

1. Introduction
In vivo medical imaging has made great progress due to advances in the engineering of imaging devices and developments in the chemistry of imaging probes. Several modalities have been utilized for medical imaging, including X-ray radiography and computed tomography (x-ray CT), radionuclide imaging using single photons and positrons, magnetic resonance imaging (MRI), ultrasonography (US), and optical imaging. In order to extract more information from imaging, “contrast agents” have been employed. For example, organic iodine compounds have been used in X-ray radiography and computed tomography, superparamagnetic or paramagnetic metals have been used in MRI, and microbubbles have been used in ultrasonography. Most of these, however, are non-targeted reagents.
Molecular imaging is widely considered the future for medical imaging. Molecular imaging has been defined as the in vivo characterization and measurement of biologic process at the cellular and molecular level1, or more broadly as a technique to directly or indirectly monitor and record the spatio-temporal distribution of molecular or cellular processes for biochemical, biologic, diagnostic, or therapeutic application2. Molecular imaging is the logical next step in the evolution of medical imaging after anatomic imaging (e.g. x-rays) and functional imaging (e.g. MRI). In order to attain truly targeted imaging of specific molecules which exist in relatively low concentrations in living tissues, the imaging techniques must be highly sensitive. Although MRI, US, and x-ray CT are often listed as molecular imaging modalities, in truth, radionuclide and optical imaging are the most practical modalities, for molecular imaging, because of their sensitivity and the specificity for target detection.
Radionuclide imaging, including gamma scintigraphy and positron emission tomography (PET), are highly sensitive, quantitative, and offer the potential for whole body scanning. However, radionuclide imaging methods have the disadvantages of poor spatial and temporal resolution3. Additionally, they require radioactive compounds which have an intrinsically limited half life, and which expose the patient and practitioner to ionizing radiation and are therefore subject to a variety of stringent safety regulations which limit their repeated use4.
Optical imaging, on the other hand, has comparable sensitivity to radionuclide imaging, and can be “targeted” if the emitting fluorophore is conjugated to a targeting ligand3. Optical imaging, by virtue of being “switchable”, can result in very high target to background ratios. “Switchable” or activatable optical probes are unique in the field of molecular imaging since these agents can be turned on in specific environments but otherwise remain undetectable. This improves the achievable target to background ratios, enabling the detection of small tumors against a dark background5,6. This advantage must be balanced against the lack of quantitation with optical imaging due to unpredictable light scattering and absorption, especially when the object of interest is deep within the tissue. Visualization through the skin is limited to superficial tissues such as the breast7-9 or lymph nodes10,11 The fluorescence signal from the bright GFP-expressing tumors can be seen in the deep organ only in the nude mice 12,13. However, optical molecular imaging can also be employed during endoscopy14 or surgery 15,16.
2. History of fluorescence probes in medical imaging
The history of fluorescence imaging in medicine is a long one. However, only 2 fluorophores are currently approved by the US Food and Drug Administration (FDA) for medical use; indocyanine green (ICG) and fluoresein. A third flurorophore, Rhodamine B, was approved for use in 1966 but was subsequently banned after 1987 after being linked to cancer in mice and rats – with an estimated risk of only 1 in 9 millon over a 70 year lifespan 17. Fluorescein has been approved for human use for over 30 years, primarily in ophthalmology, while Indocyanine green has been approved for use as an opthalmalogic agent and as a hepatic functional agent for 50 years. Today, these two agents are primarily used to obtain retinal angiograms that require fairly high doses of the agent (500mg and 40mg for a single human dose of fluorescein and ICG, respectively). Side effects such as hypersensitivity reactions can occur, however, specific organ toxicity has not been reported18,19. For molecular imaging the fluorophore dose will be much lower than the current clinical dose used in ophthalmology, and therefore, minimal toxicity is expected.
3. General requirements for fluorescence probes in medical imaging
There are several characteristics of a successful optical molecular probe for medical imaging, including wavelength, brightness, bio- and photo-stability, and pharmacokinetics including non-specific tissue accumulation.
3.1. Wavelength
Fluorophores require excitation light in order to emit light. Excitation in the ultraviolet can cause direct tissue damage20 whereas excitation in the near infrared can lead to tissue heating21. Fluorophores requiring blue and/or green excitation light are compatible with surface imaging applications, however, such wavelengths have poor tissue penetration, making them appropriate only for superficial structures or the small animal imaging. Fluorophores requiring excitation with yellow or red light (at around 600 nm) leads to excessive autofluorescence because the bulk of naturally occurring endogenous fluorophores, mostly hemoglobin and related molecules (Figure 1), are also excited in this range3. The optimal excitation wavelength of a fluorophore is in the deep red or near infrared range because of the combined virtues of good tissue penetration and low autofluorescence9,22,23.
Fig. 1.
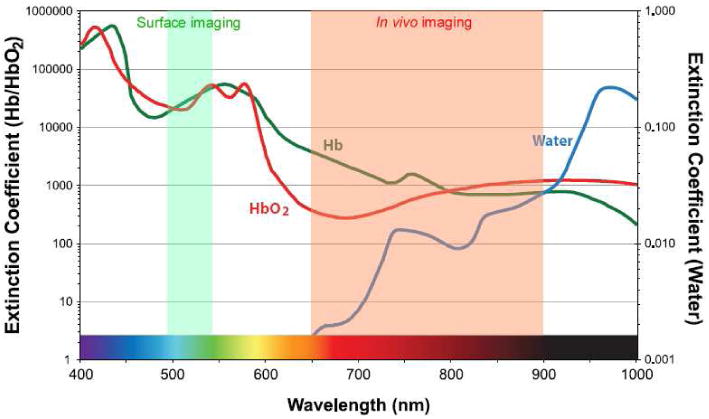
Extinction coefficient value of water, oxy- and deoxy-hemoglobin are plotted ranging from visible to near infrared wavelength.
It should be noted that the asymmetry between excitation and emission wavelengths, known as the Stokes shift, is important in designing a probe. The lower wavelength excitation light may not penetrate sufficiently into tissue to generate emissions from the desired target, so that even though the emission light wavelength is theoretically satisfactory for imaging, the excitation light may not be sufficient to attain deeper tissue penetration. Both excitation and emission wavelengths are ideally in the 650-900nm range, which describes the range encompassing the deep red and near infrared. Here, absorbance and autofluorescence are minimal, so that light penetration through tissue is maximal (Figure 1). However, the use of NIR probes requires a special camera, as the light is not visible to the naked eye or conventional video cameras. Such “night vision” cameras are widely available, although they vary greatly in their sensitivity. It should be emphasized that the emission wavelength of a fluorophore is only one consideration among many. For instance, for surface applications, such as detecting tumors on a mucosal or epithelial surface, lower wavelength (e.g. blue, green, yellow) emitters with high quantum efficiency may produce as good, or better results than NIR probes.
3.2. Brightness
A second consideration in choosing a fluorophore is its brightness. Of course, the brighter the agent the more depth penetration is expected due to higher signal to noise. The higher the quantum yield, the less excitation light is needed for fluorescence to occur. However, increased brightness often comes at the cost of increased size. For instance, the genetically encoded fluorophore, green fluorescent protein (GFP), is quite bright and capable of in vivo imaging for visualizing deep objects12, but it is also very large at 25-50kD and difficult to conjugate, making it impractical for many applications including development of exogenously injected reagents. Quantum Dots are also extremely bright 24,25 but are harder to target because of their large size. Potential toxicity issues related to their component parts such as selenium and cadmium, both heavy metals, must also be considered26.
3.3. Stability
The stability of a fluorophore in vivo is another important consideration. While these molecules are generally stable in vitro, in vivo stability, especially after intracellular internalization is usually compromised. Once internalized within the lysosome, most of the fluorophores, including fluorescein-, BODIPY-, and cyanine-derivatives except rhodamines, lose fluorescence within several days27,28. In the case of rhodamine-derivatives, fluorescence can persist for longer than a week. In addition, most of organic fluorophores suffer photo-bleaching which compromises their fluorescence. Therefore, when longitudinal observation is necessary, organic fluorescent probes must be repeatedly injected. Furthermore, in vitro success may not predict in vivo success since the agent may have too rapid a biologic half life in vivo to be useful. Of course, from a translational point of view, some level of degradation is desirable so that the product is excreted. Quantum dots, for instance, are very slowly metabolized and must be excreted intact to be clinically useful. Stability does not only pertain to the fluorophore but also to the conjugate. Once the fluorophore is conjugated to a targeting ligand via a linker molecule its stability may be compromised by alterations in pharmacokinetics followed by catabolism. For instance, a fluorophore that by itself would be rapidly excreted, when conjugated to a larger carrier molecule will be in vivo for longer and thus, be subject to unforeseen degradation with potentially deleterious effects on fluorescence.
