

Abstract
Objective
It has been shown that CD40-TRAF6 axis in leukocytes plays a significant role in neointimal formation after carotid ligation. Because CD40 and TRAF6 are expressed not only in leukocytes but also in vascular cells, we examined the role of CD40 contributed by vascular wall cells in neointimal formation after carotid ligation in an atherogenic environment.
Methods and Results
Both CD40 and TRAF6 in medial smooth muscle cells (SMCs) was upregulated significantly at 3 d and more prominently at 7 d after injury in wildtype (WT) mice, but the TRAF6 upregulation was abolished in CD40−/− mice. In vitro, TRAF6 expression was induced by cytokines (TNF-α, IL-1β) via a NF-κB-dependent manner in wildtype SMCs, but this induction was blocked in CD40-deficient SMCs. Bone marrow chimeras revealed a comparable reduction in neointimal formation and lumen stenosis in mice lacking either vascular wall- or bone marrow- associated CD40. Lacking vascular wall-associated CD40 resulted in a significant reduction in monocyte/macrophage accumulation, NF-κB activation, and multiple proinflammatory mediators (ICAM-1, VCAM-1, MCP-1, MMP-9, tissue factor). In vitro data confirmed that CD40 deficiency or TRAF6 knockdown suppressed CD40L-induced proinflammatory phenotype of SMCs by inhibition of NF-κB activation. Moreover, both in vivo and in vitro data showed that CD40 deficiency prevented injury-induced SMC apoptosis, but did not affect SMC proliferation and migration.
Conclusions
CD40 signaling through TRAF6 in vascular SMCs seems to be centrally involved in neointimal formation in a NF-κB-dependent manner. Modulating CD40 signaling on local vascular wall may become a new therapeutic target against vascular restenosis.
Keywords: CD40, TRAF, NF-kappaB, smooth muscle cells, neointimal formation
Introduction
Despite the success of antiproliferative therapies, restenosis, which is characterized primarily by neointimal formation, remains an unsolved clinical problem after vascular interventions.1 Inflammation may play a central role in the pathogenesis of neointimal formation and restenosis.2 Accumulating evidence from clinical and experimental studies suggests an important role of CD40 ligand (CD40L, CD154) and its receptor CD40 in the physiopathology of atherosclerosis and restenosis. 3,4 We and others have demonstrated that elevated soluble CD40L increases leukocyte recruitment and neointimal formation after arterial injury.5,6 However, clinical trials indicate that blockade of CD40L may not be therapeutically feasible, as it may cause thromboembolic complications, particularly because CD40L is abundantly present on activated platelets and can interact with the integrin αIIBβ3 in platelets. 7,8 In this regard, an alternative approach that has been pursued by many investigators, including ourselves, is an attempt to target CD40 and its signaling intermediates. CD40 is a receptor of the tumor necrosis factor (TNF)-receptor superfamily that by itself has no intrinsic signaling ability but requires adaptor proteins, called TNF-receptor associated factors (TRAFs), to confer signaling.9 Indeed, previous studies have revealed an important role of either CD40 or its immediate adaptor protein TRAF6 in neointimal formation after arterial injury.10–12 These findings suggest that targeting CD40 and its associated signaling intermediates may represent a potential strategy to prevent neointimal formation and restenosis after vascular interventions.
Overexpression of CD40 and its adaptor proteins TRAFs (−1,−2,−3,−5,−6) has been observed in atherosclerotic lesions and also in developed neointimal lesions after vascular injury.10,13 Lutgens and colleagues10,14 have elegantly demonstrated that the CD40-TRAF6 in leukocytes, but not CD40-TRAF2/3/5, is a major pathway required for atherosclerosis as well as neointimal formation and arterial remodeling after carotid artery ligation. However, long-term interruption of CD40 signaling pathway in immune cells could lead to severe immune deficiencies and thus impede its clinical application. Thus, we were prompted to investigate whether/how interruption of CD40 at the level of local vascular wall is sufficient to prevent CD40 to neointimal formation after vascular injury.
Vascular smooth muscle cells (SMCs) are a major contributor to neointimal formation after vascular injury.1,15 Functional CD40 and its adaptor proteins TRAFs are expressed not only by leukocytes but also by vascular SMCs and endothelial cells (ECs).13,16 In cultured SMCs, expression of CD40 and several TRAFs including TRAF6 can be stimulated by cytokines such as TNF-α and IL-1β.13 In vitro studies indicate that CD40 ligation by CD40L activates vascular SMCs by promoting the expression of molecules thought to be involved in neointimal formation, such as adhesion molecules, cytokines, chemokines, matrix metalloproteinases, and tissue factor.17–20 However, it is unknown whether such effects indicated in vitro are relevant to neointimal formation in vivo.
Using a well-established carotid artery injury model, our results indicate a strong protective effect by CD40 deficiency in vascular wall cells on neointimal formation and lumen stenosis in an atherogenic environment. These protective effects were most likely attributed to the inhibition of TRAF6 and NF-κB activation and marked downregulation of multiple NF-κB-dependent proinflammatory mediators in mice lacking vascular wall cell-associated CD40. Our study provides the first evidence that modulating CD40 signaling at the level of local vascular wall may be clinically feasible to limit neointimal formation and restenosis after vascular interventions.
Methods
Mice
Male C57BL/6 mice and CD40−/− mice (backcrossed onto a C57BL/6 background for >10 generations) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were fed with a Western-type diet (TD 88137; Harlan-Teklad, Madison, WI) containing 21% fat by weight (0.15% cholesterol and 19.5% casein without sodium cholate) for 2 week before and 3 weeks after vascular injury, as we previously described.18 Serum samples collected from each mouse were frozen (−80°C) for subsequent measurement of total cholesterol levels using a spectrophotometric assay (Suppl. Figure 1). All animal protocols were approved by the Institutional Animal Care and Use Committee.
Bone marrow transplantation and vascular injury model
Bone marrow cells were isolated from the femurs and tibias of donor mice (WT and CD40-/-) as previously described.21 Recipient mice (about 5–6 weeks of age) were lethally irradiated (2 x 525 Rads, 3 hours apart). Then, the recipient mice received bone marrow cells (2 x 106)suspended in 200 μl phosphate-buffered saline (PBS) by tail vein injection. Mice were maintained in filter-top cages under specific pathogen-free conditions and given water containing 0.2% (2.0 mg/ml) neomycin (Invitrogen) until 2 weeks after BMT. Six to 8 weeks later, flow cytometry was used to verify chimera reconstitution by staining for CD45.1 (recipient isoform of CD45) and CD45.2 (donor isoform of CD45) expression on blood leukocytes with a fluorescein isothiocyanate-labeled anti-CD45.1 antibody and a biotinylated anti-CD45.2 antibody with a streptavidin-PerCP secondary antibody (PharMingen). Successful BM reconstitution was setas >90% of the CD45.2-positive population in total leukocytes (Suppl. Figure 4) as previously described.21
A flow-restriction vascular injury was caused by ligating the bifurcation of the left common carotid artery.22 In brief, the left common carotid artery was exposed through a small midline incision in the neck and the artery was completely ligated with a 7–0 silk suture just proximal to the carotid bifurcation to disrupt blood flow.
Tissue harvesting and histologic analysis
The arterial segments were dehydrated in ethanol and xylene and embedded in paraffin. Serial cross-sections (5 μm thick) were cut beginning 100 μm proximal to the carotid ligation site. Histomorphometric analysis was performed in seven cross-section levels (with 120 μm intervals). For each level, three cross-sections were stained with Verhoeff’s Elastic stain and morphometric analysis was performed by an individual blinded to experimental design. Morphometric analysis was performed by two “blinded” researchers by a computerized image system (Image Pro Plus 5.0) and the mean of their results wan calculated. We measured the lumen area, intimal area, medial area, and total vessel area at each level as we previously described.5 Percentage stenosis was calculated as the ratio of the intimal area to the area inside the original internal elastic lamina. The mean value of neointimal area, intima/media ratio, percentage stenosis, and total vessel area was calculated over the 7 levels.
