± 1 nM/s (n = 11) for DKO cells transfected with EGFP + PS1, and 0.99 ± 0.16 nM/s (n = 10) for DKO cells transfected with EGFP + PS1-M146V (Figure 5G). Thus, the rate of passive Ca2+ leak is 5-fold higher in EGFP + PS1-transfected cells than in cells transfected with EGFP alone or with EGFP + PS1-M146V. Our results support the hypothesis that the wild-type PS1, but not the PS1-M146V mutant, is able to rescue ER Ca2+ leak function impaired in DKO cells.
In the next series of experiments, we utilized the same paradigm to evaluate the ability of other presenilin expression constructs to rescue Ca2+ signaling defects observed in DKO cells. We found that expression of PS1-DE9 and PS1-D257A mutants in DKO cells reduced the amplitude of BK-induced Ca2+ signals (Figure 6A) and the content of the ionomycin-sensitive Ca2+ pool (Figure 6B), similar to expression of wild-type PS1. Expression of wild-type PS2 reduced the amplitude of BK-induced Ca2+ signals (Figure 6A), but the PS2-N141I mutant was ineffective (Figure 6A). Cotransfection of PS1 and PS1-M146V plasmids had no effect on BK-induced Ca2+ signals (Figure 6A). Thus, we concluded that the behavior of all presenilin constructs and construct combinations in DKO fibroblast rescue experiments (Figure 6A) is consistent with the behavior observed for the same constructs in BLM reconstitution experiments (Figure 2). To rule out potential artifacts resulting from transient overexpression of PS1 and PS2, we performed a series of Ca2+ imaging experiments with stably transfected DKO fibroblasts. The expression of PS1 and PS2 rescue constructs in these stable lines was confirmed by Western blotting (Figure S6). We found that the content of ionomycin-sensitive Ca2+ stores was reduced to wild-type levels in DKO cells stably transfected with PS1 (HPS1 line), PS1-D257A, PS1-DE9, or PS2 (HPS2 line), but not with PS2-N141L (Figure 6B).
Figure 6. Summary of DKO Fibroblast Rescue Experiments.
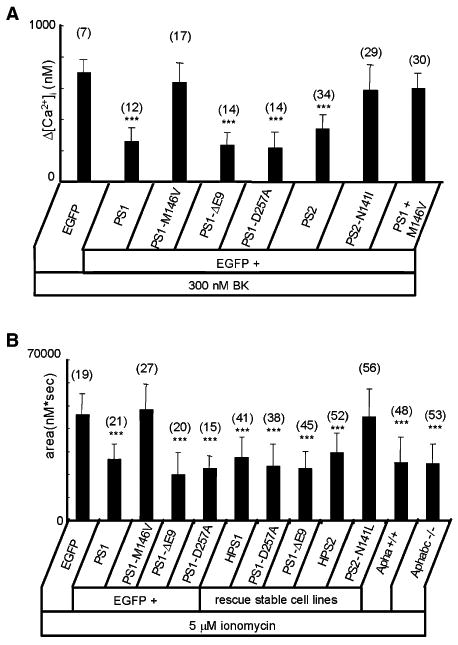
(A)The average amplitude of BK-induced Ca2+ release from DKO cells transfected with EGFP together with PS1 and PS2 rescue constructs is shown as mean ± SD (n = number of cells analyzed). When compared to DKO cells transfected with EGFP alone, the amplitude of BK-in-duced Ca2+ response is significantly (***p < 0.05) smaller in DKO cells transfected with EGFP + PS1, EGFP + PS1-DE9, EGFP + PS1-D257A, and EGFP + PS2.
(B)The average size of the IO-sensitive Ca2+ pool is shown for DKO cells transiently transfected with EGFP and PS1 rescue constructs and for stably transfected DKO cells as mean ± SD (n = number of cells analyzed). When compared to DKO cells transfected with EGFP alone, the size of the IO-releasable Ca2+ pool is significantly (***p < 0.05) smaller in DKO cells transfected with EGFP + PS1, EGFP + PS1-DE9, and EGFP + PS1-D257A. The size of the IO-releasable Ca2+ pool is also significantly (***p < 0.05) smaller in HPS1, PS1-D257A, PS1-DE9, and HPS2 cell lines.