3.4. Pharmacokinetics
Most small molecule fluorophores alter the pharmacokinetics of targeting moieties to which they are conjugated such as peptides and proteins, especially when more than one fluorophore is conjugated to each targeting ligand. For example, when several fluorophores such as rhodamineX and Cy5.5 are conjugated to a single protein molecule, the fluorophores can drastically alter the pharmacokinetics of the targeting probe leading to rapid liver accumulation before successful targeting. When these small fluorophores are conjugated with a sugar or an amino acid, the fluorophore dominates the pharmacokinetics. Quantum dots (Qdots) or other fluorescent nano-particles are even larger than antibodies, therefore, the pharmacokinetics of target-specific molecular conjugates with fluorescent nano-particles such as Qdots vary greatly with the final form of the probe.
4. Classification of Fluorophores
For purposes of discussion fluorophores can be divided into three major classes. Small synthetic fluorophores form the bulk of the commercially available fluorescent molecules. Genetically encoded proteins are often naturally occurring fluorophores produced within cells. Finally, nanocrystals are larger, solid state nano-particles generally characterized by large molecular diameter and high quantum yields.
4.1. Small Molecule Fluorophores
An array of low molecular weight synthetic fluorophores with various core structures including fluorescein-, BODIPY-, rhodamine-, cyanine-derivatives (Figure 2), ranging from 300 Dalton to 2,000 Dalton, are available from commercial providers and span the emission spectrum from blue to NIR. Generally, those that are bright, small, hydrophilic and contain no net charge are better candidates for in vivo imaging. Low molecular weight fluorophores can be designed to be sensitive to enzymatic catalysis so that they activate in specific environments6,16,29-34. Other conditions such as acidic pH and the presence of singlet oxygen or other reactive oxygen species can influence the performance of small molecule fluorophores35-37.
Fig. 2.
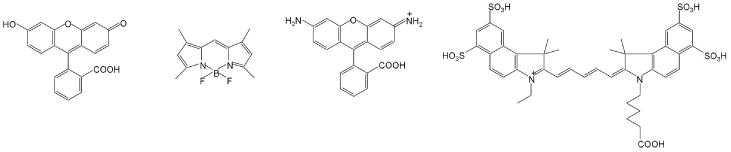
Small molecular fluorophores with representative cores including fluorescein-(fluorescein), BODIPY-(BODYPY-FL), rhodamine-(Rhodamine Green), cyanine-cores (Cy5.5).
4.2. Genetically Encoded Fluorophores
Genetically encoded fluorescent proteins come from a wide range of animals found in nature. Artificial, endogenous proteins have also been generated with emission wavelengths not found in nature, including infrared 38,39. Typically such fluorophores are genetically encoded and must be transfected within the genome of host cells. This makes clinical translation infeasible except in the context of gene therapy40. Fusion proteins with various gene products have been used to trace the fate of specific proteins in vivo. Endogenous proteins include Green fluorescent protein (GFP; Figure 3), Yellow fluorescent protein (YFP) and Red fluorescent protein (RFP). Some of them have unique features including a long Stokes shift and a switchable ability by a specific pulse of excitation light41. Newer constructs can be made conditionally activatable so that the fluorescence is only observed in precise biologic circumstances, such as during the expression of a particular gene. A limitation of such proteins is their relatively large size (30-50kD) which limits their delivery, making them impractical as injectable target-specific fluorescent protein probes.
Fig. 3.
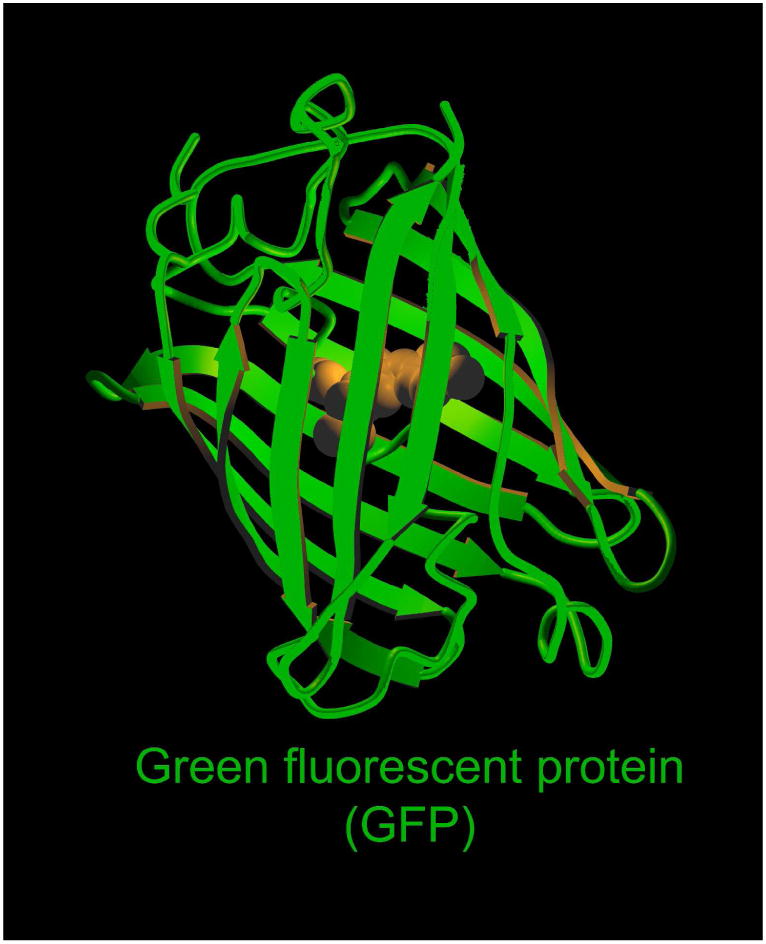
3D-structure of the green fluorescent protein.
4.3 Fluorescent nano-crystals
The synthesis of nano-crystals is currently a developing field in material science and nanotechnology (Figure 4). Numerous nano-crystals have been reported some with unique optical properties. Quantum dots (Qdot) in particular, are characterized by a broad excitation range, a narrow emission peak, resistance to photo-bleaching, and ultrahigh brightness42,43. Target-specific Qdot conjugates with monoclonal antibodies or peptides have been synthesized44,45. As a further extension of quantum dots, self-illuminating quantum dots, in which the Qdot is excited by bioluminescent enzymes (luciferase), have been developed. This is known as bioluminescence resonance energy transfer (BRET). Upon encountering the enzyme substrates (luciferin or coelenterazine) the conjugate emits light which, in turn induces the Qdot to emit a strong light 46,47.
Fig. 4.
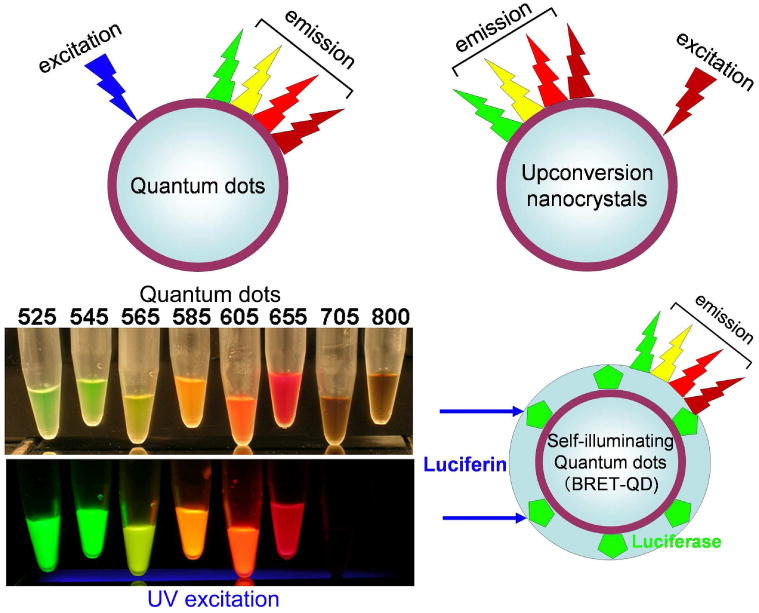
Schematic excitation and emission profiles of 3 nano-particles with heavy metal cores, which are currently employed for in vivo fluorescence imaging.