Immunohistochemistry
Paraffin-embedded sections were stained with the avidin-biotin -peroxidase method (Vector). The following primary antibodies (from Santa Cruz, unless otherwise indicated) were used: ICAM-1 (1:50), VCAM-1 (1:50), MCP-1 (1:1000), tissue factor (TF, 1:400), fibrinogen (Fgn, 1:800; Dako), MMP-9 (1:75; R&D Systems), NF-κB p65 (1:750; Abcam), Phospho-p65(Ser536) (1:50; Abcam), and Mac-2 (1:1000; M3/38, Accurate) to detect monocyte/macrophages. Isotype-matched antibodies served as negative controls. Sections were incubated with the appropriate secondary antibody (Vector) and visualized by 3,3′-diaminobenidine (DAB; Vector) and counterstained with hematoxylin. For quantitative comparison of the expression of indicated molecules, the percent of the positively stained area to the total traced area was determined in triplicate.
Double immunofluorescence staining was performed on paraffin-embedded sections using the following antibodies: CD40 (1:400), TRAF6 (1:30), PCNA (1:100), VCAM-1, MCP-1, TF, Fgn, MMP-9, CD31 (1:50; Abcam), SMα-actin conjugated with Cy3 (1:500; Sigma). Sections were incubated with the indicated antibodies overnight at 4°C. Immunoreactions were visualized using Alexa Fluor 488- or Alexa Fluor 555-conjugated secondary antibodies (1:200; Invitrogen). Apoptotic vascular cells were identified by TUNEL assay (Roche). Mounting medium containing DAPI was then applied. Images were acquired using a fluorescence microscope.
Cell culture and siRNA transfection
Aortic SMCs were obtained from thoracic aortas of 10 mice of each genotype as we previously described.23 Vascular SMCs (passage 3–6) were grown to subconfluency in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, deprived of serum for 72 hours, and then treated with siRNA oligonucleotides or other indicated reagents under defined conditions. In some experiments, NF-κB pathway was pharmacologically inhibited with NF-κB inhibitors NF-κB SN50 or BAY 11-7082 (Calbiochem).
Mouse TRAF2 siRNA (sc-36711), TRAF6 siRNA (sc-36718), nonspecific control siRNA (sc-37007), and transfection reagents were purchased from Santa Cruz Biotechnology (CA, USA). Each siRNA oligonucleotide was delivered into vascular SMCs according to the manufacturer’s instruction. In brief, vascular SMCs were seeded in 6-well plates with 2 ml of culture medium until 70% confluence was reached. Then, siRNAs with transfection reagents were added to the serum-free medium at a final concentration of 100 nM. After 24 h of transfection, the medium was replaced with fresh culture medium, and the cells were incubated at 37°C in 5% CO2 for additional 24 h before performing experiments. Specificity and efficiency of each siRNA were verified in our pre-experiments (Suppl. Figure 2).
NF-κB p65 nuclear translocation assay
The nuclear translocation of NF-κB p65 was examined using immunocytochemical staining as previously described.24,25 Vascular SMCs were plated in 4-well chamber slides (Lab-Tek II, Thermo) for overnight and then made quiescent in DMEM/0.1%BSA for 72 hours. Cells were stimulated with recombinant mouse CD40L (10 μg/ml; R&D Systems) or vehicle for 1h. In some experiments, cells were pre-transfected with siRNA as described above or pretreated with indicated kinase inhibitors (Calbiochem) before stimulation. Cells were fixed with 2% PFA, followed by permeabilization with 0.2% Triton X-100. After blocking, cells were incubated with anti-NF-κB p65 (1:100, Abcam) overnight at 4°C, followed by incubation with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:200, Invitrogen). Nuclei were stained with DAPI. Images were taken using a fluorescence microscope (Nikon, Japan) and quantification was performed using Image-Pro Plus software. Three independent experiments were performed. The results were expressed as percentage (mean ± SEM) of NF-κB p65 positive nuclei-stained cells relative to total cells that were counted at least 8 fields per group under 200 magnification.
Cell proliferation, migration, and apoptosis assay
Cell proliferation was evaluated by cell counting. Briefly, vascular SMCs were serum starved for 72 hours and plated into 24-well plates (104 cells/well) in DMEM supplemented with 10% FBS. At different time points (2, 4, 6, and 8 days), cells were trypsinized and counted in a Coulter counter. Cell migration was measured using transwell chambers with a filter membrane (8-μm pore size) (Corning Inc.). Briefly, vascular SMCs were serum starved for 72 hours and seeded into the upper chambers (105 cells/well). 10% FBS or PDGF-BB (20 ng/ml) was added to the lower chamber and incubated for 4 hours. Cells that migrated on the filter were stained with DAPI and counted under a fluorescence microscope. For cell apoptosis assay, vascular SMCs were plated into 4-well chamber slides (6 x 104 cells/well) and serum starved for 72 hours. Apoptosis was induced by treating cells with apoptosis inducer C2 ceramide (100 μM) for 5 hours. Then, cells were stained with DAPI for analysis of apoptotic nuclear morphology. Each treatment was done in triplicate wells, and at least three independent experiments were performed.
Quantitative real-time RT- PCR
Total RNA was extracted from arteries or cell cultures using TRIzol Reagent (Invitrogen) and treated with DNase I to remove genomic DNA. A quantitative real-time reverse-transcriptase (RT)-PCR was performed with a Bio-Rad thermocycler and an SYBR green kit(Bio-Rad) following manufacturer’s instructions. Sequence-specific primers used were presented in Suppl. Table 1. The relative mRNA expression was normalized to GAPDH. Data are expressed as mean ± SEM from 3 independent experiments.
Statistical analysis
Results are presented as mean ± SEM. Experimental data were analyzed by unpaired 2-tailed Student’s t test or ANOVA followed by Bonferroni multiple-comparison correction. P<0.05 was considered statistically significant.
Results
CD40 is required for injury-induced upregulation of TRAF6 in vascular SMCs
First, we investigated the expression and potential association of CD40 and TRAF-6 in the carotid artery wall in response to vascular injury. In WT mice, injury induced a significant upregulation of both CD40 and TRAF6 at 3 d and more prominently at 7d, notably with strong co-localization of both molecules with medial SMα-actin-positive SMCs (Figure 1, A-B) and CD31-positive ECs (Suppl. Figure 3). In contrast, the injury-induced TRAF6 upregulation both in medial SMCs and ECs was almost completely abolished in CD40−/− mice (Figure 1, B-D).
Figure 1. CD40 is required for injury-induced upregulation of TRAF6 in vascular SMCs.





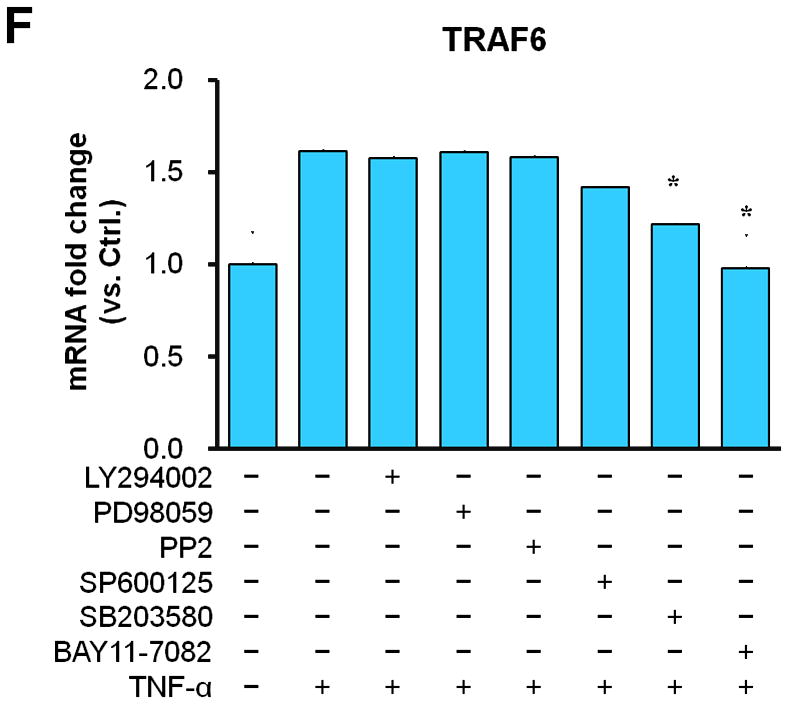
Representative images illustrating co-expression of CD40 (A) and TRAF6 (B, C) with SMα-actin in cross-sections of uninjured and injured (3d, 7d) carotid arteries from WT and CD40−/− mice. Inserts show appropriate isotype controls combined with DAPI staining. Arrows indicate the internal elastic lamina. Scale bars: 50 μm. (D) Quantitative analysis of the CD40 and TRAF6 shown in A-C. ** P<0.01 versus uninjured WT control, ## P<0.01 versus corresponding WT group. (E) mRNA levels of CD40, TRAF2, and TRAF6 were determined by quantitative RT-PCR in WT and CD40−/− SMCs stimulated with or without TNF-α (20 ng/ml) or IL-1β (10 ng/ml) for 4 hours. mRNA levels are normalized to GAPDH. * P<0.05 versus control, # P<0.05 versus corresponding WT group. (F) mRNA levels of TRAF6 were determined by quantitative RT-PCR in wildtype SMCs pretreated with LY294002 (20 μM), PD98059 (100 μM), PP2 (10 μM), SP600125 (20 μM), SB203580 (10 μM), BAY11-7082 (10 μM), or DMSO for 1 hour, followed by stimulation with or without TNF-α (20 ng/ml)for 4 hours. mRNA levels are normalized to GAPDH. * P<0.05 versus TNF-α-treated group.
