

Abstract
ATP-dependent chromatin remodeling complexes, or remodelers, are large protein assemblies that use the energy from ATP hydrolysis to non-covalently modify the structure of nucleosomes, playing a central role in the regulation of chromatin dynamics. Our understanding of the mechanism and regulation of this remodeling activity and the diversity of products that chromatin remodelers can generate remains limited, partly because very little structural data is available on these challenging samples. Electron microscopy (EM) and single-particle approaches have made inroads into the structural characterization of a number of remodeling complexes. Here I will review the work done to date, focusing on functional insights we have gained from these structures.
Introduction
ATP-dependent chromatin remodeling complexes, also known as chromatin remodelers, remodeling complexes or simply remodelers, are large, multisubunit assemblies that play a central role in the regulation of chromatin dynamics. These complexes non-covalently modify the structure of nucleosomes, the basic unit of DNA packaging. All remodelers share the presence of a core catalytic subunit whose ATPase domain is a member of the superfamily 2 (SF2) of DNA translocases [1]. Remodelers, which are conserved from yeast to humans, are classified into four families based on the nature of additional domains/motifs present in their ATPase subunits. These families are commonly referred to as (1) SWI/SNF; (2) ISWI; (3) Mi-2/CHD and (4) SWR1/INO80 [2,3].
Remodelers are capable of generating a variety of reaction products, both in vivo and in vitro. Among the reactions identified so far are sliding of nucleosomes along DNA; partial unwrapping of nucleosomal DNA; ejection of H2A/H2B histone dimers; ejection of full octamers and exchange of H2A/H2B and H2A.Z/H2B dimers, with different remodelers giving rise to one or more of these products [2,3]. Specific reactions can show further specialization depending on their biological context and the remodeler(s) responsible for them; for example, members of the SWI/SNF family slide nucleosomes along DNA to randomize their positions while some members of the ISWI family slide nucleosomes to evenly space them [2,3]. How do remodeling complexes modify nucleosome structure? How are such a variety of outcomes achieved? How are these processes regulated? All of these questions remain open and await, among other data, structural information about the remodeling complexes and their interactions with their substrates.
Remodeling complexes pose a number of challenges to classical structural analysis by X-ray crystallography. First, they are large: the simpler complexes weigh in at around 300 kDa and the larger ones reach 1.5 MDa in higher eukaryotes [2,3]. Second, their composition can be quite complex, with some remodelers containing up to 15 different subunits [2,3]. Finally, except for the smaller chromatin remodelers, or core subassemblies of the larger ones, remodelers have been refractory to reconstitution, requiring instead their purification from native sources. These properties make them particularly suitable to electron microscopy (EM) and single-particle approaches (see Box 2), where small sample size is not as limiting and large molecular weight is an advantage. It is not surprising then that all the structures available today of full remodelers have come from EM. Yet, these complexes remain a major challenge for EM; all the reconstructions published so far have resolutions lower than 20Å. Despite this limitation, which hardware and software advances in EM should soon overcome, reconstructions of chromatin remodelers have not only provided us with the first look at these essential assemblies but have also yielded important functional insights into remodeling mechanisms.
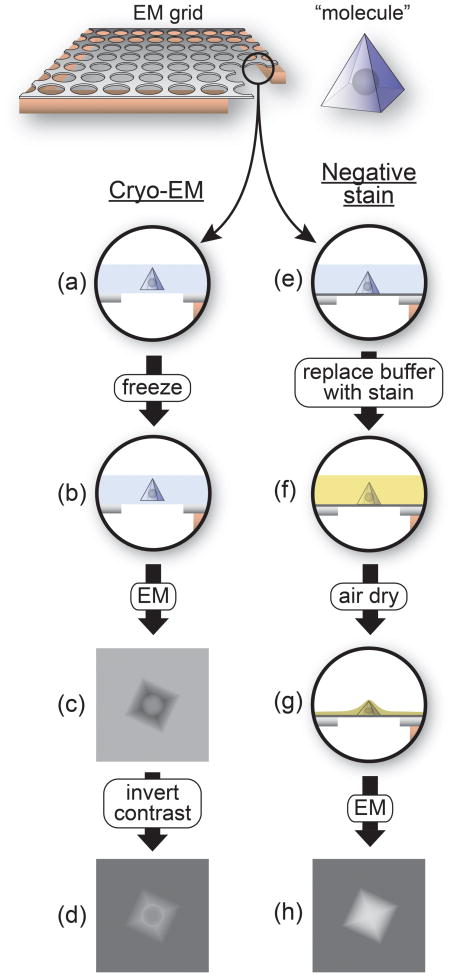
Box 1. Negatively stained and frozen-hydrated samples in electron microscopy (EM).