Another group of unique optical crystals are upconversion nano-crystals. Typical fluorophores emit light at a longer wavelength than their excitation wavelength. Upconverting nanocrystals are unique fluorophores that emit light at shorter wavelengths (visible and near infrared) after excitation in the near infrared. This dramatically reduces background autofluorescence since endogenous fluorophores are not excited by the longer excitation wavelengths. This results in high target to background ratios and good tissue penetration due to the use of NIR both for excitation and emission. 48.
A major concern for nano-materials is their toxicity. Qdots, for instance, contain heavy metals such as cadmium, selenium, in their core. These fluorescent crystals are generally larger than the renal excretion limit (<6 nm in diameter)49. Unless a nano-particle can be made especially small 50,51 and thus is excreted via the kidney, these particles typically have delayed clearance and are mostly excreted through the liver and into the bile without significant metabolism52. For developing targeted reagents, conjugation with targeting moieties only adds to the size of the particles. Therefore, a major challenge with nanoparticle-based molecular probes is synthesizing agents that are large enough to take advantage of the unique features of nano-particles (e.g. brightness) while minimizing toxicity by having particles small enough that they are excreted via the urinary tract.
5. Advanced applications of fluorescence probes in medical imaging
Targeted molecular imaging probes consist of three basic parts; a signaling payload, a carrier, and a targeting moiety, which respectively provide a signal for imaging, optimized pharmacokinetics, and binding to the receptor. When the signaling payload and the targeting moiety are small, there is freedom to choose a carrier that provides appropriate pharmacokinetics for the application. If a large molecule such as a nanocrystal for signaling or an antibody for targeting are employed the clearance of the imaging probe will be markedly prolonged. Therefore, the use of small molecule fluorescence dyes in designing imaging probes is generally advantageous as there are more choices for optimizing the delivery and one dye can be used on multiple targeting-carrier molecules. In contrast to the conventional design strategy for the “always on” monochrome imaging probes, which are used for X-ray radiography, CT, MRI, and radioisotope imaging, there are two approaches to improving the sensitivity and specificity of optical molecular probes and both involve enhancing the target-to-background ratio: 1. maximizing the signal from the target, 2. minimizing the background signal. There has been a long term focus on maximizing signal from the target, especially in the context of measuring drug delivery with radionuclide imaging for dosimetry. Less attention has been paid to reducing background signal. A unique feature of optical imaging is that optical probes can be designed to generate images that maximize the target signal while minimizing the background signal resulting in higher target/background ratios than are possible with conventional imaging methods. In this section we demonstrate the features of advanced optical probes in the context of two strategies for minimizing the background signal from an optical probe; 1. Simultaneous multicolor imaging, 2. Signal activation in the target tissue. The former strategy is based on processing the multi-parametric signals obtained by the simultaneous use of multiple color fluorophores in a mathematical way. The later signal activation strategy is based on the chemical design and synthesis of “activatable” imaging probes.
5.1 Multiple color imaging
The simultaneous imaging of multiple molecular targets with multi-color imaging is commonly employed in in vitro microscopy (e.g. genetically transfected fluorescent fusion proteins, immunofluorescence using exogenous fluorophores, conventional immunohistochemistry), and with the fluorescence-assisted cell sorter (FACS). Five or more colors have already been used during FACS studies53-55.These methods can be utilized for ex vivo analysis of biopsy specimens, but they are invasive and are time consuming because of the multiple steps needed for processing resected tissue. Nuclear imaging has the potential for the simultaneous imaging of 2 (or at most 3) molecular targets under limited conditions including different physical decays and emitting energy windows56,57, however, only optical imaging can simultaneously and independently distinguish five or more separate imaging probes in vivo10.
Spectral imaging is the most sensitive optical technique for the identification of multiple targets. With spectral imaging, several fluorescence signals can be individually detected through a process called “unmixing”, although fixed two-color imaging can be performed with minimal background signal 58 only employing appropriate an excitation light source and excitation/emission filter sets 12,59. Unmixing also removes unwanted background autofluorescence signal in various ranges of wavelengths. Furthermore, by simultaneously using multiple fluorophores with close but distinct emissions, it is possible to define the spectrum of the mixture of unbound fluorophores still circulating within the serum and subtract it from the background. At the same time one can delineate each of the bound fluorophores by their unique spectrum.
The total number of organic small molecular fluorophores that can be imaged simultaneously is still limited due to overlapping emission spectra. Typically, small, organic fluorophores have a distinct and narrow excitation spectra close to their broad peak emission spectra, and successful resolution of more than two dyes is technically challenging. Among the dyes capable of simultaneous multicolor imaging are a few organic dyes with long Stokes shift, non-organic Qdots with wide excitation ranges, and the Keima series of fluorescent proteins with various wide Stokes shifts 60. In order to overcome this limited number of fluorophores, multi-excitation spectral fluorescence imaging algorithms have been developed to distinguish two or more dyes61-63. Since the multi-excitation spectral fluorescence imaging method can serially excite each fluorophore at the appropriate wavelength, it is able to overcome the problem created by the narrow excitation bands of small organic fluorophores, which has been a major disadvantage of such dyes in the past. Therefore, this method allows more flexibility in color selection and permits the simultaneous use of greater numbers of target agents with which to perform multicolor in vivo fluorescence imaging. Several studies have successfully demonstrated the simultaneous characterization of multiple molecular targets in vivo using multi-excitation spectral methods. (Figure 5).
Fig. 5.
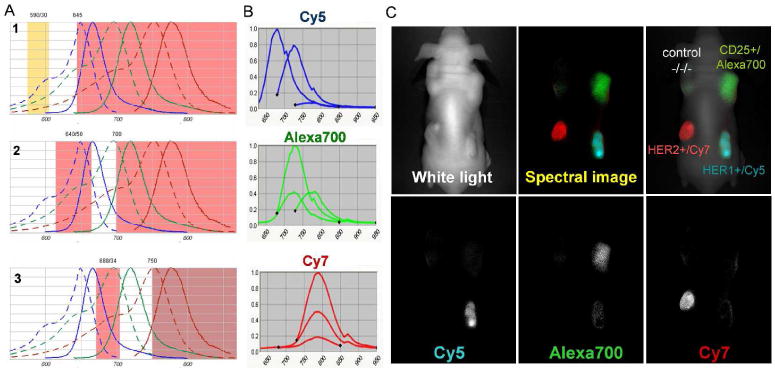
Multiple excitation multi-color spectral fluorescence imaging. A. Filter profiles, which were used for multiple excitation spectral fluorescence imaging with a combination of 3 near infrared fluorophores; Cy5, AlexaFlore700, and Cy7. B. Emission profiles of Cy5, AlexaFlore700, and Cy7 serially excited by 3 filter sets shown in A. C. An in vivo multiple excitation multi-color spectral fluorescence “in vivo immunohistochemistry” image of a mouse bearing 4 tumors; A431(HER1+), 3T3/HER2(HER2+), SP2/Tac(CD25+), and LS174T(a negative control). This image was taken 1 day after injection of 50ug cetuximab-Cy5 (anti-HER1), 50ug daclizumab-Alexa700 (anti-CD25), 50ug trastuzumab-Cy7 (anti-HER2) monoclonal antibody. Distinct antibodies were specifically accumulated in the respective tumors overexpressed with their specific antigens.
There are several obstacles facing multicolor in vivo imaging. First, totally overlapping fluorescence emissions can be difficult to delineate, even with spectral unmixing software. Fluorophores must be chosen so that their emissions are at least 20nm apart from each other, ultimately limiting the number of potential probes (currently up to 5, especially in the crowded NIR (650-800nm) range)(Figure 6). However, there are a growing number of NIR fluorophores which have been developed by several companies 64-67 and academic centers 68,69 which will enable more NIR probes to be used simultaneously. The recent development of NIR probes with emissions >800nm such as Cypate, IR800, and AlexaFluor790, will extend the range of NIR fluorescence enabling even more simultaneous NIR probes. Additionally, spectral unmixing with tunable crystal filter technology reduces the time required to obtain each frame to about 5 seconds. This temporal resolution is inadequate for fluorescence guided surgery, which requires near real time frame rates, so that multi-color surgical guidance has not been possible70. However, this limitation could be minimized or overcome by further development of spectral imaging technology.
Fig. 6.