Subsequently, we determined whether CD40 is required for induction of TRAF6 expression in vascular SMCs in vitro. We found that mRNA expression of both CD40 and TRAF6, but not TRAF2, was stimulated by proinflammatory cytokines (TNF-α, IL-1β) in wildtype SMCs (Figure 1E), but the cytokine-induced upregulation of TRAF6 was abrogated in CD40-deficient vascular SMCs (Figure 1E). Further, we examined potential signaling pathways involved by using different kinase inhibitors, and found that the TNF-α-induced TRAF6 expression was completely blocked by a NF-κB inhibitor (BAY11-7082) and partially inhibited by a p38 MAPK inhibitor (SB203580), but not affected by other kinase inhibitors including PI3-kinase, Src kinase, Erk1/Erk2 (Figure 1F). Taken together, these data suggest that CD40 and TRAF6 are among injury-responsive genes in arterial wall, and CD40 is required for injury-induced upregulation of TRAF-6 in vascular SMCs via a NF-κB-dependent mechanism.
Vascular wall cell-derived CD40 substantially contributes to neointimal formation and vascular remodeling after vascular injury
To determine whether/how vascular wall cell-derived CD40 contributes to neointimal formation, we created bone marrow chimeric mice and performed carotid artery ligation on the mice in the presence of hypercholesterolemia induced by the high-cholesterol diet (Suppl. Figure 1). We demonstrated a significant and comparable reduction in neointimal formation and lumen stenosis between the WT chimeric mice and the CD40-deficient chimeric mice reconstituted with wild-type bone marrow (i.e. lacking vascular wall cell-derived CD40) (Figure 2, A-D). Moreover, we observed that total vessel size was significantly smaller in chimeric mice lacking either vascular wall- or bone marrow-derived CD40 or both, when compared that of WT chimeric mice (Figure 2E). These observation were similar to previous report in WT and CD40-/- mice,10 indicating that CD40 deficiency impaired vascular remodeling after arterial injury. Taken together, our data suggest an important role for vascular wall cell-derived CD40 in neointimal formation and vascular remodeling in response to vascular injury.
Figure 2. Vascular wall cell-derived CD40 substantially contributes to neointimal formation and vascular remodeling after vascular injury.





(A) Representative elastic stained cross sections (level 3) of carotid arteries of chimeric mice at 21d after injury. Arrows indicate the internal elastic lamina. Scale bars: 50 μm. Intima area (B) and vessel area (E) were measured at the 7 section levels (120-μm intervals), and the mean area was calculated. Intima/media ratio (C) and lumen stenosis (D) at each level, and their mean values were determined. Numbers at the base of the bars indicate the number of mice in each group. * P<0.05 and ** P<0.01 versus corresponding WT to WT group.
Vascular wall cell-derived CD40 modulates inflammatory response to vascular injury
Next, we examined whether/how vascular wall cell--associated CD40 affects key inflammatory and prothrombotic events associated with vascular injury. Immunohistochemistry showed that absence of CD40 in the vascular wall (i.e. WT to CD40−/− group) resulted in a dramatic reduction in the content of Mac-2+ monocyte/macrophages in the neointimal lesions at 21 d (Figure 3A), which was most likely attributed to a significant reduction in adhesion molecules (ICAM-1, VCAM-1) and chemokine MCP-1 (Figure 3B). CD40 deficiency in the vascular wall also downregulated the expression of prothrombotic molecules (TF, Fgn), and MMP-9 (Figure 3C). Using double immunostaining, we further identified that CD40 deficiency in the vascular wall profoundly inhibited such proinflammatory and prothrombotic molecules in the medial SMCs and ECs at 7 d after injury (Figure 3D). These in vivo findings were further supported by in vitro observations that the CD40L-induced expression of ICAM-1, VCAM-1, and MCP-1 was blocked in CD40-deficient SMCs (Figure 3E). Taken together, these data support the importance of local vascular wall cell-derived CD40 in modulating inflammatory and prothrombotic responses to vascular injury.
Figure 3. Vascular wall cell-derived CD40 modulates inflammatory response to vascular injury.
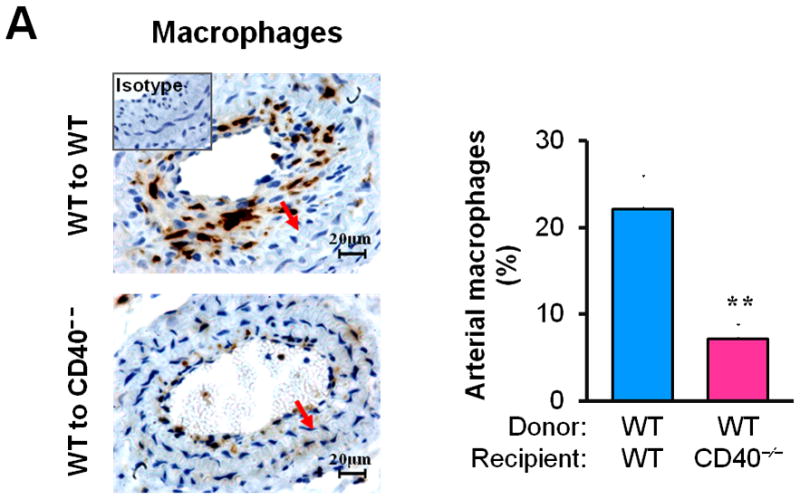




Immunohistochemical staining illustrating Mac-2-positive monocyte/macrophages (A), the expression of VCAM-1, ICAM-1, and MCP-1 (B), TF, Fgn, and MMP-9 (C), in carotid arteries of chimeric mice at 21d after ligation, as well as their quantitative analysis. Inserts show appropriate isotype controls. Arrows indicate the internal elastic lamina. Scale bars: 20 μm. n= 5 per group. ** P<0.01 versus corresponding WT to WT group. (D) Representative images showing co-expression of VCAM-1, MCP-1, TF, Fgn (7d), and MMP-9 (21d) with SMα-actin or CD31 in injured carotid arteries from chimeric mice. Inserts show appropriate isotype controls combined with DAPI staining. Scale bars: 50 μm. (E) mRNA levels of VCAM-1, ICAM-1, and MCP-1 were determined by quantitative RT-PCR in WT and CD40−/− SMCs exposed to rmCD40L (10 μg/ml) for 16 hours. mRNA levels are normalized to GAPDH. ** P<0.01 versus WTgroup.
CD40 and TRAF6 mediate proinflammatory phenotype of vascular SMCs in vitro through a NF-κB dependent mechanism
NF-κB is a known major transcriptional factor that regulates expression of a variety of proinflammatory genes.26,27 We demonstrated that the levels of total NF-κB p65 and phosphorylated NF-κB p65 (Ser536) were increased in injured carotid arteries of WT chimeric mice, mainly localized in neointimal lesions and medial layer at 21d (Figure 4A), but the levels were markedly reduced in chimeric mice lacking vascular wall cell-associated CD40 (Figure 4A).
Figure 4. CD40-induced proinflammatory phenotype of vascular SMCs is dependent on NF-κB activation.