Single-particle reconstructions are typically generated from one of two types of samples: frozen-hydrated samples, commonly referred to as “cryo-EM”, or samples embedded in a heavy-atom stain, known as “negatively stained”. The accompanying figure illustrates the properties of these two types of samples. A small area of an EM grid is shown on the left at the top of the figure with a microfabricated carbon layer containing regular holes lying on top of the copper mesh. A corner has been cut out on the right to show the profile of the grid. The black circle shows the region used throughout the rest of the figure to illustrate the different steps of sample preparation. The “molecule” is shown on the right at the top as a pyramid containing an internal spherical density. (a-d) In a cryo-EM sample, a suspension of the macromolecule of interest is applied to an EM grid and, after blotting the excess solution to leave behind a very thin layer of liquid (typically 100 nm or less) (a), the grid is rapidly plunged into liquid ethane kept at liquid nitrogen temperature (b) [27]. Freezing occurs at a high enough rate that the aqueous solution is vitrified before crystalline ice can form. Imaging results in molecules that are darker than the surrounding vitrified ice (c) due to the higher density of biological macromolecules relative to aqueous buffers. For image processing, the contrast of the images is inverted to result in lighter macromolecules on a darker background (d). Cryo-EM is arguably the most physiological technique for imaging at high resolution as it allows molecules to be studied under the same conditions used in biochemical assays. The technique preserves the structural integrity of the sample and all high-resolution EM reconstructions have been obtained from frozen-hydrated samples [25,26]. The drawback of cryo-EM samples is their low contrast (compare (d) with (h)), which arises from the relatively small difference in scattering cross-section between the macromolecule and the surrounding solution. This is particularly challenging when analyzing a novel macromolecule where no information is available to validate the results from image processing. The solution to this problem is to embed the sample in a heavy-atom salt, where the much larger difference in scattering cross-section between the macromolecule and the surrounding heavy atoms leads to higher contrast. (e-h) Negative staining typically involves adsorbing the sample to an EM grid coated with a thin layer of amorphous carbon as the support (e, dark grey line) and replacing the aqueous buffer with a heavy-atom salt solution (such as uranyl acetate or formate) (f) [28]. After blotting excess stain, the sample is allowed to air dry, resulting in a “cast” of the heavy atom salt punctuated by holes created by the macromolecules (g). The images recorded represent the shapes of these holes rather than the internal structure of the macromolecules themselves (h), hence the “negative” in negative stain. Note that the spherical density inside the “molecule”, which is not accessible to the stain, is not present in the image obtained using negative stain (h) but does appear in the cryo-EM image (d). Imaging of negatively stained samples results in molecules that are lighter than the surrounding stain layer (h) due to the higher density of the heavy atom relative to atoms present in biological macromolecules. Besides the non-physiological conditions involved in negative staining, its major drawback is the often-severe flattening of the molecules that results from the air-drying of the sample (g). This flattening can make refinement of the initial reconstructions to higher resolution difficult if not impossible. A less widely used sample preparation technique that combines the high contrast of a heavy-atom solution with the preserving nature of vitrification is cryo-negative staining, where the sample is frozen in a solution containing a heavy atom component [29].
Here I will review the major features of all the remodeler structures published, focusing in particular on their functional implications. I will divide the discussion in two sections, corresponding to the two different families of remodelers for which structures are available (SWI/SNF and ISWI). There are currently no structures for chromatin remodelers in the Mi-2/CHD and SWR1/INO80 families.
SWI/SNF family chromatin remodelers
The SWI/SNF chromatin remodeling complexes are large assemblies composed of 8 to 14 different subunits and they often exceed 1 MDa in molecular weight [2,3]. These chromatin remodelers exist in two forms in most eukaryotes, with related “cores” containing the catalytic subunit and other conserved components [2,3]. The SWI/SNF family of remodelers can catalyze a number of different remodeling events, including octamer sliding and ejection, in a variety of chromatin contexts and for different purposes [2,3]. In contrast to ISWI remodeling complexes (see below) SWI/SNF remodelers are not involved in chromatin assembly.
Seven of the ten chromatin remodeler structures reported to date are for members of the SWI/SNF family, the first group of remodeling complexes identified [4-6]. Of these seven structures, two are of the yeast S.cerevisiae SWI/SNF complex [7,8], four are of its close relative the yeast chromatin remodeler RSC [9-12] and the remaining one is of the human RSC ortholog, PBAF [13].
SWI/SNF (SWItching and Sucrose Non-Fermenting) and RSC (Remodel the Structure of Chromatin) are the two forms of SWI/SNF remodeling complex found in the yeast S.cerevisiae. RSC, but not SWI/SNF, is essential and about 10 times more abundant [2,3]. Both complexes contain a core catalytic subunit characterized by the Swi2/Snf2 ATPase domain conserved among all chromatin remodeler families as well as two additional domains (a bromodomain and a helicase-SANT domain) that define this specific family of remodelers [2,3]. RSC and its orthologs in flies and humans stand out for containing six bromodomains, either in a single subunit (flies and humans) or distributed among several (yeast) [2,3]. Both RSC and SWI/SNF contain the actin-related proteins Arp7 and Arp9 [2,3]. The total number of subunits found in RSC and SWI/SNF is 11 and 17, respectively, and both complexes have molecular weights around 1.2 MDa [2,3]. RSC and SWI/SNF have been implicated in several DNA transactions, including replication, transcription and DNA repair but RSC appears to also play a role in aspects of chromosome segregation [2,3]. PBAF (Polybromo and BRG1-Associated Factors), the human ortholog of RSC, has been implicated in a number of roles, including differentiation, viral integration and tumor suppression [2].
Overall architecture
All four reconstructions of the RSC complex show a similar C-shaped arrangement of four large globular protein densities. These densities, typically linked by narrower connections, surround a large central cavity with dimensions compatible with those of a nucleosome (Figure 1A-D). Three of these structures [9-11] were solved from negatively stained samples (see Box 1) while the fourth [12*] was obtained by refining one of the original negative stain structures against data from a frozen-hydrated, or “cryo-EM”, sample (see Box 1). Different reconstruction methods have been used as well: two initial structures were generated using random conical tilt (RCT) [9,11] and one using the orthogonal tilt reconstruction (OTR) method [10] (see Box 2). The main difference among the negative stain reconstructions of RSC is that those obtained with the RCT method show the four densities occupying a single plane (Figure 1A and C) while they occupy different ones in the structure obtained using OTR (Figure 1B). The cryo-EM reconstruction of RSC, obtained by refining the structure in Figure 1A against data from a frozen-hydrated sample, shows increased dimensionality with densities appearing outside the single plane observed initially (compare Figures 1A and 1D).
Figure 1. Electron microscopy reconstructions of SWI/SNF family chromatin remodelers.