The ability of the PS1-D257A mutant to rescue BK- and ionomycin-induced Ca2+ responses in DKO cells (Figures 6A and 6B) suggested that γ-secretase function of PS1 is dispensable for Ca2+ signaling function studied in our experiments. To further explore this issue, we performed Ca2+ imaging experiments with MEFs generated from Aph-1abc−/− triple knockout mice and control Aph-1a+/+ (Aph-1bc−/−) MEFs. In separate experiments, we demonstrated that γ-secretase activity is completely absent in Aph-1abc−/− cells and that PS1 protein is present as a holoprotein in these cells at levels similar to those in wild-type MEFs (L.S. and B.D.S., unpublished data). In experiments with ionomycin, we found that the size of the ER Ca2+ pool was similar to that in wild-type MEFs for both Aph-1a+/+ and Aph-1abc−/− cells (Figure 6B).
Presenilins and ER Ca2+ Homeostasis
The results obtained above are consistent with a role of presenilins as passive ER Ca2+ leak channels. As an additional test of this hypothesis, we performed a series of Ca2+ flux measurements with isolated ER microsomes. In these experiments, Ca2+ accumulated in microsomes as a result of Ca2+ pump activity and then was released via a passive leak mechanism following addition of thapsigargin. We discovered that the rate of thapsigargin-induced Ca2+ leak was equal to 0.41 ± 0.06 nM Ca2+/s (n = 3) for microsomes from noninfected Sf9 cells, 1.1 ± 0.24 nM Ca2+/s (n = 5) for PS1-containing microsomes, and 0.35 ± 0.07 nM Ca2+/s (n = 3) for PS1-M146V-containing microsomes (Figure S7). In complementary experiments, we found that the rate of thapsigargin-induced Ca2+ leak was equal to 1.82 ± 0.32 nM Ca2+/s (n = 5) for MEF microsomes, 0.45 ± 0.09 nM Ca2+/s (n = 4) for DKO microsomes, and 1.51 ± 0.27 nM Ca2+/s (n = 4) for HPS1 microsomes (Figure S8).
To further test our hypothesis, we directly measured Ca2+ concentration in the ER ([Ca2+]ER) of wild-type and DKO MEFs with the low-affinity Ca2+ imaging dye Mag-Fura-2 (Hofer, 1999). Consistent with our expectations, we found that [Ca2+]ER is elevated ∼2-fold in DKO cells (Figure 7A). On average, [Ca2+]ER was equal to 87 ± 16 mM (n = 20) in MEF cells and 190 ± 48 mM (n = 28) in DKO cells (Figure 7B). The transient transfection of DKO cells with EGFP or EGFP + PS1-M146V had no significant effect on [Ca2+]ER (Figure 7B). In contrast, transfection of DKO cells with PS1, PS1-D257A, or PS1-AE9 constructs reduced [Ca2+]ER to wild-type levels (Figure 7B). Consistent with transient transfection results, [Ca2+]ER was reduced to wild-type levels in HPS1, PS1-D257A, PS1-AE9, or HPS2 cell lines but remained elevated in PS2-N141L cells (Figure 7B). [Ca2+]ER in both Aph-1a+/+ and Aph-1abc−/− MEFs was the same as in wild-type MEFs (Figure 7B), further demonstrating that γ-secretase activity is not required for proper control of ER Ca2+ homeostasis.
Figure 7. Presenilins and ER Ca2+ Homeostasis.