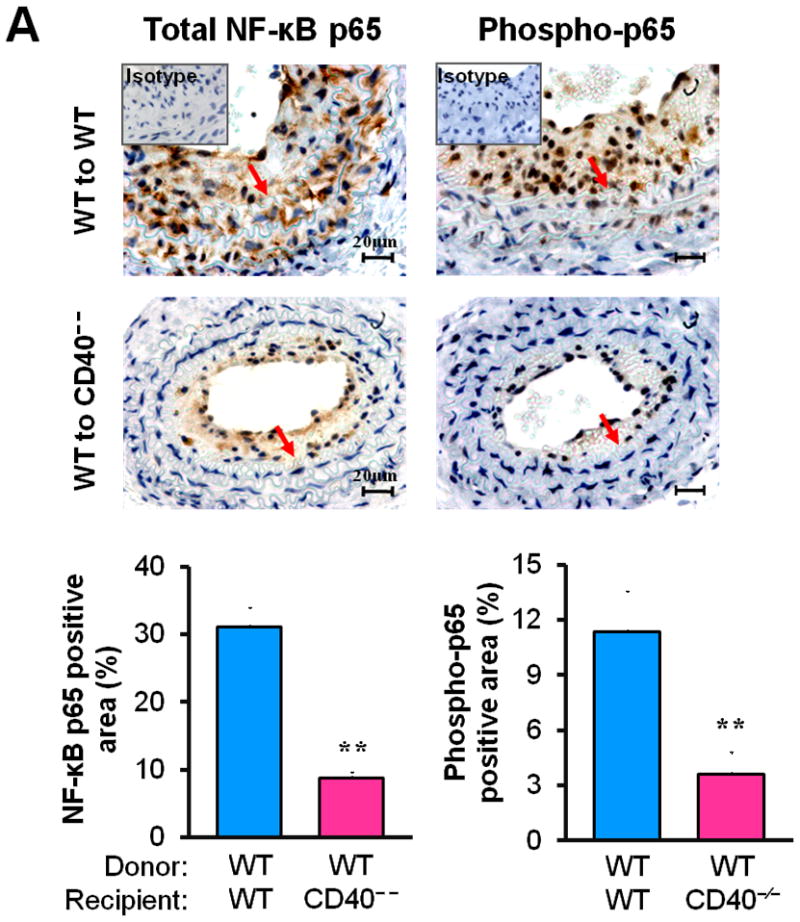
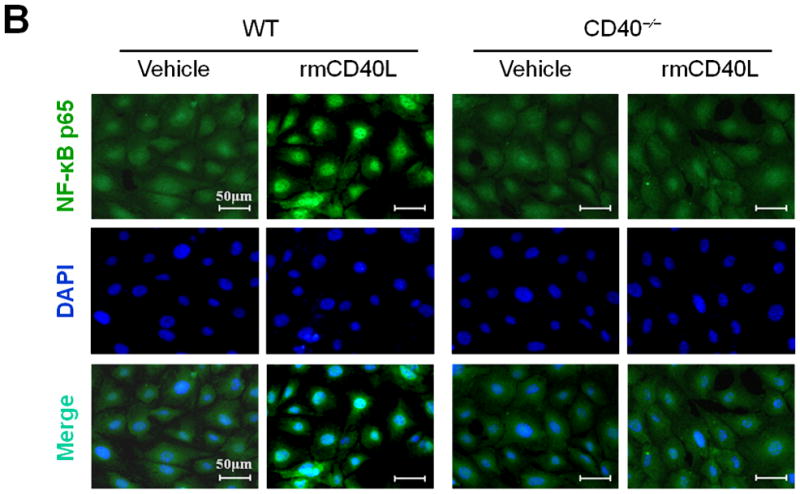
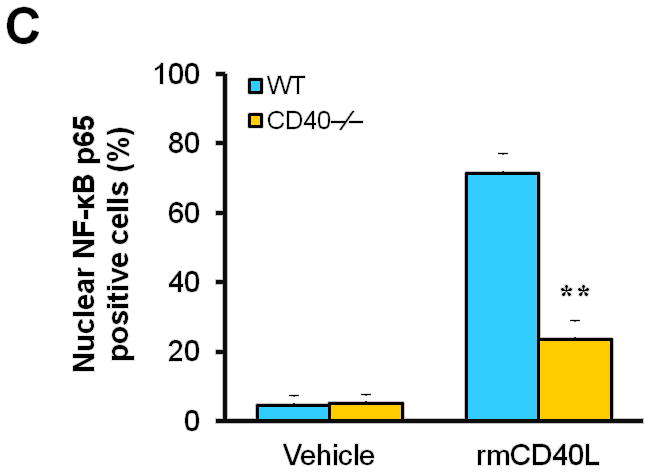

(A) Representative images showing the expression of total NF-κB p65 and activated phospho-p65 in the carotid arteries of chimeric mice at 21d after ligation, as well as their quantitative analysis. Inserts show appropriate isotype controls. Arrows indicate the internal elastic lamina. Scale bars: 20 μm. n=5 per group. ** P<0.01 versus WT to WT group. (B) Immunofluorescence staining of NF-κB p65 (green) showing its nuclear translocation in WT and CD40−/− SMCs exposed to rmCD40L (10 μg/ml) or vehicle for 1 h. Cell nuclei were detected by DAPI (blue). Scale bars: 50 μm. (C) Quantification of NF-κB nuclear translocation shown in B is expressed as the percentage of p65 nuclei-positively stained cells to the total cells. ** P<0.01 versus WT group. (D) mRNA levels of VCAM-1, ICAM-1, and MCP-1 were determined by quantitative RT-PCR in wildtype SMCs pretreated with or without NF-κB SN50 (2.5 μM) or BAY 11-7082 (10 μM) for 45 min, followed by stimulation with rmCD40L (10 μg/ml) for 16 hours. mRNA levels are normalized to GAPDH. ** P<0.01 versus rmCD40L-treated group.
Subsequently, we performed in vitro experiments to determine how CD40 affects NF-κB activation and proinflammatory phenotype of vascular SMCs. Our findings include 1) CD40L induced NF-κB p65 translocation from the cytoplasm to the nucleus that is a key step in activation of NF-κB pathway in wildtype SMCs, but this effects was abrogated in CD40-deficient SMCs (Figure 4, B and C); 2) CD40L induced expression of ICAM-1, VCAM-1, and MCP-1 in wildtype SMCs, but this effect was almost completely blocked by inhibition of NF-κB with the BAY11-7082, a selective inhibitor of IκBα phosphorylation, or SN50, a peptide inhibitor of NF-κB translocation and activation (Figure 4D).
Next, we determined how TRAF6 affects NF-κB activation and proinflammatory phenotype of vascular SMCs. We performed siRNA-mediated knockdown of TRAF6 and TRAF2 in vitro. Compared with vascular SMCs transfected with control siRNA, siRNA knockdown of TRAF6, but not TRAF2, significantly reduced NF-κB p65 nuclear translocation induced by CD40L (Figure 5, A and B), with a significant reduction in the expression of ICAM-1, VCAM-1, and MCP-1 (Figure 5C). Further, we examined the downstream signaling pathways responsible for CD40L-induced NF-κB activation in vascular SMCs. NF-κB p65 nuclear translocation was assessed using different kinase inhibitors. We demonstrated that CD40L-induced NF-κB p65 nuclear translocation was completely blocked by the NF-κB inhibitor (BAY11-7082), and also largely inhibited by the PI3K inhibitor (LY294002) and Erk1/Erk2 inhibitor (PD98059) (Figure 5, D and E). Taken together, these data suggest that CD40 and TRAF6 mediate proinflammatory phenotype of vascular SMCs in vitro through a NF-κB dependent mechanism.
Figure 5. TRAF6, PI3K activity, and MAPK activity are required for CD40-induced NF-κB proinflammatory signaling in vascular SMCs.