This figure shows reconstructions for three remodelers in the SWI/SNF family: RSC (S.cerevisiae) in (A) [9]; (B) [10]; (C) [11] and (D) [12*]; SWI/SNF (S.cerevisiae) in (F) [7] and PBAF (human) in (E) [13]. The figure is divided into three panels, one for each type of remodeler. The reconstructions shown in shades of blue were obtained from negatively stained samples (Box 1). Those shown in yellow (D) are from frozen-hydrated samples. The circular images are experimental “class averages” (Box 2) corresponding to the view of the reconstruction shown below them. The grey arrows point to densities that were seen to be conformationally flexibile in the different reconstructions. The orange asterisk indicates the proposed binding sites for the nucleosome in the different reconstructions. [Sth1 ] marks the densities where the ATPase subunit of RSC (Sth1) was predicted to be located (B and D). The blue density in the reconstruction on the right in (D) is the result of a difference map calculated between reconstructions with and without the nucleosome present [12*]. Scale bars are shown for each panel. Images adapted with permission from Macmillan Publishers Ltd: Nature Structural Biology (2003) [7]; Nature Structural and Molecular Biology (2007) [12*]; from Elsevier (2005) [13]; from the American Society for Biochemistry and Molecular Biology (2007) [11]; Copyright (2002) [9] and (2007) [10], National Academy of Sciences, USA.
Highlights.
Remodelers are large protein complexes that modify the structure of nucleosomes.
Electron microscopy (EM) has provided the only structures of full remodelers.
EM reconstructions have provided insights into nucleosome-binding sites
EM reconstructions have provided insights into remodeling mechanisms.
The PBAF complex, the human ortholog of RSC, also showed a C-shaped architecture (Figure 1E). Although some of its densities are more continuous with each other, there are still four major features arranged around a large central cavity (Figure 1E) [13].
Surprisingly, the structures available for SWI/SNF bear no similarity to RSC, despite the presence of several homologous subunits [2,3] (Figure 1F). This striking dissimilarity, observed both in the original negative stain reconstruction of SWI/SNF [7] (Figure 1F) and a subsequent refinement of this structure using data from frozen-hydrated samples [8], remains puzzling. In fact, one of the characteristic views (“class averages”, see Box 2) reported for the SWI/SNF complex (top of Figure 1F) shows significant similarity to those reported for RSC (top of Figures 1A; B; C and D), with the characteristic four large densities surrounding a central cavity. The similarity is even stronger between the SWI/SNF and PBAF class averages (compare the top of Figures 1E and F). It remains to be seen whether the apparent discrepancy between the 2D images and the 3D structures is the result of truly different structures giving rise to similar-looking views or an indication that actual structural similarities were lost during 3D reconstruction due to the different approaches used.
Nucleosome binding
The prominent cavities seen in the earlier RSC and PBAF reconstructions (Figure 1A-C and E) seemed sterically well suited to fit a nucleosome and were proposed to be its binding site [9-11,13]. In the case of RSC, the large potential interaction area between the remodeling complex and the nucleosome would account for the protection provided by the former in DNase I footprinting assays [9,14]. Direct evidence for nucleosome binding in the cavity came first in the form of 2D image analysis of PBAF-nucleosome complexes [13] and eventually in 3D when the first cryo-EM (see Box 1) reconstructions of both RSC and a RSC-nucleosome complex were obtained by refining the original negative stain structure (Figure 1A) [9] using cryo-EM data (Figure 1D) [12*]. The extra density observed in the RSC-nucleosome reconstruction (Figure 1D, right) could not account for a full nucleosome, showing partial density for both the DNA and one of the H2A/H2B dimers [12*]. It was suggested that this partially resolved density might be a result of the nucleosome's structure being altered by its interactions with RSC, even under the no-ATP conditions used for this work [12*]. The general orientation of the nucleosome in the cryo-EM reconstruction was similar to that proposed in previous models [9-11]. Furthermore, this orientation suggested that the ATPase subunit of RSC, Sth1, would be located in the upper-left density feature in Figure 1D, in agreement with the location proposed for a different RSC reconstruction, based on modeling of a RSC-nucleosome complex (Figure 1B) [10].
The SWI/SNF complex lacks any cavities and nucleosome binding was proposed to occur on a large depression on its surface whose dimensions could accommodate a nucleosome [7,8] (Figure 1F).
Conformational flexibility
A remarkable common feature of the RSC and PBAF reconstructions is the conformational flexibility exhibited by these chromatin remodelers. In every single reconstruction a domain was seen to adopt two or more conformations relative to the rest of the complex. In the case of RSC, flexibility was observed in the bottom domain in the reconstructions shown in Figure 1A, C and D [9,11,12] and in the right-most domain in the reconstruction shown in Figure 1B [10]. In the case of PBAF, the top-left density shown in Figure 1E was the flexible one [13]. (In all cases the flexible density is indicated with a grey arrow.)
Although the biological significance of this conformational plasticity remains a largely unexplored question, some evidence suggests it may be involved in the regulation of remodeling. The “closed” conformation of RSC shown in Figure 1C, where the flexible bottom density moved closer to the others, was seen to be favored in the presence of peptides mimicking an acetylated histone 3 tail [11], a known activator of remodeling by RSC [15,16]. It was suggested that this closing of the structure may increase the affinity of RSC for the nucleosome in response to the appropriate acetylation marks [11].