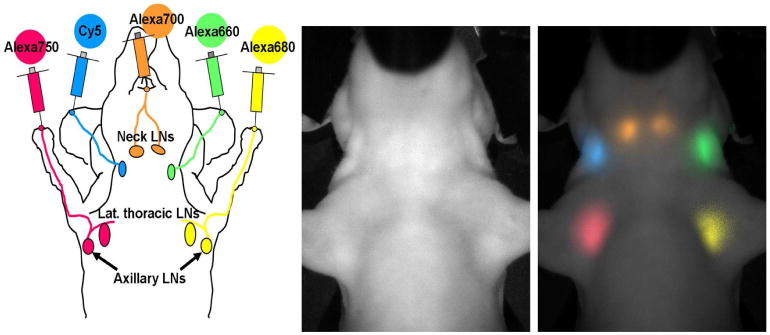
Simultaneous 5-color lympharic image in the head and neck region of a mouse injected with 5 different near infrared fluorophore-labeled half-coated generation-6 dendrimers is shown. A schema of injected sites and expected lymphatic drainage (left), a white light (left), and a 5-color spectrally resolved in vivo lymphatic drainage image (right) are shown.
Multi-color imaging has three advantages over conventional imaging; 1. Simultaneous imaging of more than one physiological or pathological condition. 2. Simultaneous evaluation of the pharmacokinetics and in vivo interaction of more than one drug. 3. Noninvasive profiling of multiple receptors expressed on target cells especially for cancer diagnosis and therapy.
In the case of interstitially injected optical probes, it is possible to simultaneously image multiple lymphatic basins draining cancers as well as normal tissue. This could help minimize lymphatic complications after the surgery, such as lymphedema 71,10. Lymphatic imaging using NIR fluorophores can be used to map lymphatic drainage patterns through the skin due to the better depth penetration of NIR light 61. However, even deep penetration is possible with ultrabright Qdots that emit in the visible range which enable simultaneous and direct visualization of the multiple lymphatic channels as well as sentinel lymph nodes with naked eyes (Supplemental video1)72.
The measurement of pharmacokinetics of several drugs at the same time is difficult with imaging. However, by using two NIR-labeled reagents it is possible to distinguish the clearance of both agents during short and long term 73 pharmacokinetic studies74. This may be useful for drug development and testing in small animals as well as a possible means of noninvasive monitoring the blood concentration of reagents in vivo.
Tumors often demonstrate a diversity of cell surface and proteomic targets. The simultaneous imaging of multiple molecular targets with multiple optical agents is commonplace in in vitro microscopy (e.g. immunohistochemistry, immunofluorescence), and fluorescence-assisted cell sorting (FACS). These methods can also be utilized for ex vivo analysis of biopsy specimens or cytology, but they are, by definition, invasive and time intensive. However, the real time characterization of an expression pattern in vivo is an opportunity to understand the biology of a living malignancy in real time without having to remove the tumor itself. Multi-color in vivo imaging is capable of performing noninvasive characterization of the expression of multiple receptors on target cancer cells63,75. This technique can not only determine the receptor profile of tumors analogous to “noninvasive in vivo immunohistochemistry” (see also Figure 5) but also can assist in the development of multi-color fluorescence guided biopsy/surgery in real time 70,76
5.2 Activatable imaging probes
Based on pharmacokinetics, the “perfect” in vivo targeting agent has not yet been developed. Macromolecular targeting moieties such as monoclonal antibodies are highly specific and can be delivered in high concentrations to the target, but have the undesirable feature of prolonged clearance of unbound molecules, leading to high background signal. On the other hand, small molecules are rapidly cleared, thus improving the target-to-background ratio but reducing the absolute amount of accumulation within the target while increasing the accumulation in the excretion pathways, typically the hepato-biliary system or urinary tract. Therefore, to combine the desirable features of both approaches, an antibody based targeting molecule is employed to achieve highly specific delivery with a signaling molecule that is only activated within the target cancer cells, an approach that contrasts with enzyme activatable probes which are activated in the interstitium, thus reducing non-specific background signal.
From the chemistry point of view, there are three categories of activation mechanisms for optical probes; 1. Small molecules with their own intrinsic activating mechanisms. 2. Large molecule self-quenching probes that activate upon disaggregation of the component molecules 3. Large molecular probes, linked to small molecular activatable probes. The third design is optimal for target cell-specific probes because of the flexibile combinations of large targeting and small activatable signaling molecules for targeted activation at wide varieties of molecules.
5.2.1. Small molecule activatable fluorescent probes
Most existing small molecule probes have been developed for fluorescence microscopy. Currently available fluorescence probes can be classified into two groups, autofluorescent protein (AFP)-based probes and small organic probes. Both probes react or bind specifically to receptors on the cell leading to marked changes in optical properties such as the wavelength (colour) or the intensity of emitted light. Several photochemical mechanisms can be employed to achieve these results. One mechanism, fluorescence resonance energy transfer (FRET), is frequently used for AFP-based probes. For example, calcium ion sensitive AFP-based probes such as Cameleon are comprised of two different AFPs bound by a linker and a calcium-binding motif77. Upon binding to a calcium ion, the orientation or the distance between the two AFPs is altered leading to changes in the efficiency of the resonance energy transfer which is reflected in the relative ratio of the two emission peaks, thus providing a direct read-out of the concentration of calcium ion. This mechanism is quite suitable for AFP-based probes, and indeed a wide variety of probes with specific photochemical sensitivities have been developed. For small molecule probes, the FRET mechanism can also be employed, however, because two fluorophores are necessary for this type of probe, the overall molecular size is relatively large which sometimes impedes efficient cellular uptake of the probe. So, other mechanisms are frequently used to develop small molecule fluorescence probes. A classic strategy is to develop small optical probes that alter their absorption spectra upon reaction with a target analyte. For instance, aminomethylcoumarin(AMC) is one of the most frequently used scaffolds for protease probes. Peptidyl-AMC has its absorbance maximum around 320 nm, however, after cleavage of the amide bond by the target enzyme, AMC, which has an absorbance maximum around 340 nm, is released. As peptidyl-AMCs have minimal absorbance and AMC absorbs well at 380 nm, the activity of the target enzymes can be monitored by determining light intensity after excitation at 380 nm and emission at 460 nm.
Another strategy for developing activatable fluorescence probes is based on xanthene fluorophores such as fluorescein and rhodamine 110. The former is frequently used as a scaffold of probes for various esterases and phosphatases, and the latter for various peptidases. Diacetylated fluorescein (FDA) has no color and no fluorescence due to its intramolecular lactone structure78. Upon reaction with esterase, two acetyl groups are cleaved to release the fluorescein which then fluoresces strongly. Thus, the activity of the target esterase can be monitored by exciting at 490 nm and monitoring for the emission light at 515 nm. Almost the same strategy can be applied to the rhodamine 110-based protease probe. Dipeptidyl-rhodamine 110 has no color, and upon cleavage of two amide bonds by the target peptidase, highly fluorescent rhodamine 110 is produced79. These probes are highly sensitive and can be used to image live cells, however, a pitfall is that the increase in fluorescence is not directly proportional to enzymatic activity because the reactions require two step cleavages to yield the highly fluorescent product.
Recently, a novel strategy utilizing intramolecular spirocyclization was reported. X-ray crystallography demonstrates that specific rhodamine derivatives which bear a hydroxymethyl or a mercaptomethyl group instead of the original carboxy group show unique intramolecular spirocyclic structures. By regulating the spirocyclization and modulating the absorbance and fluorescence of the rhodamine derivatives before and after the reaction with target analytes, it was possible to develop a new activation strategy for fluorescence probes. For instance, a new fluorescence probe which specifically detects hypochlorous acid, an acid found in phagosomes, was successfully developed using this strategy. This probe, dubbed HySOx, has no color and no fluorescence in aqueous solution at pH 7.4 due to a mercaptomethyl group which results in a spirocyclic structure. HySOx reacts with hypochlorous acid to yield a highly fluorescent rhodamine by the oxidation of sulphur (Figure 7). Indeed, the production of hypochlorous acid inside phagosomes of living neutrophils could be successfully monitored in real time with using HySOx80 (Figure 7b).
Fig. 7.