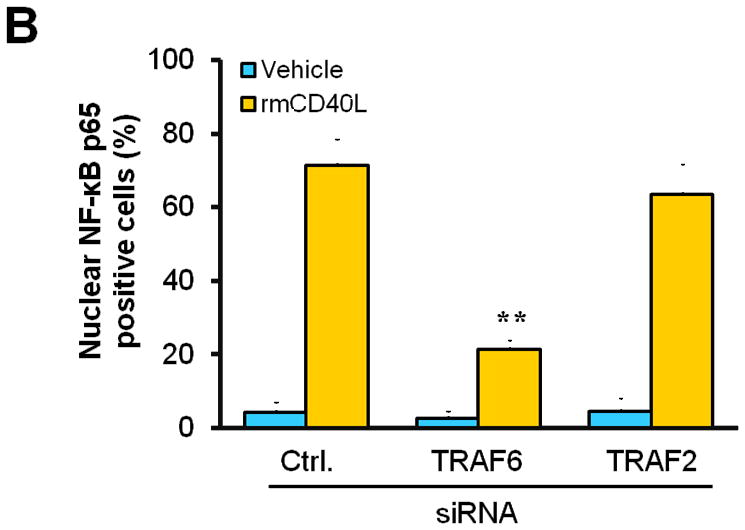


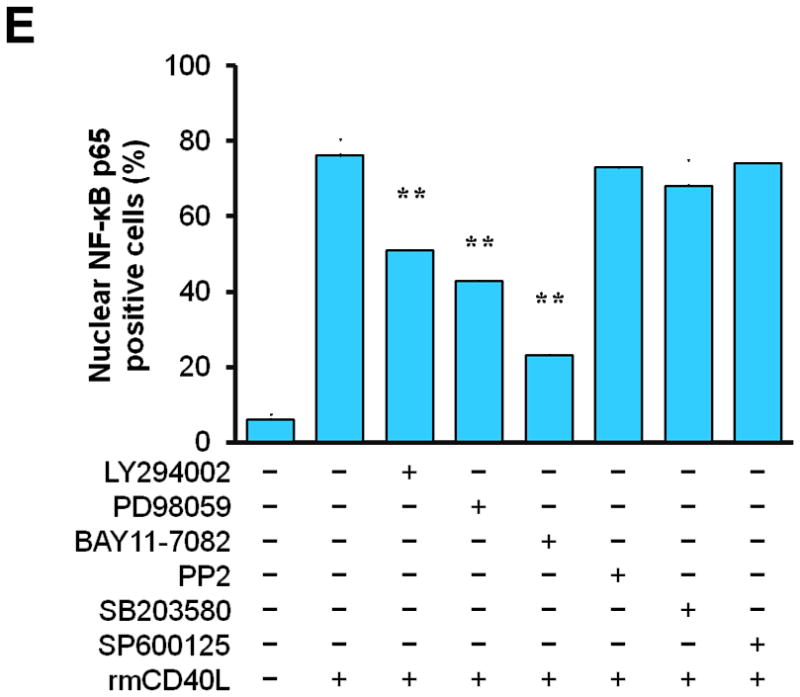
(A) Detection of NF-κB nuclear translocation by immunofluorescence staining for NF-κB p65 (green) in wildtype SMCs transfected with TRAF6 siRNA, TRAF2 siRNA, or control (Ctrl.) siRNA exposed to rmCD40L (10 μg/ml) for 1 h. Scale bars: 50 μm. (B) Quantification of NF-κB nuclear translocation shown in A. ** P<0.01 versus corresponding control siRNA group. (C) mRNA levels of VCAM-1, ICAM-1, and MCP-1 were measured by quantitative RT-PCR in wildtype SMCs transfected with TRAF2 siRNA, TRAF6 siRNA or Ctrl. siRNA exposed to rmCD40L (10 μg/ml) for 16 hours. mRNA levels are normalized to GAPDH. ** P<0.01 versus control siRNA. (D) NF-κB nuclear translocation was assessed by immunofluorescence staining for NF-κB p65 (green) in wildtype SMCs pretreated with LY294002 (20 μM), PD98059 (100 μM), BAY11-7082 (10 μM), PP2 (10 μM), SB203580 (10 μM), SP600125 (20 μM), or DMSO, followed by stimulation with or without rmCD40L (10 μg/ml) for 1 h. Scale bars: 50 μm. (E) Quantification of NF-κB nuclear translocation shown in C. ** P<0.01 versus rmCD40L-treated group.
CD40 mediates vascular SMC apoptosis but has no effect on SMC migration and proliferation in vivo and in vitro
Vascular SMC migration and proliferation as well as apoptosis contribute to the development of neointimal formation.28–30 However, the exact role of the receptor CD40 in vascular SMC biology has not been established. Using combined bone marrow chimeras and double immunostaining, we investigated the effects of CD40 deficiency in the vascular wall on medial SMC proliferation (by PCNA stain) and apoptosis (by TUNEL stain). We observed that CD40 deficiency in the vascular wall did not affect medial SMC proliferation (Figure 6A), but significantly reduced medial SMC apoptosis (Figure 6B), at 7 days after carotid ligation. Further, these in vivo observations were verified by our in vitro data. We found no difference in proliferation and migration between WT and CD40-deficient SMC (Figure 6, C and D). In contrast, the apoptosis induced by C2 ceramide was significantly reduced in CD40-deficient versus wildtype SMCs (Figure 6E).
Figure 6. CD40 mediates vascular SMC apoptosis but has no effect on SMC proliferation and migration in vivo and in vitro.


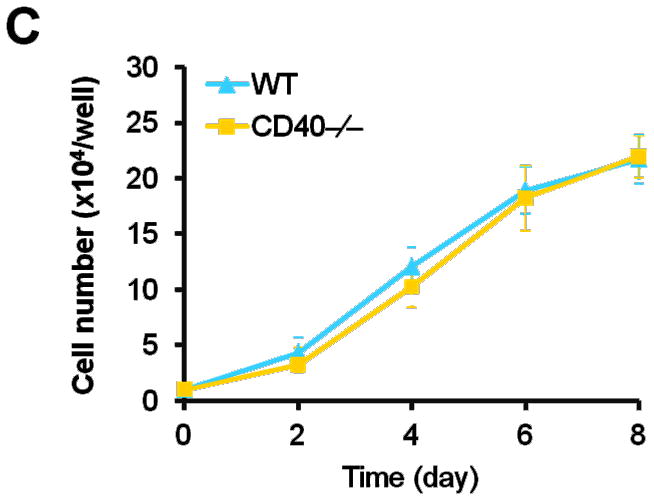


Representative images showing double-staining of PCNA (A) and TUNEL (B) with SMα-actin in carotid arteries of chimeric mice at 7d after ligation, as well as their quantitative analysis (n=5 per group). Insert shows appropriate isotype control combined with DAPI staining. Scale bars: 50 μm. ** P<0.01 versus WT to WT group. (C) Cell proliferation measured by cell counting in WT and CD40−/− SMCs exposed to 10% FBS at the indicted time points. (D) Cell migration toward 10% FBS or PDGF-BB (20 ng/ml) measured by transwell chambers assay in WT and CD40−/− SMCs. Representative images of filters with DAPI-stained cells are shown (upper panel). Scale bar: 50 μm. Quantification of migrated cells is shown (lower panel). (E) Representative photomicrographs of DAPI staining in WT and CD40−/− SMCs treated with apoptosis inducer C2 ceramide (100 μM) for 5 hours, as well as quantitative analysis of cell apoptosis index (%). Arrows indicate the condensed or fragmented nuclei of apoptotic cells. Scale bars: 50 μm. ** P<0.01 versus WT group.
Discussion
The present study presents several novel important findings as follows: First, both CD40 and its adaptor protein TRAF6 are robustly co-upregulated in the medial SMCs in the early phase (3d, 7d) after carotid ligation, which substantially contribute to vascular inflammatory response and proinflammatory phenotype of SMCs, and eventually to neointimal formation. Second, CD40 is required for the vascular injury- and cytokine-induced TRAF6 upregulation via a NF-κB dependent mechanism. Third, the CD40-TRAF6 mediates proinflammatory phenotype of vascular SMCs in vitro through a NF-κB dependent mechanism. Forth, modulating SMC apoptosis might be a potential novel mechanism by which CD40 contributes to neointimal formation following vascular injury.
CD40 is expressed not only on circulating inflammatory cells but also on local vascular wall cells, both of which are active players in the response to vascular injury. Bone marrow-derived CD40 has previously been shown to contribute to neointimal formation.10 However, the role of local vascular wall cell-derived CD40 in neointimal formation remain undefined. The present study provides the first direct evidence, to our knowledge, that vascular wall cell-derived CD40 also plays a critical role in the vascular response to injury.