ISWI family chromatin remodelers
ISWI chromatin remodelers are significantly smaller than those in the SWI/SNF family, containing only 2-4 subunits and having much lower molecular weights [2,3]. Some ISWI family members, though not all, have the ability to evenly space nucleosomes in an array, in contrast to the randomizing activity of SWI/SNF remodelers; as a result of this property, these ISWI remodeling complexes promote chromatin condensation and gene silencing [2,3]. ISWI remodelers are characterized by the presence of one of two different catalytic subunits, both containing the conserved Swi2/Snf2 ATPase domain as well as the SANT and SLIDE domains that define the family [2,3]. Higher eukaryotes form several ISWI-family remodeling complexes by using a variety of accessory subunits [2,3]. In addition to chromatin assembly, ISWI remodelers have been implicated in both replication and transcriptional regulation [2,3].
The three available reconstructions for members of the ISWI family of chromatin remodelers are from subassemblies of the yeast ISW1a (Imitation SWitch 1a) and human ACF (ATP-dependent Chromatin assembly Factor) remodelers and for the entire human CHRAC (CHRomatin Accessibility Complex) complex. The native ACF and ISW1a complexes are made up of two subunits each and have molecular weights of approximately 310 kDa and 220 kDa, respectively [2,3]. CHRAC shares two subunits with ACF (the catalytic subunit plus an accessory one) and contains two additional small components for a total molecular weight of approximately 345 kDa [2,3]. ACF has been shown to play roles in replication; ISW1a in transcription and CHRAC in chromatin assembly [2,3].
Some of the most exciting results in terms of the mechanistic insights they have provided have come from recent reconstructions of members of this family.
Nucleosome binding
Two structures of ISWI family members bound to nucleosomes have been solved: (1) SNF2h, the catalytic subunit of the human ACF complex [17**] and (2) a sub-complex of the yeast ISW1a chromatin remodeler, termed ISW1a(ΔATPase), consisting of the catalytic subunit Isw1 without its ATPase domain and its accessory subunit Ioc3 [18**] (Figure 2A and C). Although the full complexes will have more interactions with the nucleosome than those seen in the current structures, it is unlikely that these will be as extensive as those observed or modeled for SWI/SNF remodelers given the much larger size of the latter.
Figure 2. Electron microscopy reconstructions of ISWI family chromatin remodelers.

This figure shows reconstructions for three remodelers in the ISWI family: (A) SNF2h, the ATPase subunit of ACF (human) [17**]; (C) a version of ISW1a (S.cerevisiae) lacking the ATPase domain of Isw1, its calytic subunit [18**] and (E) CHRAC (human) [20]. The figure also illustrates mechanisms proposed for SNF2h/ACF (B) and ISW1a (D) based on structural and other biochemical data [18**]. The figure is divided into three panels, one for each type of remodeling complex. The structures of SNF2h (ACF panel) and CHRAC were obtained from negatively stained samples while those of ISW1a(ΔATPase) are from frozen-hydrated samples (Box 1). The circular images are experimental “class averages” (Box 2), corresponding to the view of the reconstruction shown below them. (A, bottom row) Two views are shown of a reconstruction consisting of two SNF2h monomers bound to a nucleosome. The atomic structure of the nucleosome (PDB 1KX5) was fitted into the density by the authors [17**]. The SNF2h monomers bind to an area surrounding the superhelical locations (SHL) −2 and +2. (B) A proposed mechanism for ACF's nucleosome centering activity (compare (1) with (6)) (adapted from [17**]). The histone octamer is depicted as a yellow sectioned disk and DNA as black/grey tubing. The orange lines represent the histone H4 N-terminal tails, which have been shown to be recognized by and modulate the activity of ISWI-type ATPases ([35-41]). SNF2h is shown in either light or dark blue, representing inactive (nucleotide-free) and active (nucleotide-bound) states, respectively. The expected location of the ATPase domain in the active monomer is marked with an asterisk. Red arrows indicate the direction of DNA motion. Green arrows mark the position, and direction, of DNA translocation within the nucleosome. (2) Two ACF monomers bind to a nucleosome; if a monomer containing ATP binds to its cognate histone H4 N-terminal tail and interacts with a longer DNA linker, its translocation activity is stimulated, leading to DNA being drawn into the nucleosome from that linker. (3) DNA diffuses through the nucleosome and emerges at the other end. (4) As the opposite linker becomes longer, the translocation activity of the second SNF2h is stimulated, resulting in translocation in the opposite direction (5). This tug-of-war between the two SNF2h monomers results in a histone octamer centered in its DNA (6). The contact between the two SNF2h monomers is hypothetical and was only included to symbolize communication between them. An alternative mechanism is discussed in [17**]. (C, bottom row) Two different structures were solved from frozen-hydrated samples containing nucleosomes and “ISW1a(ΔATPase)”, a complex consisting of the C-terminal HAND-SANT-SLIDE (HSS) portion of the ATPase Isw1 and the accessory subunit Ioc3 . In the first structure (left), the nucleosome contained DNA linkers of 45 and 29 base pairs. A density was seen bridging these two DNA extensions where ISW1a(ΔATPase) could be docked. The second structure (right) was obtained with nucleosomes containing a single 45 bp linker. In this case, a two-fold symmetric structure was observed, mediated by two copies of ISW1a(ΔATPase) that had adopted a new orientation relative to the nucleosome. The nucleosome dyad axis is shown with a green rod; its superhelical axis with a red rod and the two-fold dyad axis of the dimeric particle with a black rod. (D) The two modes of interaction between ISW1a(ΔATPase) and a nucleosome observed in the cryo-EM reconstructions as well as the interactions observed in the ISW1a(ΔATPase)-DNA co-crystal structure were used to propose a model for ISW1a's ability to take an array of randomly distributed nucleosomes (1) and evenly space them (5). The depiction of nucleosomes and the meaning of the red and green arrows are the same as in (B). Isw1 is represented in blue, with its HAND-SANT-SLIDE portion binding to linker DNA and its ATPase domain (not present in the cryo-EM reconstructions) modeled as bound at superhelical location (SHL) −2/+2, as previously mapped ([19,42,43]). The accessory Ioc3 subunit is shown in orange. The yellow arrows indicate nucleosome movement. (2) ISW1a engages its substrate with a number of interactions: the ATPase domain binds to the nucleosome where translocation will occur (the “mobile” nucleosome); the HSS portion of Isw1 binds to the DNA linking this nucleosome to the neighboring one (the “static” nucleosome, where no translocation occurs) and the Ioc3 subunit bridges this same DNA to the distal linker in the static nucleosome while at the same time interacting with HSS. (2 and 3) The Isw1 ATPase translocates DNA from the linker joining the mobile and static nucleosomes and this DNA emerges at the distal linker in the mobile nucleosome [brown segment in (4)]. Once the static nucleosome reaches the Ioc3 subunit (4) further translocation is blocked, resulting in a fixed DNA linker length between the two nucleosomes (5). (E) A view of the reconstruction of CHRAC. The dashed circle represents the contour of a nucleosome and indicates the proposed interaction between CHRAC and one of the disk-like histone faces of the nucleosome. Images adapted with permission from Macmillan Publishers Ltd: Nature (2009) [17**] and (2011) [18**].