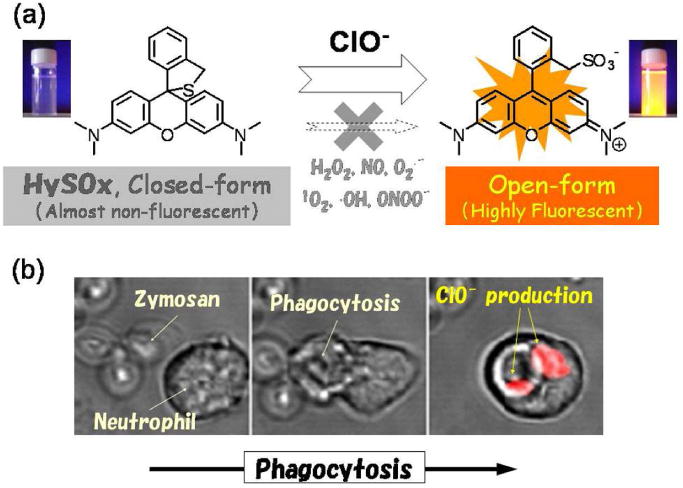
a. A schema of HySOx activation reacted with the hypochlorous acid. b. Serial images of cells, which produced hypochlorite, monitoring with HySOx in real-time.
Another, more widely applicable mechanism for developing small molecule-based probes is “photoinduced electron transfer” (PeT). PeT is a widely accepted mechanism for fluorescence quenching, in which electron transfer from the PeT donor to the excited fluorophore diminishes the fluorescence of the fluorophore. Recently, it was reported that fluorescein, could be considered to be part of a directly linked electron donor – fluorophore acceptor system. In other words, the fluorescence properties of fluorescein derivatives could be precisely controlled by intramolecular PeT. However, until recently, PeT was not thought to be possible in long-wavelength fluorophores, and indeed almost all the photochemical reports of PeT utilized UV-excitable fluorophores such as anthracene etc. Urano and Nagano et al. found that when the fluorescein structure was deconstructed into two parts, i.e., the benzoic acid moiety as the PeT donor and the xanthene ring as the fluorophore, only small alterations in absorbance were observed among fluorescein and its derivatives. Moreover, the dihedral angle between the benzoic acid moiety and the xanthene ring is almost 90°, which suggests that there is little ground-state interaction between these two parts. They found that when the HOMO energy of the benzoic acid moiety was higher than a certain threshold, PeT occured efficiently resulting in little fluorescence81. On the other hand, when the HOMO energy is lower than the threshold, as is the case for generic fluorescein, the rate of PeT slows and the molecule becomes highly fluorescent. PeT-based quenching was evidenced by observing transient absorption spectra in laser flash photolysis experiments, i.e., bands corresponding to the radical cation of the electron donor and the radical anion of the xanthene moiety were successfully detected. The fact that PeT rates and the rates of back electron transfer follow the Marcus parabolic dependence of electron transfer rate with a driving force provides the basis for rational design of high efficiency, quantitative, modulation of the fluorescence properties of fluorescein-based probes. Further, this strategy has not only been used with fluorescein but also with a wide range of long-wavelength excitable families of fluorophores such as BODIPYs, rhodamines, and the cyanines.
Just as PeT represents the flow of electrons from donor to fluorophore, the transfer in the opposite direction provides another fluorescence switch in visible light-excitable fluorophores: the fluorescence properties can be modulated via the PeT process from the excited fluorophore to a reducible benzene moiety (donor-excited PeT; d-PeT). In other words, when the LUMO energy of the benzoic acid moiety is lower than a certain threshold, the rate of d-PeT is quite fast and hence this derivative will be non-fluorescent. This finding also provides a basis for a new and practical strategy for the rational design of novel functional fluorescence probes82.
Based on these PeT-based strategies, various novel fluorescence probes were successfully developed (Figure 8); (a) DPAXs and DMAXs for singlet oxygen, (b) DAFs and DAMBOs for nitric oxide, (c) HPF and APF for highly reactive oxygen species (hROS), (d) MitoHR and MitoAR for hROS stress in mitochondria, (e) NiSPYs for peroxynitrite, (f) DACals and APC as highly sensitive probes for nitric oxide and hROS which are highly retained in living cells, (g) DNAT-Me for the activity of glutathione S-transferase (GST), (h) acidic pH-sensitive BODIPY-based probes, among others.
Fig. 8.
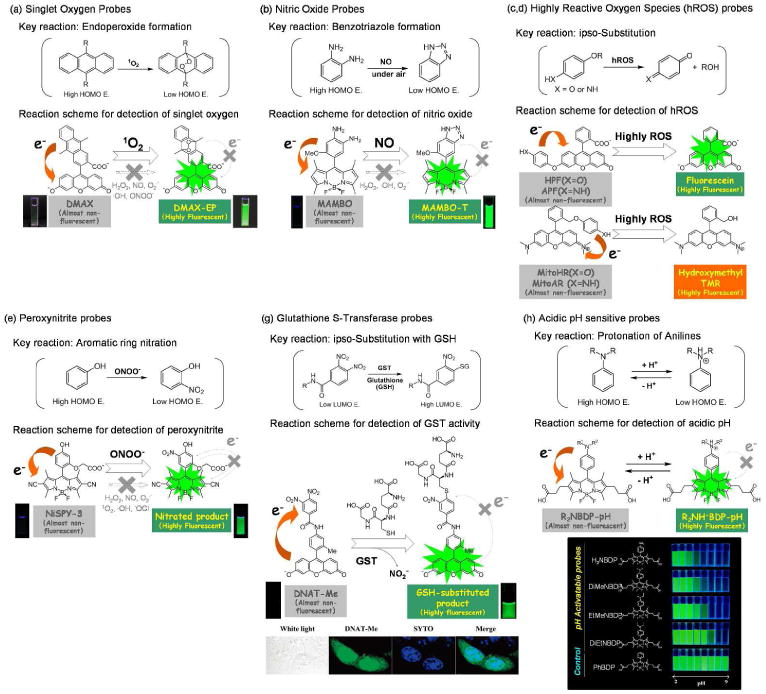
Schemas for the fluorescent signal activation of fluorescein-based activatable probes reacted with the singlet oxygen (a), nitric oxide (b), highly reactive oxygen species (hROS) (c, d), peroxynitrite (e), glutathione S-transferase (g), and acidic pH (h) based on the photon-induced electron transfer (PeT) theory.
To develop singlet oxygen-sensitive fluorescence probes the endo-addition of singlet oxygen to anthracene was utilized which yields the corresponding endoperoxide product whose HOMO energy is much lower than that of the starting anthracene. Thus, the initial fluorescence of DPAXs83and DMAXs84was efficiently quenched by PeT, but was recovered by the reaction with singlet oxygen because the rate of PeT is quite slow due to the lowered HOMO energy of endoperoxides (Figure 8a). Further, the endo-addition reaction is specific for singlet oxygen making these probes quite selective for sensing singlet oxygen.
Nitric oxide sensitive fluorescence probes such as DAFs85, DAMBOs86, DARs87, DACs88 were successfully developed by utilizing the reaction of NO with o-phenylenediamines to yield corresponding benzotriazoles (Figure 8b). The HOMO energy of phenylenediamines is quite high due to the existence of two strong electron donating amino groups, which leads to efficient quenching of adjacent fluorophores by PeT. The PeT mechanism is applicable to a wide range of fluorophores such as fluoresceins, rhodamines, BODIPYs, cyanines and so on. On the other hand, because the HOMO energy of benzotriazoles is quite low, the resulting products of these probes when bound to NO were highly fluorescent. So, these probes work quite well as selective and sensitive fluorescence probes for NO. However, users of these probes must be cautious that these probes are not reactive with NO itself, but rather, reactive for NO+ or its equivalent species. NO is easily oxidized by dioxygen molecule to yield NO+ or its equivalent species, so these compounds can be used as NO probes under aerobic conditions. But if these probes are used under anaerobic conditions, no fluorescence increase will be observed whether in the presence of NO or not.
Numerous reactive oxygen species (ROS) are known to occur, however the reactivity of each ROS differs substantially. For example, while the hydroxyl radical is quite short-lived and reactive, thus promoting oxidation of alkanes and arenes, hydrogen peroxide is relatively inert and stable. Thus, it is of interest to generate in vivo probes that detect highly reactive oxygen species (hROS) such as hydroxyl radical, peroxynitrite, and hypochlorite. For selective monitoring of hROS production in cells, HPF and APF were developed89 (Figure 8c, d). These compounds are based on fluorescein but utilize the O-dearylation reaction of aryloxyphenols and aryloxyanilines to create selective reactions catalyzed by hROS. HPF and APF were almost non-fluorescent due to the intramolecular PeT from the hydroxy- or aminophenoxy- group attached to the xanthene ring of fluorescein. These electron donor moieties are cleaved off by the reaction with hROS to yield highly fluorescent fluorescein. HPF can selectively detect hydroxyl radical and peroxynitrite among various ROS, and APF detects hypochlorite also. Further, these probes are completely resistant to autoxidation which is a well known pitfall of fluorescence probes for ROS such as DCFH and DHR123.