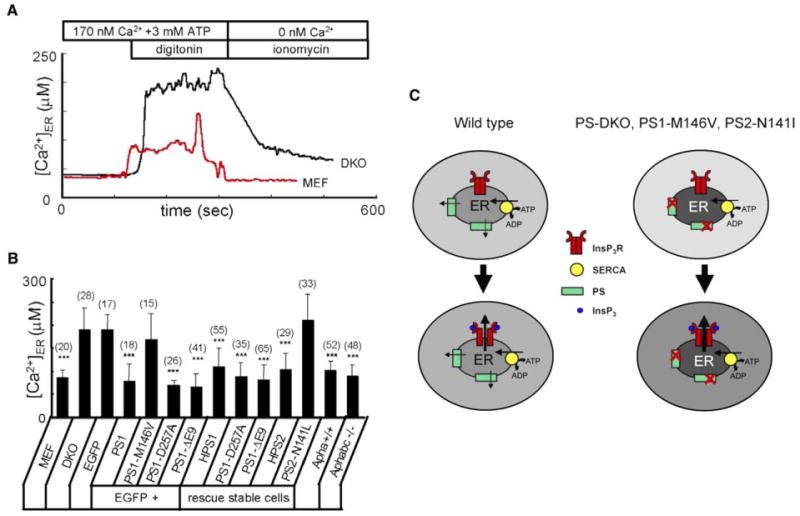
(A) Representative ER Ca2+ traces recorded by ER-loaded Mag-Fura-2 in wild-type (red line) and DKO (black line) MEFs. Cells were loaded with Mag-Fura-2 and permeabilized by 10 μM digitonin in buffer containing 170 nM Ca2+ and 3 mM ATP. At the end of the experiment, the ER membrane was permeabilized with 5 μM ionomycin in the Ca2+-free buffer. 340/380 Mag-Fura-2 ratios were converted to [Ca2+]ER.
(B) The average ER Ca2+ concentration determined as shown in (A) is presented for wild-type MEF cells, DKO cells transiently transfected with EGFP and PS1 rescue constructs, and stably transfected DKO rescue cells as mean ± SD (n = number of cells). When compared to DKO cells transfected with EGFP alone, the ER Ca2+ concentration is significantly (***p < 0.05) lower in DKO cells transfected with EGFP + PS1, EGFP + PS1-D257A, and EGFP + PS1-DE9. The ER Ca2+ concentration is also significantly (***p < 0.05) smaller in HPS1, PS1-D257A, PS1-DE9, and HPS2 stable lines.
(C) Model of Ca2+ homeostasis in wild-type and PS mutant cells. In wild-type cells (left), steady-state ER intraluminal Ca2+ levels are determined by a balance between SERCA pump activity and PS-facilitated passive Ca2+ leak from the ER. In PS-DKO cells or in cells expressing PS1-M146V or PS2- N141I FAD mutants, ER Ca2+ leak function of presenilins is impaired, resulting in higher steady-state intraluminal Ca2+ levels and lower cytosolic Ca2+ levels. Following generation of InsP3 and opening of InsP3R, the amplitude of Ca2+ response is higher in mutant cells (right) than in wild-type cells (left) due to the larger driving force for Ca2+ ions exiting from the ER to the cytoplasm.
Discussion
Presenilins as ER Ca2+ Leak Channels
What determines a steady-state level of intraluminal Ca2+ in the ER? The calculations predict that, under physiological conditions, the SERCA pump reaches thermodynamical equilibrium when intraluminal [Ca2+]ER is equal to 2.4 mM (see Supplemental Data for calculations). However, direct imaging of intraluminal [Ca2+]ER resulted in estimates in the 100-500 μM range (Hofer, 1999) (Figure 7A). The most likely explanation for this difference is the leakiness of the ER membrane for Ca2+ ions. According to this idea (Figure 7C, left panel), the steady-state ER intraluminal Ca2+ level is determined by an equilibrium between SERCA-mediated movement of Ca2+ from the cytosol into the ER lumen and the passive leak of Ca2+ from ER into the cytosol. Experimental estimates for the rate of Ca2+ leak vary from 19 μM ER Ca2+/min in acinar cells to 90 μM ER Ca2+/min in neurons (Camello et al., 2002).