In the case of ACF, a small remodeler consisting of the ATPase SNF2h and the accessory subunit Acf1, the reconstruction, obtained from negatively stained samples (Box 1) using the RCT method (Box 2), revealed two SNF2h monomers bound to the nucleosome, following its pseudo two-fold symmetry (Figure 2A). As it had been predicted from biochemical studies with the related yeast ISW2 ATPase [19], each SNF2h monomer bound two turns of DNA away from the dyad axis of the nucleosome, at the superhelical (SHL) positions −2 or +2. The C-terminal portion of SNF2h containing the HAND-SANT-SLIDE domain, a signature motif of ISWI family ATPases involved in binding both histones and DNA [2,3], was not visible in the structure and neither were the DNA linkers to which it binds [17**]. The SNF2h-nucleosome reconstruction was accompanied by extensive biochemical and biophysical studies that indicated that several properties of SNF2h (and ACF) were dependent on its nucleotide state: its interaction with the histone H4 N-terminal tail, a known ligand; the binding of the ATPase to the SHL +/− 2 positions and cooperativity of nucleosome binding by the two SNF2h monomers [17**]. The combination of the structure and the biochemical and biophysical studies suggested a mechanism that accounts for ACF's ability to generate arrays of evenly spaced nucleosomes (discussed in the next section).
The work on ISW1a involved a series of crystallographic and cryo-EM structures: crystal structures were solved for ISW1a(ΔATPase) alone and bound to DNA and cryo-EM reconstructions were obtained for ISW1a(ΔATPase) bound to two types of nucleosomes differing in their DNA linkers [18**]. The most striking observation was that ISW1a(ΔATPase) interacted with the nucleosome in different ways depending on these linkers. The sample containing a nucleosome with two linkers (of 45 and 29 bp) resulted in a monomeric ISW1a(ΔATPase)-nucleosome species (Figure 2C, left side) while that containing a nucleosome with a single linker (of 45 bp) resulted in a dimeric species where two nucleosomes were bridged by two copies of ISW1a(ΔATPase) (Figure 2C, right side). Surprisingly, in this configuration ISW1a(ΔATPase) adopted an orientation relative to its proximal nucleosome reversed from that seen in the monomeric species (Figure 2C, bottom). In both structures, the interactions between ISW1a(ΔATPase) and the nucleosome are restricted to the linker DNA and the region immediately adjacent to the DNA entry site (Figure 2C, bottom).
The cryo-EM reconstructions suggested that ISW1a(ΔATPase) was capable of two modes of interaction with a nucleosome and, because of the bridging observed in both the ISW1a(ΔATPase)-DNA co-crystal structure and the dimeric cryo-EM reconstruction (Figure 2C, right), that it could bind two DNA linkers at once. Based on these and other biochemical data [18**], a mechanism has been proposed for ISW1a's nucleosome-spacing activity that uses a fundamentally different strategy from that of ACF (discussed in the next section).
The third member of the ISWI family for which a structure has been reported is human CHRAC [20] (Figure 2E). This reconstruction was obtained in the absence of a nucleosome, from a negatively stained sample (Box 1) and using in part a common lines approach to generate the initial model (Box 2). The CHRAC remodeling complex contains four different subunits: SNF2h; Acf1; CHRAC15 and CHRAC17 [2,3]. The reconstruction shows a solid volume with no cavities or holes (Figure 2E). Based on the surface complementarity between the reconstruction and a nucleosome and other steric considerations, it was suggested that one of the larger surfaces of the complex would bind to the nucleosome via one of its flat histone faces (Figure 2E, dashed circle). This contrasts with the mode of binding observed with SNF2h and ISW1a(ΔATPase), where contacts are mainly limited to the DNA edges rather than the flat histone faces (Figure 2A and C). Given that ACF and CHRAC share both SNF2h and Acf1, it is most likely that they will have similar interactions with their substrate. Which portion of the CHRAC complex is involved in interacting with the nucleosome therefore remains to be established.
Different strategies for spacing nucleosomes
The remodeling mechanisms proposed for SNF2h [17**] and ISW1a [18**] both achieve the same goal—to space nucleosomes evenly—but involve very different strategies (Figure 2B and D).
In the SNF2h model, two SNF2h monomers bind to each nucleosome but only one of them contains ATP; this monomer in turn binds to its cognate histone H4 N-terminal tail and to its corresponding DNA linker (Figure 2B, (2) – (5)). One of the cornerstones of this model is that longer DNA linkers stimulate the ATPase/translocation activity of SNF2h[21-24]. This results in a tug-of-war between the two SNF2h monomers; while monomers continuously sample their DNA linkers, DNA is translocated mainly from the longer one as a consequence of its activating its associated ATPase. The end product of this arrangement is a nucleosome with DNA linkers of similar length or an array of evenly spaced nucleosomes (Figure 2B, (6)).