MitoHR and MitoAR were developed as another type of hROS selective probe based on tetramethylrhodamine as a fluorescent scaffold35. The same O-dearylation reaction used in HPF and APF was utilized as the key reaction with hROS, and mitoHR and mitoAR are almost non-fluorescent because of the fast rate of intramolecular PeT from the benzene-bound hydroxy- or aminophenoxy group attached to tetramethylrhodamine. These probes can selectively detect damage to the mitochondria caused by hROS, because the probes accumulate exclusively in mitochondria. With these probes, it was possible to successfully observe that as little as 10 μM of hydrogen peroxide can cause hROS damage to mitochondria in living HL60 cells.
Peroxynitrite, an hROS, can be selectively detected with recently developed fluorescence probes, named NiSPYs90 (Figure 8e). NiSPYs are the first fluorescent probes which show marked fluorescence increase after nitration by peroxynitrite. Because the nitro group was believed to be a strong and general quencher of fluorescence, it was not thought possible to develop fluorescence probes for peroxynitrite which show increases in fluorescence. Ueno et al. reported that the nitrobenzene derivatives caused efficient quenching due to d-PeT originating from the excited fluorophore moiety due to the strong electron withdrawing effect of the nitro group. They developed a family of novel fluorescence probes based on dicyanoBODIPY, which are sensitive to peroxynitrite by adjusting HOMO and LUMO energies of the fluorophore and the reactive moiety for peroxynitrite. NiSPYs were almost completely non-fluorescent in the absence of peroxynitrite but became highly fluorescent upon reaction with peroxynitrite.
Fluorescein-based probes generally diffuse uniformly in the cytosol which is advantageous for detecting cytosolic ROS production. However, fluorescein derivatives are well known to leak out from living cells through anion transporters expressed on the cell membrane, which hampers sensitive and quantitative detection of target analytes due to the decreasing concentration of probes in cells. Calcein is a very hydrophilic fluorescent dye, and is well known not to leak from living cells. Thus, calcein-based fluorescence probes could be more sensitive and reliable probes especially for long term observations. Recently, it was reported that the fluorescence property of calcein can be also controlled precisely by the PeT mechanism, and calcein-based fluorescence probes for nitric oxide, named DACals, and hROS, named APC, were developed 91.
Acetoxymethyl(AM)-ester derivatives of these probes can permeate the cell membrane, and become retained in the cytosol after cleavage of the AM-ester by ubiquitous esterases found in living cells. DACals and APC were developed using the same strategies employed in the design of DAFs and APF, respectively. In the absence of their respective analyte, they were almost non-fluorescent, but became highly fluorescent upon reaction with nitric oxide and hROS, respectively. After activation, they were retained in living cells, leading to amplification of signal. As a result, DACals and APC were more sensitive to low concentrations of nitric oxide and hROS in living cells.
The concept of PeT is not restricted to developing fluorescence probes for ROS, but is also applicable for a wide range of enzymes. Glutathione S-transferase is a generic term for a family of enzymes mainly involved in the detoxification and metabolism of endogenous and exogenous compounds, including drugs. Despite the medical significance of this enzyme family, thus far, no fluorescence probes have been developed which offer selective in vivo imaging of GST activity. Recently, a novel fluorescence probe for GST activity, DNAT-Me, was successfully developed using 3,4-dinitrobenzanilide (NNBA) as a specific substrate and d-PeT as a mechanism for fluorescence quenching92 (Figure 8g). DNAT-Me is switched “on” after GST-catalyzed glutathionylation. This agent has excellent kinetic parameters for monitoring the enzymatic activity in living cells. Indeed, nuclear localization of GSH/GST activity in HuCCT1 cell lines was successfully visualized with the use of DNAT-Me. These results indicate that DNAT-Me should not only be useful for high-throughput GST-inhibitor screening but also for studies on the mechanisms of drug resistance in cancer cells.
Almost all currently known fluorescein derivatives contain a carboxylic group. However, the carboxylic group can be replaced with another functional group. Recently, Urano and Nagano et al. reported the novel fluorescein derivative, called TokyoGreen (TG), which replaces the carboxylic group with a methyl or methoxy group93. TG dyes are easily synthesized in high yields by the conventional C-C bond coupling reaction (Figure 9).
Fig. 9.
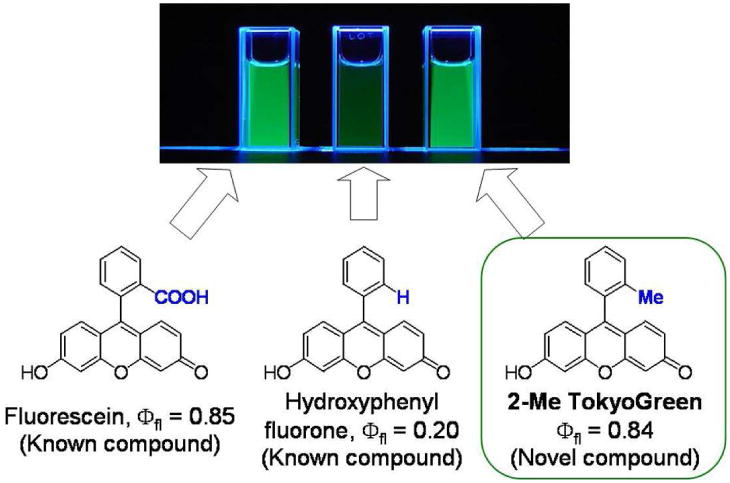
A schema of the development of Tokyo Green derived from fluorescein.
Further, by precisely controlling the HOMO energy of the benzene moiety, they were able to construct another rational design strategy for novel fluorescence probes (Figure 10). The value of this approach is exemplified by a novel, highly sensitive and membrane-permeable fluorescence probe for β-galactosidase called TG-βGal 93 (Figure 11a). Further, based on the same strategy, TG-phos94 for alkaline phosphatase, which is also a widely used reporter enzyme, and TG-NPE30 (Figure 11b) as highly efficient caged fluorophores were successfully developed. Using TG-βGal, an application of optical imaging for the detection of small cancer implants with high signal contrast was recently reported. Peritoneal dissemination of cancer is known to be difficult to treat and is often fatal. During surgery, it is also difficult to detect small tumors implants in the peritoneum due to the complex anatomy and the poor visual contrast between the tumor and normal tissue using white light imaging. Even non-activatable imaging probes have been successful in this model. Recently, Kobayashi et al. found that a lectin (asialo-receptor family)-targeted optical imaging technique which utilizes a fluorescein-avidin conjugate was effective for the visualization of peritoneal implants in a mouse ovarian cancer model15. Using a combination of lectin-targeting and activatable fluorescence probes, however, Kamiya, Kobayashi and Urano et al. demonstrated an even more sensitive and selective imaging method31. Using a two step procedure in which lectin was first used to localize β-galactosidase on cancer cells, and then was subsequently targeted by the administration of a highly-sensitive fluorescence probe very small implants with low background signal was achieved. The presence of β-galactosidase led to target-specific fluorescence activation within tumor. The net result was a novel activatable fluorescence probe for targeting tumor (AM-TG-βGal) that dramatically increases fluorescence emission when the tumor cells are pre-labeled with β-galactosidase as an activating enzyme (Figure 12). Since the tumor-targeted enzyme can catalyze numerous substrate turnovers, a great number of fluorescent molecules could be activated leading to a high tumor-to-background ratio. It was possible to see a strong fluorescence signal within prelabeled tumor cells after only one hour of incubation with AM-TG-βGal. Moreover, using the same probe with fluorescence microscopy revealed that cancer microfoci as small as 200 μm could be visualized.
Fig. 10.