A number of candidates have been previously considered to play a role as ER Ca2+ leak channels, such as the ribosome-translocon complex (Lomax et al., 2002; Van Coppenolle et al., 2004), the antiapoptotic protein bcl-2 (Pinton et al., 2000), and InsP3R (Oakes et al., 2005). However, the exact identity of ER Ca2+ leak channels still largely remains an “enigma of Ca2+ signaling” (Camello et al., 2002). Prior to endoproteolytic cleavage, presenilins are localized to the ER membrane (Annaert et al., 1999). Here we propose that the holoprotein form of presenilins functions as an ER Ca2+ leak channel in cells (Figure 7C, left panel). This hypothesis is consistent with the ability of PS1 and PS2 proteins to form low-conductance divalent-cation-permeable channels in the BLM (Figure 2) and with Ca2+ signaling abnormalities in DKO cells (Figure 4). Moreover, [Ca2+]ER in DKO cells is 2-fold higher than in wild-type MEFs (Figure 7A), directly supporting our hypothesis. Importantly, cytosolic and ER Ca2+ signaling defects in DKO fibroblasts can be rescued by expression of PS1 and PS2 constructs (Figure 5, Figure 6, and Figure 7). From these results, we conclude that, in MEF fibroblasts, presenilins account for ∼80% of ER Ca2+ leak activity. Notably, our conclusions differ from the recent study of Ca2+ signaling in DKO MEFs performed by a different methodology (Kasri et al., 2006).
The main known function of presenilins is to act as a catalytic subunit of the γ-secretase complex (De Strooper et al., 1998; Wolfe et al., 1999). Here we propose that presenilins also serve as ER Ca2+ leak channels. Moreover, our results indicate that γ-secretase function of presenilins is dispensable for ER Ca2+ leak function. In our experiments, the catalytic mutant PS1-D257A formed channels in the bilayers (Figure 2) and rescued all Ca2+ signaling defects in DKO cells (Figure 6 and Figure 7). Moreover, the filling state of ER Ca2+ stores was normal in Aph-1abc−/− MEF cells (Figure 6B and Figure 7B), which completely lack γ-secretase activity (Serneels et al., 2005). Interestingly, only the “mature” (cleaved) form of presenilins can function as γ-secretase (Tandon and Fraser, 2002), whereas in our experiments, ER Ca2+ leak function was supported by the holoprotein form of presenilins.
Deranged Ca2+ Signaling and AD
Ca2+ signaling defects have been observed in studies with fibroblasts from FAD patients (Ito et al., 1994), in FAD cellular and animal models (LaFerla, 2002; Smith et al., 2005), and in cells from PS1 and PS2 knockout mice (Hermsetal., 2003; Ris et al., 2003; Takeda et al., 2005; Yoo et al., 2000). A number of hypotheses have been previously considered to explain these phenomena. It has been proposed that Aβ-peptides associate into small aggregates that form Ca2+-permeable channels in the plasma membrane of neurons, causing Ca2+ influx (Arispe et al., 1993). Another hypothesis states that the APP intracellular domain (AICD) released by γ-secretase cleavage affects Ca2+ signaling by regulating expression of neuronal Ca2+ signaling proteins (Leissring et al., 2002). A correlation between the PS1-M146V mutation and enhanced function of neuronal ryanodine receptors has been recently described (Stutzmann et al., 2006). Here we propose that PS1-M146V and PS2-N141I FAD mutations in presenilins affect neuronal Ca2+ signaling by disrupting the ER Ca2+ leak pathway (Figure 7C, right panel). Our model is supported by the inability of PS1-M146V and PS2-N141I mutants to form divalent-cation-permeable channels in the BLM (Figure 2) and the failure of these mutants to rescue cytosolic and ER Ca2+ signaling defects in PS1/2 DKO MEFs (Figure 5, Figure 6, and Figure 7). When coexpressed with the PS1-M146V mutant, wild-type PS1 did not form cation-permeable channels in BLM (Figure 2A) and failed to restore the amplitude of BK-induced Ca2+ release in DKO fibroblasts (Figure 6A). Thus, we propose that PS1-M146V and PS2-N141I mutants exert a dominant-negative effect on PS-supported ER Ca2+ leak function, resulting in intraluminal ER Ca2+ overload at the steady state (Figure 7C, right panel). We further propose that ER Ca2+ overload in cells expressing PS1-M146V and PS2-N141I mutants leads to supranormal Ca2+ release following activation of InsP3R (Figure 7C, right panel). The proposed model (Figure 7C) is consistent with increased InsP3-induced Ca2+ release in Xenopus oocytes expressing PS1-M146V and PS2-N141I FAD mutants (Leissring et al., 1999a, 1999b, 2001), in synaptosomes and cortical neurons from PS1-M146V mutant knockin mice (Begley et al., 1999; Stutzmann et al., 2004), and in hippocampal neurons from PS2-N141I transgenic mice (Schneider et al., 2001). It is highly likely that the abnormal intraluminal ER Ca2+ levels predicted by our model (Figure 7C) may also be responsible for Ca2+ signaling defects observed in cells from PS1 and PS2 knockout mice (Herms et al., 2003; Ris et al., 2003; Takeda et al., 2005; Yoo et al., 2000).