In contrast to the competition model proposed for SNF2h, the ISW1a model involves a direct measurement of the distance between nucleosomes by the remodeling complex, using its own physical dimensions as the ruler (Figure 2D). This is accomplished by having both the HAND-SANT-SLIDE domain of the Isw1 ATPase as well as its associated subunit Ioc3 bind to the linker connecting the nucleosome where DNA translocation takes place to its neighbor, which is being drawn closer as a consequence of this translocation (Figure 2D, (2) – (4)). Once this linker reaches the target length, ISW1a becomes its own physical barrier to further translocation (Figure 2D, (4)).
Concluding remarks
Chromatin remodeling complexes are challenging samples for structural characterization due to a combination of size, complexity, conformational flexibility and a limitation in the amount of material available due to the frequent need to purify them from native sources. Electron microscopy and single-particle methods have made inroads into this area by providing the first views of the general architecture of several of these large assemblies. In some cases, particularly with the ISWI family chromatin remodelers, a combination of revealing structures and functional assays has yielded important insights into remodeling mechanisms.
Among the landmarks in EM reconstruction of remodeling complexes one would hope to see in the near future are: (1) Obtaining structures that reach “molecular” resolution (better than 10Å) where secondary structure elements become visible and make structure validation easier by docking available crystal structures into EM densities; (2) In the case of the larger complexes, mapping of the different components into the EM maps where their locations, relative to each other and the nucleosomal substrate, will provide new functional information; (3) Analyzing, qualitatively and quantitatively, under cryo-EM conditions, the conformational flexibility some of these complexes appear to exhibit and its dependence on nucleotide state and substrate characteristics to begin elucidating its functional significance and (4) Generating the first reconstructions of chromatin remodelers in the Mi-2/CHD and SWR1/INO80 families. The accelerating pace of technical improvements in EM, both at the hardware and software levels, which have already made atomic resolution a reality [25,26] should make many of these goals attainable.
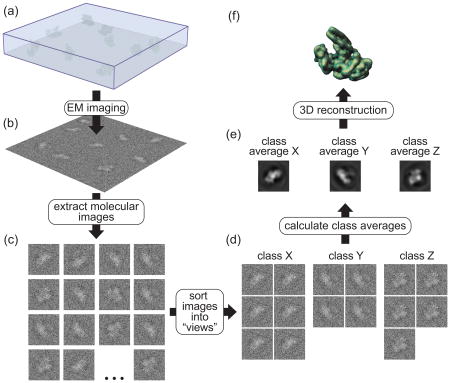
Box 2. Three-dimensional (3D) reconstruction from electron microscopy (EM) images.
The process of 3D reconstruction from EM images can be roughly divided into three steps: (1) data acquisition, (2) 2D image processing and (3) 3D reconstruction. The accompanying figure illustrates, schematically, these different steps. A cryo-EM sample (a) (see Box 1) is imaged in the electron microscope and micrographs are recorded (b). Images of individual macromolecules are extracted from the micrographs (c) and, through an iterative process, are sorted out into groups (“classes”) representing the different characteristic views of the molecule (d). The images within a class can now be averaged to generate “class averages” that have a much higher signal-to-noise ratio (e). In the final step, 3D reconstructions (f) are generated either from each individual class average or by combining a number of class averages together. A final step, the refinement of an initial 3D reconstruction to higher resolution, is not shown in this figure. How can 3D structures be obtained from 2D images? The combination of the large depth of field of EMs and the thinness of the samples used means that the images recorded are projections, i.e. an integration of the density of the sample along the direction of the electron beam. As a consequence, structural EM can take advantage of the central section theorem, which states that the Fourier transform of a projection is equivalent to a central section, perpendicular to the projection direction, through the 3D Fourier transform of the structure giving rise to that projection (see [30,31] for in-depth discussions of this principle). Stated in a different way, any EM image of a macromolecule is equivalent to sampling a section of the 3D Fourier transform of that molecule. It follows that if we have images representing many views of the molecule we can fully sample its 3D Fourier transform and therefore obtain its structure through an inverse Fourier transform. The main challenge we face is determining the relative orientations of all the images collected, which arise from macromolecules often adopting (near) random orientations on the EM grid. Different reconstruction methods use different approaches to get this information. I will briefly describe the three methods available to obtain 3D reconstructions of single particles (“single particles” refers to molecules that do not arrange themselves into higher-order symmetric structures).
(1) Angular reconstitution (AR) [32], sometimes also referred to as “common lines”. AR is a mathematical method that relies directly on the central section theorem: given that every projection is equivalent to a central section through the 3D Fourier transform of the molecule, it follows that the Fourier transforms of any two projections must share one line in common, where they intersect. This line can be found analytically. AR uses a real space approach to find this orientation, relying on the fact that any two 2D projections of a given molecule will give rise to the same 1D projection when projected in a direction perpendicular to their common line. This common line can therefore be found by rotating one projection relative to the other until their 1D projections match. AR typically uses class averages (e) as the input data due to their higher signal-to-noise ratio. One of the main challenges with AR is that its implementation assumes that the projections whose orientations must be determined are different views of the same molecular species, an assumption that breaks down when analyzing samples that suffer from any type of heterogeneity, unless independent information is available to sort those species out.