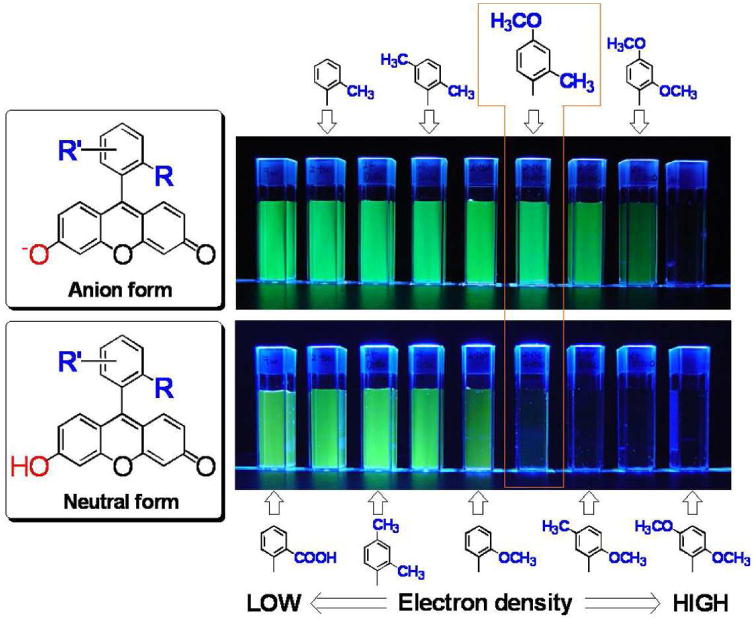
A schema of the photon-induced electron transfer (PeT) effects to the fluorescent signal of Tokyo Green-derivatives to demonstrate that 2-Me-4-OMe Tokyo Green can be a versatile scaffold for designing hydrolase probes
Fig. 11.
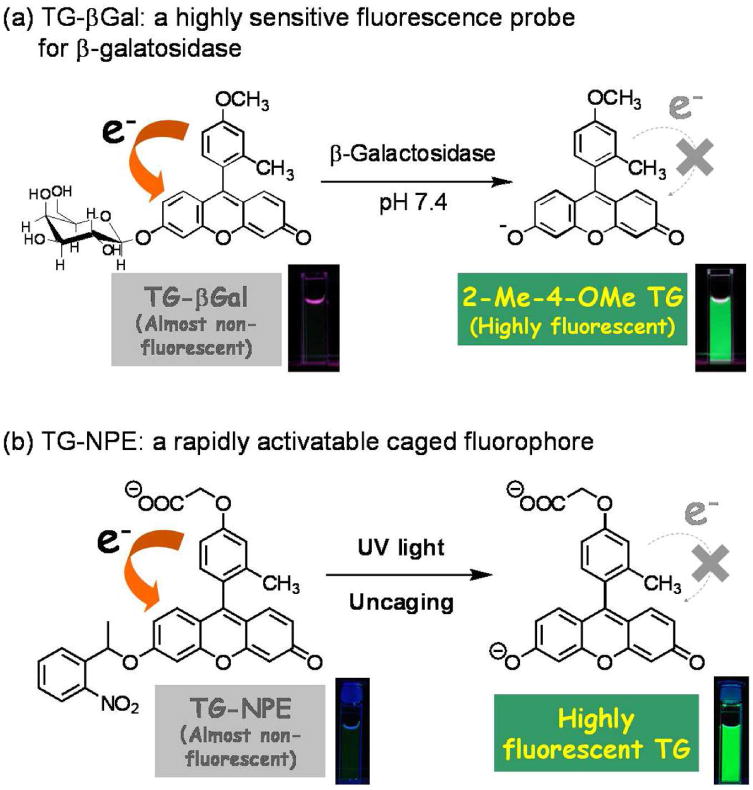
Schemas for the fluorescent signal activation of Tokyo Green-based activatable probes reacted with the β-galatosidase (a) and UV irradiation (caged) (b) probes
Fig. 12.
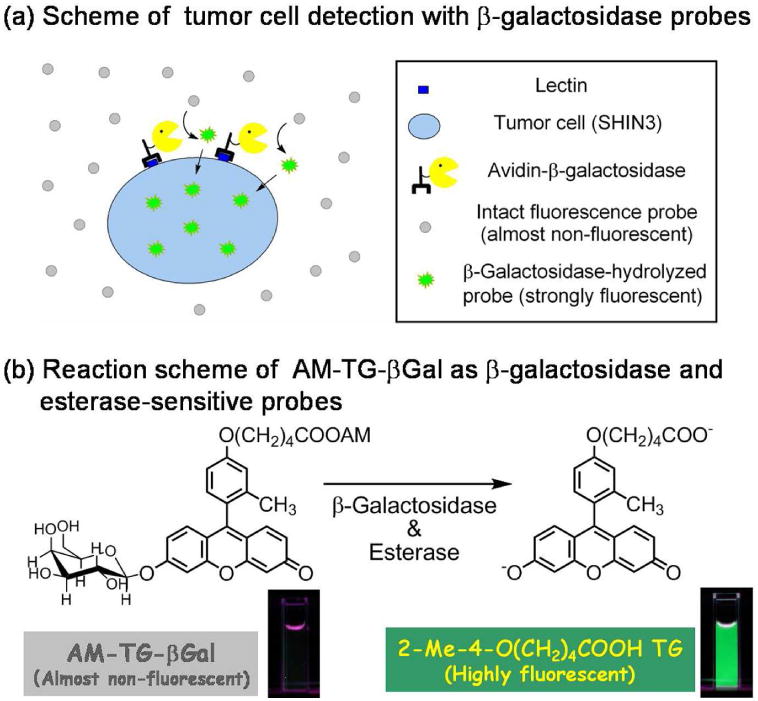
Delivery (a) and reaction (b) schemes of specific tumor cell detection with both esterase and β-galactosidase probes.
5.2.2 High molecular weight activatable fluorescent probes
High molecular weight activatable fluorescent probes depend on the intermolecular relationship between two or more fluorophores or fluorescence quenchers, and typically rely upon one or more strategies such as fluorescence resonance energy transfer (FRET), dimer formation (H-type or J-type), or photon-induced electron transfer (PeT). These probes can be categorized as; 1. Enzyme-specific probes, which are activated by enzymes in proximity to a target cell 2. Targeted-cell specific probes, which bind to the specific cell-surface target molecules, are internalized and activated by lysosomal conditions including, low pH, oxidation, unfolding, catabolism, or protein cleavage by lysosomal enzymes. The fluorescence signal arising from activated target-cell specific probes should, therefore, only occur in the target cells. In contrast, enzyme specific probes are able to activate in a broader spectrum of pathologic conditions such as inflammation and neoplasia, and are, therefore less specific than targeted-cell specific probes.
5.2.2.1 Enzyme specific activation
Activatable fluorescent probes are commonly used for molecular biology in vitro. Fluorescence resonance energy transfer (FRET) is another of the methods used to switch “on” fluorescence and can be used to detect enzymatic activity 95. FRET uses two different color fluorescent proteins or fluorophores, where the energy absorbed by one fluorophore is transferred to the second. FRET systems are designed such that the emission spectrum of the donor fluorophore overlaps the acceptor fluorophore's absorbance spectrum. Because the energy transfer takes place only when the donor and the acceptor molecules are less than 10 nm, the distance of two molecules can be monitored by measuring the fluorescence spectrum changes. Thus, FRET-based activation has been used in vitro for DNA biosensors and investigation of protein conformation change 96-98.
Excimer formation is another activating mechanism that is also used to detect DNA, RNaseH activity analysis, and protein detection 99-101. An excimer is a complex of ground state and excited state fluorophores, which emit longer and broader wavelength fluorescence than do monomers 102. Since the complex can be formed only when the molecules are close to each other, this mechanism is useful in detecting when compounds are in the monomer (separated) or excimer (in close proximity). This can be useful in determining specific enzymatic activity as the monomer state is more likely after enzymatic catalysis.
In addition, protein kinase sensors that fluoresce in upon exposure to phosphrylated compounds can be used to assess specific protein kinase activities 103,104. Such sensors, which are applicable to a wide range of kinases, depend on protein kinase substrate peptides that are combined with a sensing fluorophore, a fluorophore-quencher pair, or a self-quenched fluorophore. Fluorescence is activated upon the phospholylation of the substrate peptide.
5.2.2.1.1 Fluorophore-fluorophore activation (self-quenching)
Self-quenching is a mechanism of fluorescence switching in which excited fluorophores of similar type absorb the energy from each other that would otherwise have lead to an emitted photon, thus diminishing the fluorescence of the entire compound. This can only take place when multiple fluorophores are located close each other. Weissleder et al. have developed self-quenched but enzymatically activatable probes. They conjugated multiple NIR fluorophores, (Cy5.5) on a synthetic graft copolymer 6. The polymer consists of poly-l-lysine linked by multiple methoxypolyethylene glycol (MPEG) to extend the circulation time in vivo. The probe is supposed to be retained in the blood pool for a long period of time, and gradually leak through tumor neovasculature by enhanced permeability and retention (EPR) 105,106. The poly-L-lysine chain is then cleaved by a lysosomal protease, and the Cy5.5 fluorophores are separated from each other resulting in dequenching (fluorescence activation). This system worked in vivo as well as in vitro, with visualization of mouse tumors after intravenous injection.