In our bilayer experiments, we found that the PS1-ΔE9 FAD mutant had increased ER Ca2+ leak channel activity when compared to wild-type PS1 (Figure 2). These results suggest that PS1-ΔE9 is a gain-of-function ER Ca2+ leak mutant, whereas PS1-M146V and PS2-N141I are dominant-negative ER Ca2+ leak mutants. The gain-of-function Ca2+ leak phenotype of the PS1-ΔE9 mutant is consistent with an earlier observation of elevated basal Ca2+ levels in SH-SY5Y cells transfected with PS1-ΔE9 expression construct (Cedazo-Minguez et al., 2002).
A growing list of PS1 and PS2 FAD mutants linked to abnormal Ca2+ signaling has been compiled in a recent review article (Smith et al., 2005). In addition to PS1-M146V/L, PS2-N141I/L, and PS1-ΔE9, these mutants also include PS1-H163R, PS1-A246Q/E, PS1-L286V, and PS2-M239V/I (Smith et al., 2005). It will be interesting to evaluate ER Ca2+ leak function of these FAD mutants. The “loss of presenilin function” hypothesis of FAD was put forward based on the analysis of age-dependent neurodegeneration in PS1/2 cDKO mice (Saura et al., 2004). Our observation of loss of ER Ca2+ leak channel function in PS1-M146V and PS2-N141I mutants is in general agreement with the “loss of presenilin function” hypothesis of FAD (Saura et al., 2004). Additional studies will also be required to investigate the connection between defective Ca2+ signaling induced by PS FAD mutations, abnormal γ-secretase activity, and neurodegeneration in AD.
Experimental Procedures
Expression Constructs and Recombinant Baculoviruses
The following human PS1 and PS2 expression constructs were generated in pFastBac1 baculovirus vector (Invitrogen) and pcDNA3 mammalian expression vector (Invitrogen): PS1 = M1-I467, PS1-M146V, PS1-D257A, PS1-ΔE9 (M1-I467, del T291-S319); PS2 = M1-I448, PS2-N141I. Recombinant baculoviruses were generated using the Bac-to-Bac system (Invitrogen) as previously described (Shah et al., 2005; Tu et al., 2005a, 2005b). Expression of presenilins in Sf9 cells was confirmed by Western blotting with anti-PS1 (MAB5232, Chemicon) and anti-PS2 mAb. The samples used for Western blotting were maintained at 37°C prior to loading on the gel.
Planar Lipid Bilayer Experiments
Planar lipid bilayer (BLM) recordings of PS-supported currents were performed as previously described for studies of InsP3R (Tu et al., 2005a, 2005b) (see Supplemental Data for details). The ion currents across the BLM were amplified (OC-725C, Warner Instruments), filtered at 5 kHz, digitized (Digidata 1200, Axon Instruments), and stored on a computer hard drive and recordable optical discs. For presentation, the current traces were digitally filtered at 200 Hz (pClamp 6.0, Axon Instruments). For offline computer analysis, the stationary noise analysis method was used (see Supplemental Data for details).
Purification and Reconstitution of His-PS1
His-PS1 and His-PS1-M146V baculoviruses were generated as described above. The recombinant His-PS1 and His-PS1-M146V proteins were solubilized in 1 % CHAPS and purified by affinity chromatography on Ni-NTA agarose (Shah et al., 2005). The purified His-PS1 and His-PS1-M146V proteins were reconstituted into liposomes composed of phospholipids, ergosterol, and nystatin and used in the planar lipid bilayer experiments by following published procedures (Woodbury and Miller, 1990) (see Supplemental Data for details).