(2) Random conical tilt (RCT) [33]. RCT is a geometric method that relies on our ability to tilt the stage of an electron microscope and obtain two views of each molecule present in the sample, with these views related by an angle that is set (and thus known) by the user. An image is typically collected first at a high tilt (50°-60°), followed by a second one, of the same area, taken at 0°. (In the figure, this means we would have a second set of molecular images where each is a tilted counterpart of the images shown in (c)). Individual molecular images from the 0° micrographs are used to generate class averages (e). As a result of the rotations required to bring images into alignment within a given class average, the high-tilt counterparts of the 0° images effectively fan out in space and sample, due to the central section theorem, a large portion of the 3D Fourier transform of the molecule. A major limitation of RCT lies in our inability to collect data beyond a certain tilt angle of the stage. As a result, a cone-shaped portion of Fourier space, determined by the tilt angle used, contains no information. This “missing cone” problem results in artifacts in the final reconstruction.
(3) Orthogonal tilt reconstruction (OTR) [34]. OTR uses a modified data collection scheme to avoid the missing cone problem. The first image is collected at 45° and the second one at −45°, effectively obtaining two views of each molecule related by a 90° angle. Once one set of images is aligned and sorted out into the characteristic views of the sample, the other images effectively constitute a set of 90° views, which fully sample the 3D Fourier transform of the molecule and thus give rise to reconstructions with no missing data.
Acknowledgments
I thank Samara Reck-Peterson, Rogelio Hernandez-Lopez and Bret Redwine for helpful comments on the manuscript. I gratefully acknowledge support form the National Institutes of Health (R01 GM092895A) and a Research Fellowship from the Alfred P. Sloan Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annual review of biochemistry. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 3.Gangaraju VK, Bartholomew B. Mechanisms of ATP dependent chromatin remodeling. Mutat Res. 2007;618:3–17. doi: 10.1016/j.mrfmmm.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khavari PA, Peterson CL, Tamkun JW, Mendel DB, Crabtree GR. BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature. 1993;366:170–174. doi: 10.1038/366170a0. [DOI] [PubMed] [Google Scholar]
- 5.Cairns BR, Kim YJ, Sayre MH, Laurent BC, Kornberg RD. A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5, and SNF6 gene products isolated from yeast. Proc Natl Acad Sci U S A. 1994;91:1950–1954. doi: 10.1073/pnas.91.5.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peterson CL, Dingwall A, Scott MP. Five SWI/SNF gene products are components of a large multisubunit complex required for transcriptional enhancement. Proc Natl Acad Sci U S A. 1994;91:2905–2908. doi: 10.1073/pnas.91.8.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith CL, Horowitz-Scherer R, Flanagan JF, Woodcock CL, Peterson CL. Structural analysis of the yeast SWI/SNF chromatin remodeling complex. Nat Struct Biol. 2003;10:141–145. doi: 10.1038/nsb888. [DOI] [PubMed] [Google Scholar]
- 8.Dechassa ML, Zhang B, Horowitz-Scherer R, Persinger J, Woodcock CL, Peterson CL, Bartholomew B. Architecture of the SWI/SNF-Nucleosome Complex. Mol Cell Biol. 2008 doi: 10.1128/MCB.00693-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asturias FJ, Chung WH, Kornberg RD, Lorch Y. Structural analysis of the RSC chromatin-remodeling complex. Proc Natl Acad Sci U S A. 2002;99:13477–13480. doi: 10.1073/pnas.162504299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leschziner AE, Saha A, Wittmeyer J, Zhang Y, Bustamante C, Cairns BR, Nogales E. Conformational flexibility in the chromatin remodeler RSC observed by electron microscopy and the orthogonal tilt reconstruction method. Proc Natl Acad Sci U S A. 2007;104:4913–4918. doi: 10.1073/pnas.0700706104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skiniotis G, Moazed D, Walz T. Acetylated histone tail peptides induce structural rearrangements in the RSC chromatin remodeling complex. J Biol Chem. 2007;282:20804–20808. doi: 10.1074/jbc.C700081200. [DOI] [PubMed] [Google Scholar]
- 12*.Chaban Y, Ezeokonkwo C, Chung WH, Zhang F, Kornberg RD, Maier-Davis B, Lorch Y, Asturias FJ. Structure of a RSC-nucleosome complex and insights into chromatin remodeling. Nat Struct Mol Biol. 2008;15:1272–1277. doi: 10.1038/nsmb.1524. This paper presented the first cryo-EM reconstruction of a remodeling complex bound to a nucleosome, confirming that the large central cavity observed in RSC is the nucleosome binding site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leschziner AE, Lemon B, Tjian R, Nogales E. Structural studies of the human PBAF chromatin-remodeling complex. Structure (Camb) 2005;13:267–275. doi: 10.1016/j.str.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Saha A, Wittmeyer J, Cairns BR. Chromatin remodeling through directional DNA translocation from an internal nucleosomal site. Nat Struct Mol Biol. 2005 doi: 10.1038/nsmb973. [DOI] [PubMed] [Google Scholar]