The present study demonstrated that vascular injury induced a robust upregulation of both CD40 and TRAF6 in the arterial wall in the early phase (3d, 7d), predominantly in medial SMCs, suggesting the involvement of medial SMC CD40 and TRAF6 in the process of vascular injury response. Notably, we demonstrated that CD40 is required for the injury-induced TRAF6 expression in SMCs both in vivo and in vitro. This finding is of significant interest because TRAF6 is an important regulator of vascular SMC physiology.31 It has previously been shown that blockage of local vascular TRAF6 diminished vascular inflammation and intimal lesion formation upon injury.31,32 Apart from linking CD40 to downstream signaling events, TRAF6 also serves as adaptor protein for other cell receptors in SMCs, including TNFR, TLR, IL-1R, and angiotensin II AT1R,33–35 whereby it regulates the biological functions of a variety of proinflammatory cytokines. Thus, CD40-dependent expression of TRAF6 in SMCs may serve as a feedback mechanism to modulate itself or other TRAF6 dependent pathways that contribute to vascular response to injury and the development of neointimal formation. Also, we observed a significant injury-induced upregulation of TRAF6 in the endothelium in wildtype artery, but not in CD40-deficient artery. This finding is agreement with previous in vitro report that CD40 is required for the cytokine-induced TRAF6 expression in ECs.13
The adhesion and recruitment of inflammatory cells to the site of vascular injury is critical for the initiation and progression of neointimal formation.2,36,37 Modulation of the medial SMC phenotype has recently emerged as a key event in the vascular response to injury.38 Following vascular injury, SMCs may take on a proinflammatory phenotype, characterized by enhanced expression of various proinflammatory mediators, including cytokines, chemokines, adhesion molecules, matrix metalloproteinases, and prothrombotic molecules,15,39–41 which may functionally promote leukocyte recruitment to the arterial wall. Although CD40 signaling has been implicated in inflammatory response, its role on proinflammatory phenotype of SMCs is unclear. The present study identified CD40 as a key regulator for proinflammatory phenotype of SMCs following vascular injury and serves to govern injury-induced synthesis of a variety of proinflammtory mediators in the arterial wall, including chemokines (e.g. MCP-1), adhesion molecules (e.g. VCAM-1, ICAM-1), matrix metalloproteinases (e.g. MMP-9), and prothrombotic molecules (e.g. TF, Fgn). Therefore, our results help to explain the mechanism by which CD40 expressed on vascular wall cells contributes to vascular inflammatory response and neointimal progression after vascular injury.
Activation of transcription factor NF-κB plays a key role in regulating the expression of various proinflammatory mediators involved in atherosclerosis and neointimal formation after vascular injury.27,42,43 However, the molecular mechanism whereby vascular injury induces NF-κB activation in the vessel wall remains largely unclear. The present study demonstrated that CD40 deficiency in vascular wall cells markedly inhibits NF-κB activation after carotid ligation. It has been shown that CD40 modulates activation of NF-κB in different cell types via distinct signaling mechanisms in vitro. For example, in endothelial cells, CD40 ligation elicits activation of NF-κB through PI3K/Akt and p38 MAPK signaling.44,45 However, it is unknown how CD40 ligation activates NF-κB in vascular SMCs. The present study demonstrated that either genetic deficiency of CD40 or siRNA knockdown of TRAF6 (but not TRAF2) leads to impaired NF-κB activation in cultured SMCs induced by CD40L. CD40-mediated NF-κB activation also showed dependence upon PI3K activity and MAPK activity. Thus, we assume that CD40 is required for NF-κB activation in vascular SMCs through TRAF6-mediated, PI3K-and MAPK-dependent pathways.
Numerous studies have shown that vascular SMC proliferation and migration substantially contribute to the development of neointimal formation in relation to restenosis and atherosclerosis.28,29 Although functional CD40 has been shown to be expressed on SMCs, little is known about its role in biological functions of SMCs. There are contradictory reports about the role of CD40L in SMC proliferation and migration in vitro. Hermann et al46 showed that CD40L does not stimulate SMC proliferation and migration, whereas Chai et al47 reported that CD40L increases SMC proliferation and migration. It should be noted that both previous studies examined the role of CD40L, rather than CD40. Increasing evidence has shown that the CD40L-mediated effects are not necessarily identical to the CD40-mediated effects. This is possibly because that CD40 is not the only receptor for CD40L, e.g. CD40L can bind to other receptors such as GPIIb/IIIa in platelets and Mac-1 in leukocytes.3 Likewise, CD40L is not the only protein that interacts with CD40, e.g. other proteins such as HSP70 can interact with CD40.48,49 The present study demonstrated for the first time that CD40 deficiency does not affect vascular SMC proliferation and migration, but intriguingly, it reduces SMC apoptosis in vivo and in vitro. Vascular SMC apoptosis is known to be involved in the pathological mechanism of neointimal formation.30,50 CD40 has been shown to transduce apoptotic signals in several cell types, including vascular ECs51 and carcinoma cells.52 But its role in vascular SMC apoptosis is unknown. The present study indicated that CD40 deficiency in vascular wall cells protects medial SMCs from injury-induced apoptosis in the early stage (at 7d) after vascular injury. This effect was verified by in vitro experiments demonstrating that CD40 deficiency leads to SMC resistance to apoptosis. Medial SMC apoptosis has been associated with subsequent neointimal progression in vascular injury models.22,53,54 It has been suggested that the early medial SMC apoptosis could exacerbate neointimal formation by provoking a greater wound healing response to overcome the cellular deficit.15,55 In support of this concept, apoptotic SMCs has been shown to release cytokines (e.g. IL-1α), which may stimulate the surrounding viable SMCs to produce proinflammatory cytokines such as IL-6 and MCP-1, leading to amplification of vascular inflammation.56 Furthermore, emerging evidence suggests that inhibition of medial SMC apoptosis acts to prevent neointimal formation after vascular injury.57–59 Thus, modulating SMC apoptosis might be a potential novel mechanism by which CD40 contributes to neointimal formation following vascular injury.
Study Limitations
Different TRAFs exhibit specific biological functions. The role of individual TRAFs in the activation of different CD40-dependent signaling pathways has not yet been fully defined.9 Although the previous work10 and our present data support a key role for CD40 signaling through TRAF6 in neointimal formation, a role for CD40 mediated by other TRAF proteins cannot be fully excluded. At present, it is difficult to differentiate individual functions of other distinct TRAF proteins in animal studies in vivo, because TRAF6 has a distinct binding site on CD40, but TRAF-2,-3,-5 have overlapping binding sites.9,10 In addition, there is evidence which indicates functional difference between a specific TRAF and a specific CD40-associated TRAF signaling, because in addition to CD40 signaling TRAFs are also involved in signaling of other TNF superfamily members. For example, although CD40 mutation with an impaired TRAF6 prevents atherosclerosis in mice,14 in contrast, Stachon et al60 reported that TRAF6 is not required for atherosclerogenesis in mice and does not associate with atherosclerosis in humans. Moreover, animal studies of the role of distinct TRAF proteins in CD40 effector functions have also been hampered by the poor viability of most TRAF-/- mice.9,10 Another limitation of this study is that it is unclear about the role for CD40 through specific TRAF signaling in the endothelium, especially involved in the process of reendothelialization after endothelial denudation injury in vivo. Future studies are required to address these issues.
Conclusions
Our results demonstrate that CD40 signaling through TRAF6 in vascular SMCs seems to be centrally involved in the development of neointimal formation in a NF-κB-dependent manner. Our findings may have potential implications for developing novel therapeutic approach as adjuvant therapy for vascular interventions. Given that long-term inhibition of CD40 signaling in immune cells (leukocytes) may be not therapeutically feasible, as it may compromise systemic immune response, modulating CD40 in the vascular wall cells may provide a new therapeutic approach to prevent neointimal formation hopefully with less side effects. This new target strategy would be feasible, in particular, for local vascular wall drug delivery, e.g. angioplasty plus drug-eluting stents against vascular restenosis.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grant HL087990 (G.L.) and by a scientist development grant (0530166N, G.L.) from the American Heart Association.
Footnotes
Disclosures
None.