MEF Ca2+ Imaging Experiments
DKOand MEF fibroblasts cultured on poly-D-lysine (Sigma) coated 12 mm round glass coverslips were established as described (Herreman et al., 2000). Cytosolic Ca2+ imaging experiments with DKO and MEF cells were performed as previously described for medium spiny neuron (MSN) primary cultures (Tang et al., 2003) (see Supplemental Data for details). In rescue experiments, DKO cells were transfected using Lipofectamine (Invitrogen) with pEGFP-C3 plasmid (Clontech) or with a 1:3 mixture of pEGFP-C3 and PS expression plasmids (in pcDNA3) as indicated in the text. The Ca2+ imaging experiments were performed 48 hr after transfection. The transfected cells were identified by GFP imaging. The stable DKO rescue lines and Aph-1abc−/− MEF lines were previously described (Nyabi et al., 2003; Serneels et al., 2005). For ER Ca2+ measurements with Mag-Fura-2 dye, we used procedures previously described for BHK fibroblasts (Hofer, 1999) (see Supplemental Data for details).
Microsomal Ca2+ Flux Measurements
ER microsomes were isolated by gradient centrifugation from noninfected Sf9 cells; from Sf9 cells infected with PS1 and PS1-M146 baculoviruses; and from wild-type, DKO, and HPS1 MEFs. The isolated microsomes were used for Ca2+ flux measurement with Fura-2 (see Supplemental Data for details). The initial rate of thapsigargin-induced Ca2+ flux was measured for each type of microsome as explained in the text.
Supplementary Material
Acknowledgments
We are grateful to Donald Hilgemann for advice on thermodynamical calculations and for comments on the paper, to Tie-Shan Tang for advice on Ca2+ imaging experiments, to Malu Tansey for helping with DKO and MEF cells, to Tianhua Lei for help with Sf9 cell culture, and to Janet Young for administrative assistance. I.B. is supported by the Robert A. Welch Foundation, NINDS R01 NS38082, UT Southwestern Medical Center Alzheimer's Disease Center grant NIA P30 AG12300, and Alzheimer's Association award IIRG-06-24703. G.Y. is supported by the Robert A. Welch Foundation and NIA R01 AG023104. B.D.S. is supported by a Pioneer award from the Alzheimer's Association, the KU Leuven (GOA 2004/12), and the Federal Office for Scientific Affairs, Belgium (IUAP P5/19).
Footnotes
Supplemental Data. Supplemental Data include Supplemental Experimental Procedures, Supplemental References, and eight figures and can be found with this article online at http://www.cell.com/cgi/content/full/126/5/981/DC1/.
References
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–294. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci USA. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley JG, Duan W, Chan S, Duff K, Mattson MP. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J Neurochem. 1999;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- Camello C, Lomax R, Petersen OH, Tepikin AV. Calcium leak from intracellular stores-the enigma of calcium signalling. Cell Calcium. 2002;32:355–361. doi: 10.1016/s0143416002001926. [DOI] [PubMed] [Google Scholar]
- Cedazo-Minguez A, Popescu BO, Ankarcrona M, Nishimura T, Cowburn RF. The presenilin 1 deltaE9 mutation gives enhanced basal phospholipase C activity and a resultant increase in intracellular calcium concentrations. J Biol Chem. 2002;277:36646–36655. doi: 10.1074/jbc.M112117200. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- Herms J, Schneider I, Dewachter I, Caluwaerts N, Kretzschmar H, Van Leuven F. Capacitive calcium entry is directly attenuated by mutant presenilin-1, independent of the expression of the amyloid precursor protein. J Biol Chem. 2003;278:2484–2489. doi: 10.1074/jbc.M206769200. [DOI] [PubMed] [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol. 2000;2:461–462. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- Hofer AM. Measurement of free [Ca2+] changes in agonist-sensitive internal stores using compartmentalized fluorescent indicators. Methods Mol Biol. 1999;114:249–265. doi: 10.1385/1-59259-250-3:249. [DOI] [PubMed] [Google Scholar]
- Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasri NN, Kocks SL, Verbert L, Hebert SS, Callewaert G, Parys JB, Missiaen L, De Smedt H. Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 is responsible for a decreased endoplasmic-reticulum Ca(2+) content in presenilin double knock-out cells. Cell Calcium. 2006;40:41–51. doi: 10.1016/j.ceca.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann N Y Acad Sci. 1989;568:1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x. [DOI] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Laudon H, Hansson EM, Melen K, Bergman A, Farmery MR, Winblad B, Lendahl U, von Heijne G, Naslund J. A nine-transmembrane domain topology for presenilin 1. J Biol Chem. 2005;280:35352–35360. doi: 10.1074/jbc.M507217200. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Parker I, LaFerla FM. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem. 1999a;274:32535–32538. doi: 10.1074/jbc.274.46.32535. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM. Alzheimer's presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999b;72:1061–1068. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- Leissring MA, LaFerla FM, Callamaras N, Parker I. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol Dis. 2001;8:469–478. doi: 10.1006/nbdi.2001.0382. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Murphy MP, Mead TR, Akbari Y, Sugarman MC, Jannatipour M, Anliker B, Muller U, Saftig P, De Strooper B, et al. A physiologic signaling role for the gamma -secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA. 2002;99:4697–4702. doi: 10.1073/pnas.072033799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax RB, Camello C, Van Coppenolle F, Petersen OH, Tepikin AV. Basal and physiological Ca(2+) leak from the endoplasmic reticulum of pancreatic acinar cells. Second messenger-activated channels and translocons. J Biol Chem. 2002;277:26479–26485. doi: 10.1074/jbc.M201845200. [DOI] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD. Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000;23:222–229. doi: 10.1016/s0166-2236(00)01548-4. [DOI] [PubMed] [Google Scholar]
- Nyabi O, Bentahir M, Horre K, Herreman A, Gottardi-Littell N, Van Broeckhoven C, Merchiers P, Spittaels K, Annaert W, De Strooper B. Presenilins mutated at Asp-257 or Asp-385 restore Pen-2 expression and Nicastrin glycosylation but remain catalytically inactive in the absence of wild type Presenilin. J Biol Chem. 2003;278:43430–43436. doi: 10.1074/jbc.M306957200. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ris L, Dewachter I, Reverse D, Godaux E, Van Leuven F. Capacitative calcium entry induces hippocampal long term potentiation in the absence of presenilin-1. J Biol Chem. 2003;278:44393–44399. doi: 10.1074/jbc.M300971200. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, III, Kandel ER, Duff K, et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Schneider I, Reverse D, Dewachter I, Ris L, Caluwaerts N, Kuiperi C, Gilis M, Geerts H, Kretzschmar H, Godaux E, et al. Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem. 2001;276:11539–11544. doi: 10.1074/jbc.M010977200. [DOI] [PubMed] [Google Scholar]
- Serneels L, Dejaegere T, Craessaerts K, Horre K, Jorissen E, Tousseyn T, Hebert S, Coolen M, Martens G, Zwijsen A, et al. Differential contribution of the three Aph1 genes to gamma-secretase activity in vivo. Proc Natl Acad Sci USA. 2005;102:1719–1724. doi: 10.1073/pnas.0408901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, III, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005;122:435–447. doi: 10.1016/j.cell.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer's disease: recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427–437. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda T, Asahi M, Yamaguchi O, Hikoso S, Nakayama H, Kusakari Y, Kawai M, Hongo K, Higuchi Y, Kashiwase K, et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+ signaling. FASEB J. 2005;19:2069–2071. doi: 10.1096/fj.05-3744fje. [DOI] [PubMed] [Google Scholar]
- Tandon A, Fraser P. The presenilins. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-11-reviews3014. reviews3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Wang Z, Bezprozvanny I. Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys J. 2005a;88:1056–1069. doi: 10.1529/biophysj.104.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Wang Z, Nosyreva E, De Smedt H, Bezprozvanny I. Functional characterization of mammalian inositol 1,4,5-trisphosphate receptor isoforms. Biophys J. 2005b;88:1046–1055. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Coppenolle F, Vanden Abeele F, Slomianny C, Flourakis M, Hesketh J, Dewailly E, Prevarskaya N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J Cell Sci. 2004;117:4135–4142. doi: 10.1242/jcs.01274. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Woodbury DJ, Miller C. Nystatin-induced liposome fusion: a versatile approach to ion channel reconstitution into planar bilayers. Biophys J. 1990;58:833–839. doi: 10.1016/S0006-3495(90)82429-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. The mitogen-regulated calcium current of T lymphocytes is activated by depletion of intracellular calcium stores. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.