- 15.Kasten M, Szerlong H, Erdjument-Bromage H, Tempst P, Werner M, Cairns BR. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. Embo J. 2004;23:1348–1359. doi: 10.1038/sj.emboj.7600143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.VanDemark AP, Kasten MM, Ferris E, Heroux A, Hill CP, Cairns BR. Autoregulation of the rsc4 tandem bromodomain by gcn5 acetylation. Mol Cell. 2007;27:817–828. doi: 10.1016/j.molcel.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17**.Racki LR, Yang JG, Naber N, Partensky PD, Acevedo A, Purcell TJ, Cooke R, Cheng Y, Narlikar GJ. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature. 2009;462:1016–1021. doi: 10.1038/nature08621. The first view of an ISWI ATPase bound to a nucleosome and direct evidence that ACF functions as a dimer, leading to a mechanistic model for its nucleosome spacing activity, distinct from that proposed for ISW1a (see [18**]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Yamada K, Frouws TD, Angst B, Fitzgerald DJ, DeLuca C, Schimmele K, Sargent DF, Richmond TJ. Structure and mechanism of the chromatin remodelling factor ISW1a. Nature. 2011;472:448–453. doi: 10.1038/nature09947. A combination of crystallographic and cryo-EM studies of the ISW1a remodeling complex suggesting that its substrate is a dinucleosome. A mechanism for nucleosome spacing, distinct from that proposed for ACF (see [17**]) is presented. [DOI] [PubMed] [Google Scholar]
- 19.Kagalwala MN, Glaus BJ, Dang W, Zofall M, Bartholomew B. Topography of the ISW2-nucleosome complex: insights into nucleosome spacing and chromatin remodeling. Embo J. 2004;23:2092–2104. doi: 10.1038/sj.emboj.7600220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu M, Zhang YB, Qian L, Brinas RP, Kuznetsova L, Hainfeld JF. Three-dimensional structure of human chromatin accessibility complex hCHRAC by electron microscopy. J Struct Biol. 2008;164:263–269. doi: 10.1016/j.jsb.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem. 2006;281:16279–16288. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitehouse I, Stockdale C, Flaus A, Szczelkun MD, Owen-Hughes T. Evidence for DNA translocation by the ISWI chromatin-remodeling enzyme. Mol Cell Biol. 2003;23:1935–1945. doi: 10.1128/MCB.23.6.1935-1945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang JG, Madrid TS, Sevastopoulos E, Narlikar GJ. The chromatin-remodeling enzyme ACF is an ATP-dependent DNA length sensor that regulates nucleosome spacing. Nat Struct Mol Biol. 2006;13:1078–1083. doi: 10.1038/nsmb1170. [DOI] [PubMed] [Google Scholar]
- 24.Zofall M, Persinger J, Kassabov SR, Bartholomew B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat Struct Mol Biol. 2006;13:339–346. doi: 10.1038/nsmb1071. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Y, Walz T. The advent of near-atomic resolution in single-particle electron microscopy. Annu Rev Biochem. 2009;78:723–742. doi: 10.1146/annurev.biochem.78.070507.140543. [DOI] [PubMed] [Google Scholar]
- 26.Grigorieff N, Harrison SC. Near-atomic resolution reconstructions of icosahedral viruses from electron cryo-microscopy. Curr Opin Struct Biol. 2011;21:265–273. doi: 10.1016/j.sbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobro MJ, Melanson LA, Jensen GJ, McDowall AW. Plunge freezing for electron cryomicroscopy. Methods in enzymology. 2010;481:63–82. doi: 10.1016/S0076-6879(10)81003-1. [DOI] [PubMed] [Google Scholar]
- 28.Ohi M, Li Y, Cheng Y, Walz T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Carlo S, Stark H. Cryonegative staining of macromolecular assemblies. Methods in enzymology. 2010;481:127–145. doi: 10.1016/S0076-6879(10)81006-7. [DOI] [PubMed] [Google Scholar]
- 30.Frank J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. 2nd. New York, New York: Oxford University Press; 2006. [Google Scholar]
- 31.Glaeser RM, Downing KH, DeRosier D, Chiu W, Frank J. Electron crystallography of biological macromolecules. 1st. New York, New York: Oxford University Press; 2007. [Google Scholar]
- 32.Van Heel M. Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy. 1987;21:111–123. doi: 10.1016/0304-3991(87)90078-7. [DOI] [PubMed] [Google Scholar]
- 33.Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J Microsc. 1987;146(Pt 2):113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- 34.Leschziner AE, Nogales E. The orthogonal tilt reconstruction method: an approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J Struct Biol. 2006;153:284–299. doi: 10.1016/j.jsb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 35.Clapier CR, Langst G, Corona DF, Becker PB, Nightingale KP. Critical role for the histone H4 N terminus in nucleosome remodeling by ISWI. Mol Cell Biol. 2001;21:875–883. doi: 10.1128/MCB.21.3.875-883.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clapier CR, Nightingale KP, Becker PB. A critical epitope for substrate recognition by the nucleosome remodeling ATPase ISWI. Nucleic Acids Res. 2002;30:649–655. doi: 10.1093/nar/30.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dang W, Kagalwala MN, Bartholomew B. Regulation of ISW2 by concerted action of histone H4 tail and extranucleosomal DNA. Mol Cell Biol. 2006;26:7388–7396. doi: 10.1128/MCB.01159-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fazzio TG, Gelbart ME, Tsukiyama T. Two distinct mechanisms of chromatin interaction by the Isw2 chromatin remodeling complex in vivo. Mol Cell Biol. 2005;25:9165–9174. doi: 10.1128/MCB.25.21.9165-9174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreira H, Flaus A, Owen-Hughes T. Histone modifications influence the action of Snf2 family remodelling enzymes by different mechanisms. J Mol Biol. 2007;374:563–579. doi: 10.1016/j.jmb.2007.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamiche A, Kang JG, Dennis C, Xiao H, Wu C. Histone tails modulate nucleosome mobility and regulate ATP-dependent nucleosome sliding by NURF. Proc Natl Acad Sci U S A. 2001;98:14316–14321. doi: 10.1073/pnas.251421398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 42.Dang W, Bartholomew B. Domain architecture of the catalytic subunit in the ISW2-nucleosome complex. Mol Cell Biol. 2007;27:8306–8317. doi: 10.1128/MCB.01351-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwanbeck R, Xiao H, Wu C. Spatial contacts and nucleosome step movements induced by the NURF chromatin remodeling complex. J Biol Chem. 2004;279:39933–39941. doi: 10.1074/jbc.M406060200. [DOI] [PubMed] [Google Scholar]