References
- 1.Zargham R. Preventing restenosis after angioplasty: a multistage approach. Clin Sci (Lond) 2008;114:257–264. doi: 10.1042/CS20070228. [DOI] [PubMed] [Google Scholar]
- 2.Donners MM, Daemen MJ, Cleutjens KB, Heeneman S. Inflammation and restenosis: implications for therapy. Ann Med. 2003;35:523–531. doi: 10.1080/07853890310014876. [DOI] [PubMed] [Google Scholar]
- 3.Lievens D, Eijgelaar WJ, Biessen EA, Daemen MJ, Lutgens E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost. 2009;102:206–214. doi: 10.1160/TH09-01-0029. [DOI] [PubMed] [Google Scholar]
- 4.Santilli F, Basili S, Ferroni P, Davì G. CD40/CD40L system and vascular disease. Intern Emerg Med. 2007;2:256–268. doi: 10.1007/s11739-007-0076-0. [DOI] [PubMed] [Google Scholar]
- 5.Li G, Sanders JM, Bevard MH, Sun Z, Chumley JW, Galkina EV, Ley K, Sarembock IJ. CD40 ligand promotes Mac-1 expression, leukocyte recruitment, and neointima formation after vascular injury. Am J Pathol. 2008;172:1141–1152. doi: 10.2353/ajpath.2008.070633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hristov M, Gümbel D, Lutgens E, Zernecke A, Weber C. Soluble CD40 ligand impairs the function of peripheral blood angiogenic outgrowth cells and increases neointimal formation after arterial injury. Circulation. 2010;121:315–324. doi: 10.1161/CIRCULATIONAHA.109.862771. [DOI] [PubMed] [Google Scholar]
- 7.Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6:114. doi: 10.1038/72162. [DOI] [PubMed] [Google Scholar]
- 8.Prasad KS, Andre P, Yan Y, Phillips DR. The platelet CD40L/GP IIb-IIIa axis in atherothrombotic disease. Curr Opin Hematol. 2003;10:356–361. doi: 10.1097/00062752-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Bishop GA, Moore CR, Xie P, Stunz LL, Kraus ZJ. TRAF proteins in CD40 signaling. Adv Exp Med Biol. 2007;597:131–151. doi: 10.1007/978-0-387-70630-6_11. [DOI] [PubMed] [Google Scholar]
- 10.Donners MM, Beckers L, Lievens D, Munnix I, Heemskerk J, Janssen BJ, Wijnands E, Cleutjens J, Zernecke A, Weber C, Ahonen CL, Benbow U, Newby AC, Noelle RJ, Daemen MJ, Lutgens E. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood. 2008;111:4596–4604. doi: 10.1182/blood-2007-05-088906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyahara T, Koyama H, Miyata T, Shigematsu H, Inoue J, Takato T, Nagawa H. Inflammatory signaling pathway containing TRAF6 contributes to neointimal formation via diverse mechanisms. Cardiovasc Res. 2004;64:154–164. doi: 10.1016/j.cardiores.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 12.Miyahara T, Koyama H, Miyata T, Shigematsu H, Inoue J, Takato T, Nagawa H. Inflammatory responses involving tumor necrosis factor receptor-associated factor 6 contribute to in-stent lesion formation in a stent implantation model of rabbit carotid artery. J Vasc Surg. 2006;43:592–600. doi: 10.1016/j.jvs.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Zirlik A, Bavendiek U, Libby P, MacFarlane L, Gerdes N, Jagielska J, Ernst S, Aikawa M, Nakano H, Tsitsikov E, Schönbeck U. TRAF-1, −2, −3, −5, and −6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40L in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:1101–1107. doi: 10.1161/ATVBAHA.107.140566. [DOI] [PubMed] [Google Scholar]
- 14.Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A, Seijkens T, Engel D, Cleutjens J, Keller AM, Naik SH, Boon L, Oufella HA, Mallat Z, Ahonen CL, Noelle RJ, de Winther MP, Daemen MJ, Biessen EA, Weber C. CD40 deficiency-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an anti inflammatory profile. J Exp Med. 2010;207:391–404. doi: 10.1084/jem.20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schober A. Chemokines in vascular dysfunction and remodeling. Arterioscler Thromb Vasc Biol. 2008;28:1950–1959. doi: 10.1161/ATVBAHA.107.161224. [DOI] [PubMed] [Google Scholar]
- 16.Mach F, Schonbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A. 1997;94:1931–1936. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schönbeck U, Mach F, Sukhova GK, Herman M, Graber P, Kehry MR, Libby P. CD40 ligation induces tissue factor expression in human vascular smooth muscle cells. Am J Pathol. 2000;156:7–14. doi: 10.1016/S0002-9440(10)64699-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schönbeck U, Mach F, Bonnefoy JY, Loppnow H, Flad HD, Libby P. Ligation of CD40 activates interleukin 1beta-converting enzyme (caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin 1beta. J Biol Chem. 1997;272:19569–19574. doi: 10.1074/jbc.272.31.19569. [DOI] [PubMed] [Google Scholar]
- 19.Krzesz R, Wagner AH, Cattaruzza M, Hecker M. Cytokine-inducible CD40 gene expression in vascular smooth muscle cells is mediated by nuclear factor kappaB and signal transducer and activation of transcription-1. FEBS Lett. 1999;453:191–196. doi: 10.1016/s0014-5793(99)00683-3. [DOI] [PubMed] [Google Scholar]
- 20.Stojakovic M, Krzesz R, Wagner AH, Hecker M. CD154-stimulated GM-CSF release by vascular smooth muscle cells elicits monocyte activation--role in atherogenesis. J Mol Med. 2007;85:1229–1238. doi: 10.1007/s00109-007-0225-y. [DOI] [PubMed] [Google Scholar]
- 21.Krieglstein CF, Anthoni C, Cerwinka WH, Stokes KY, Russell J, Grisham MB, Granger DN. Role of blood- and tissue-associated inducible nitric-oxide synthase in colonic inflammation. Am J Pathol. 2007;170:490–496. doi: 10.2353/ajpath.2007.060594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 23.Li G, Jin R, Norris RA, Zhang L, Yu S, Wu F, Markwald RR, Nanda A, Conway SJ, Smyth SS, Granger DN. Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis. 2010;208:358–365. doi: 10.1016/j.atherosclerosis.2009.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mistry P, Deacon K, Mistry S, Blank J, Patel R. NF-kappaB promotes survival during mitotic cell cycle arrest. J Biol Chem. 2004;279:1482–1490. doi: 10.1074/jbc.M310413200. [DOI] [PubMed] [Google Scholar]
- 25.Takada Y, Aggarwal BB. Flavopiridol inhibits NF-kappaB activation induced by various carcinogens and inflammatory agents through inhibition of IkappaBalpha kinase and p65 phosphorylation: abrogation of cyclin D1, cyclooxygenase-2, and matrix metalloprotease-9. J Biol Chem. 2004;279:4750–4759. doi: 10.1074/jbc.M304546200. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh S, Hayden MS. New regulators of NF-κB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 27.Brasier AR. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res. 2010;86:211–218. doi: 10.1093/cvr/cvq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferns GA, Avades TY. The mechanisms of coronary restenosis: insights from experimental models. Int J Exp Pathol. 2000;81:63–88. doi: 10.1046/j.1365-2613.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrés V. Control of vascular cell proliferation and migration by cyclin-dependent kinase signalling: new perspectives and therapeutic potential. Cardiovasc Res. 2004;63:11–21. doi: 10.1016/j.cardiores.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Mayr M, Xu Q. Smooth muscle cell apoptosis in arteriosclerosis. Exp Gerontol. 2001;36:969–987. doi: 10.1016/s0531-5565(01)00090-0. [DOI] [PubMed] [Google Scholar]
- 31.Miyahara T, Koyama H, Miyata T, Shigematsu H, Inoue J, Takato T, Nagawa H. Inflammatory signaling pathway containing TRAF6 contributes to neointimal formation via diverse mechanisms. Cardiovasc Res. 2004;64:154–164. doi: 10.1016/j.cardiores.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Miyahara T, Koyama H, Miyata T, Shigematsu H, Inoue J, Takato T, Nagawa H. Inflammatory responses involving tumor necrosis factor receptor-associated factor 6 contribute to in-stent lesion formation in a stent implantation model of rabbit carotid artery. J Vasc Surg. 2006;43:592–600. doi: 10.1016/j.jvs.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 33.Kawamura A, Baitsch D, Telgmann R, Feuerborn R, Weissen-Plenz G, Hagedorn C, Saku K, Brand-Herrmann SM, von Eckardstein A, Assmann G, Nofer JR. Apolipoprotein E interrupts interleukin-1beta signaling in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2007;27:1610–1617. doi: 10.1161/ATVBAHA.106.129957. [DOI] [PubMed] [Google Scholar]
- 34.Chandrasekar B, Mummidi S, Valente AJ, Patel DN, Bailey SR, Freeman GL, Hatano M, Tokuhisa T, Jensen LE. The pro-atherogenic cytokine interleukin-18 induces CXCL16 expression in rat aortic smooth muscle cells via MyD88, interleukin-1 receptor-associated kinase, tumor necrosis factor receptor-associated factor 6, c-Src, phosphatidylinositol 3-kinase, Akt, c-Jun N-terminal kinase, and activator protein-1 signaling. J Biol Chem. 2005;280:26263–26277. doi: 10.1074/jbc.M502586200. [DOI] [PubMed] [Google Scholar]
- 35.Doyon P, Servant MJ. Tumor necrosis factor receptor-associated factor-6 and ribosomal S6 kinase intracellular pathways link the angiotensin II AT1 receptor to the phosphorylation and activation of the IkappaB kinase complex in vascular smooth muscle cells. J Biol Chem. 2010;285:30708–30718. doi: 10.1074/jbc.M110.126433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopp CW, Hölzenbein T, Steiner S, Marculescu R, Bergmeister H, Seidinger D, Mosberger I, Kaun C, Cejna M, Horvat R, Wojta J, Maurer G, Binder BR, Breuss JM, Ecker RC, de Martin R, Minar E. Inhibition of restenosis by tissue factor pathway inhibitor: in vivo and in vitro evidence for suppressed monocyte chemoattraction and reduced gelatinolytic activity. Blood. 2004;103:1653–1661. doi: 10.1182/blood-2003-04-1148. [DOI] [PubMed] [Google Scholar]
- 37.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 38.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 39.Zeiffer U, Schober A, Lietz M, Liehn EA, Erl W, Emans N, Yan ZQ, Weber C. Neointimal smooth muscle cells display a proinflammatory phenotype resulting in increased leukocyte recruitment mediated by P-selectin and chemokines. Circ Res. 2004;94:776–784. doi: 10.1161/01.RES.0000121105.72718.5C. [DOI] [PubMed] [Google Scholar]
- 40.Furgeson SB, Simpson PA, Park I, Vanputten V, Horita H, Kontos CD, Nemenoff RA, Weiser-Evans MC. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation. Cardiovasc Res. 2010;86:274–282. doi: 10.1093/cvr/cvp425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. 2007;101:234–247. doi: 10.1161/CIRCRESAHA.107.151860b. [DOI] [PubMed] [Google Scholar]
- 42.Landry DB, Couper LL, Bryant SR, Lindner V. Activation of the NF-kappa B and I kappa B system in smooth muscle cells after rat arterial injury. Induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am J Pathol. 1997;151:1085–1095. [PMC free article] [PubMed] [Google Scholar]
- 43.Bu DX, Erl W, de Martin R, Hansson GK, Yan ZQ. IKKbeta-dependent NF-kappaB pathway controls vascular inflammation and intimal hyperplasia. FASEB J. 2005;19:1293–1295. doi: 10.1096/fj.04-2645fje. [DOI] [PubMed] [Google Scholar]
- 44.Chakrabarti S, Blair P, Freedman JE. CD40-40L signaling in vascular inflammation. J Biol Chem. 2007;282:18307–18317. doi: 10.1074/jbc.M700211200. [DOI] [PubMed] [Google Scholar]
- 45.Xia M, Li G, Ma J, Ling W. Phosphoinositide 3-kinase mediates CD40 ligand-induced oxidative stress and endothelial dysfunction via Rac1 and NADPH oxidase 2. J Thromb Haemost. 2010;8:397–406. doi: 10.1111/j.1538-7836.2009.03683.x. [DOI] [PubMed] [Google Scholar]
- 46.Hermann A, Schrör K, Weber AA. CD40 ligand (CD40L) does not stimulate proliferation of vascular smooth muscle cells. Eur J Cell Biol. 2002;81:213–221. doi: 10.1078/0171-9335-00240. [DOI] [PubMed] [Google Scholar]
- 47.Chai H, Aghaie K, Zhou W. Soluble CD40 ligand induces human coronary artery smooth muscle cells proliferation and migration. Surgery. 2009;146:5–11. doi: 10.1016/j.surg.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 48.Becker T, Hartl FU, Wieland F. CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J Cell Biol. 2002;158:1277–1285. doi: 10.1083/jcb.200208083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Kelly CG, Karttunen JT, Whittall T, Lehner PJ, Duncan L, MacAry P, Younson JS, Singh M, Oehlmann W, Cheng G, Bergmeier L, Lehner T. CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC-chemokines. Immunity. 2001;15:971–983. doi: 10.1016/s1074-7613(01)00242-4. [DOI] [PubMed] [Google Scholar]
- 50.Malik N, Francis SE, Holt CM, Gunn J, Thomas GL, Shepherd L, Chamberlain J, Newman CM, Cumberland DC, Crossman DC. Apoptosis and cell proliferation after porcine coronary angioplasty. Circulation. 1998;98:1657–1665. doi: 10.1161/01.cir.98.16.1657. [DOI] [PubMed] [Google Scholar]
- 51.Longo CR, Arvelo MB, Patel VI, Daniel S, Mahiou J, Grey ST, Ferran C. A20 protects from CD40-CD40 ligand-mediated endothelial cell activation and apoptosis. Circulation. 2003;108:1113–1118. doi: 10.1161/01.CIR.0000083718.76889.D0. [DOI] [PubMed] [Google Scholar]
- 52.Eliopoulos AG, Davies C, Knox PG, Gallagher NJ, Afford SC, Adams DH, Young LS. CD40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol Cell Biol. 2000;20:5503–5515. doi: 10.1128/mcb.20.15.5503-5515.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han DK, Haudenschild CC, Hong MK, Tinkle BT, Leon MB, Liau G. Evidence for apoptosis in human atherogenesis and in a rat vascular injury model. Am J Pathol. 1995;147:267–277. [PMC free article] [PubMed] [Google Scholar]
- 54.Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, Omata M, Makuuchi M, Hirata Y, Nagai R. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32:2097–2104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- 55.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Möpps B, Mericskay M, Gierschik P, Biessen EA, Weber C. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res. 2005;96:784–791. doi: 10.1161/01.RES.0000162100.52009.38. [DOI] [PubMed] [Google Scholar]
- 56.Clarke MC, Talib S, Figg NL, Bennett MR. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ Res. 2010;106:363–372. doi: 10.1161/CIRCRESAHA.109.208389. [DOI] [PubMed] [Google Scholar]
- 57.Jagadeesha DK, Lindley TE, Deleon J, Sharma RV, Miller F, Bhalla RC. Tempol therapy attenuates medial smooth muscle cell apoptosis and neointima formation after balloon catheter injury in carotid artery of diabetic rats. Am J Physiol Heart Circ Physiol. 2005;289:H1047–1053. doi: 10.1152/ajpheart.01071.2004. [DOI] [PubMed] [Google Scholar]
- 58.Beohar N, Flaherty JD, Davidson CJ, Maynard RC, Robbins JD, Shah AP, Choi JW, MacDonald LA, Jorgensen JP, Pinto JV, Chandra S, Klaus HM, Wang NC, Harris KR, Decker R, Bonow RO. Antirestenotic effects of a locally delivered caspase inhibitor in a balloon injury model. Circulation. 2004;109:108–113. doi: 10.1161/01.CIR.0000105724.30980.CD. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki T, Sawaki D, Aizawa K, Munemasa Y, Matsumura T, Ishida J, Nagai R. Kruppel-like factor 5 shows proliferation-specific roles in vascular remodeling, direct stimulation of cell growth, and inhibition of apoptosis. J Biol Chem. 2009;284:9549–9557. doi: 10.1074/jbc.M806230200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stachon P, Missiou A, Walter C, Varo N, Colberg C, Wolf D, Buchner M, von Zur Mühlen C, Zirlik K, Bode C, Zirlik A. Tumor necrosis factor receptor associated factor 6 is not required for atherogenesis in mice and does not associate with atherosclerosis in humans. PLoS One. 2010;5:e11589. doi: 10.1371/journal.pone.0011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.