The expression of specific enzyme activity is often elevated both in tumors and inflammatory conditions and often relates to the disease severity rather than providing specificity as to the process. Therefore, the utilization of more specific enzymes for fluorescence activation instead of broad proteases found in the lysosome should increase the specificity to the target tissue and aid in the characterization of the pathological condition.
For example, cathepsins are related to several diseases processes, e.g. cancer, atherosclerosis, Alzheimer's disease 107-111. Tung et al. conjugated cathepsin D substrate peptide and multiple Cy5.5 molecules, and MPEG to poly-l-lysine 112. This probe was thought to be activated only after cathepsin D substrate peptide cleavage which released Cy5.5 molecules. From the same group, Bremer et al. applied this method to cathepsin B substrate, and succeeded in detecting tumors in vivo 113. They also developed a cathepsin K specific probe for atherosclerosis imaging and osteoclast activity measurement 114,115.
Matrix metalloproteinase (MMP) is another potential target enzyme. MMPs are expressed in cancers in higher levels than in normal tissue, and play important roles in metastasis and angiogenesis 116,117. Bremer et al. combined MMP-2 substrate peptide, with multiple Cy5.5, MPEG to poly-l-lysine to form a single probe 118,119. Successful results were obtained both in vitro and in vivo. The degree of fluorescence activation reflected MMP enzyme activities, and the fluorescence activation was inhibited by an MMP inhibitor. During in vivo imaging studies, tumors located in the mammary fat pad were visualized. Since MMPs are highly expressed in atherosclerotic plaques, they may also be useful in identifying vulnerable plaque 120. MMPs-sensitive deliverable fluorescent probes have recently synthesized based on the activatable cell-penetrating peptides for the use of detecting cancer, inflammation, and atherosclerotic plaques 121.
5.2.2.1.2 Fluorophore-quencher activation
Self-quenching involving only fluorophores still yields weak fluorescence even in the quenched state. An alternative to fluorophore-fluorophore quenching, is to use a fluorophore-quencher combinations in which the quencher is non-fluorescent.
Farber et al. developed a probe to image phospholipase A2 (PLA2) activity. They employed BODIPY as a fluorophore and dinitrophenyl as the quencher, and combined them with phospholipid substrate analogs. In the zebrafish model, effective quenching and dequenching was observed with and without phospholipid 122. Bullok et al. applied a different fluorophore-quencher to detect apoptosis 123. AlexaFluor 647 was used as a fluorophore and QSY21 as a quencher. AlexaFluor 647 and QSY21 were conjugated across a caspase recognition sequence, which was combined with a Tat-peptide to enhance permeation. Caspase activity produced fluorescence detectable at microscopy. A cathepsin sensitive probe using a fluorophore-quencher pair was reported by Bulm et al.124. They selected the peptide acyloxymethylketones (AOMKs) as a cathepsin substrate. Then, both Cy5 (fluorophore) and QSY21 (quencher) were linked to AOMK across the cathepsin cleavage point. They tested this probe in tumor bearing mice, and compared it with the, unquenched fluorophore. Unlike the always-on probe, the quenched probe produced a specific signal in the tumors and showed virtually no background signal. The tumor signal increased with time and reached a maximum 6-8 h after probe injection.
5.2.2.2 Targeted-cell specific activation
Another strategy to increase target specificity is to utilize target specific carrier molecules followed by binding to the target cell surface molecules, internalization and activation of the fluorescence signal within the target cells. In vivo molecular imaging with always-ON probes has been performed using antibodies, proteins, and peptides as targeting molecules. Integrins are cell adhesion molecules that are overexpressed during tumor growth and metastasis. Cyclic RGD peptides are known to bind to αvβ3 integrin. When the Cy5.5 conjugated RGD peptide was delivered for in vivo imaging, the tumors were visualized with this probe; however, the background fluorescent signal from whole body was high due to the always-ON nature of the probe 125,126. Similar results were obtained with epidermal growth factor (EGF) conjugated Cy5.5 or Quantum dots 127,128.
However, there are a growing number of humanized monoclonal antibodies and other targeting molecules directed against tumor specific cell surface antigens and epitopes, which are now used for cancer therapies, and therefore, molecular imaging with antibodies has great potential to detect and characterize cancers. Several groups have tried in vivo tumor imaging with fluorophores conjugated to antibodies129-131. Unfortunately, as has been confirmed with radiolabeled monoclonal antibodies, antibody imaging is limited by slow background clearance leading to high background signals as well as non-specific uptake due to enhanced permeability and retention (EPR) caused by the leaky tumor vasculature75. To overcome these limitations, several types of antibody fragments have been engineered to improve the pharmacokinetics by reducing the molecular size of the targeting moiety132. These engineered antibodies are usually cleared rapidly from the circulation and therefore, do not accumulate as much in tumors. Using smaller molecule targeting ligands, the background signal is low enough to be neglected in in vivo imaging. Therefore, despite the pharmacokinetic advantages of smaller molecules, the whole IgG is likely the best platform for activatable antibody probes among existing engineered antibodies and antibody-derived fragments since the absolute accumulation in the target tissue can be maximized. In vivo molecular imaging with activatable antibody probes using conjugates should have a great potential for sensitive and specific target detection.
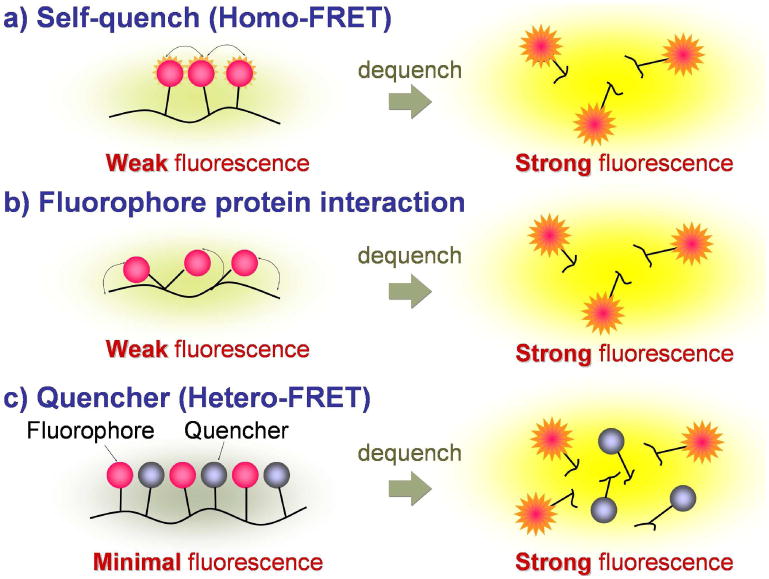
To maximize the activation efficacy, fluorescence signal activation based on targeting-internalization in which fluorophores are delivered to the target tissue by a carrier molecule (such as a monoclonal antibody) and activated once inside the target cells, results in high target to background ratios. In general, the two quench-activation mechanisms are self-quenching, which operates only with single or multiple distinct fluorophore molecules, and fluorophore-quencher combinations, which operate based on the interaction between two different molecules (Figure 13). In both cases, fluorophore(s) can be quenched upon conjugation to the targeting moiety and stay quenched in the circulation, and then can be dequenched by certain processes in the lysosome of the target cell.
Fig. 13.
A schema for quenching/de-quenching (activation) of macromolecular probes conjugated with multiple fluorophore/quencher molecules.
5.2.2.2.1 Fluorophore-fluorophore activation in the targeted cells (self-quenching)
In the same manner as the enzyme-specific activation probe, self-quenching is another mechanism by which target-cell-specific activation can be achieved. Self quenching occurs when multiple fluorophores are conjugated to a single targeting protein, e.g. antibodies, receptor ligands and the fluorophores become crowded on the molecule (Figure 13a). Alternatively, even a single fluorophore molecule such as ICG or BODIPY-FL can be quenched by the interaction with certain side chains of amino acids at the conjugation with some targeting proteins (Figure 13b) such as humanized monoclonal antibodies74 or avidin derivatives133, respectively.
For instance, multiple copies of carboxy-X-rhodamine (ROX) conjugated to avidin is an effective activatable probe that utilizes the self-quenching mechanism134. Avidin is a noncovalently bound homotetrameric glycoprotein and it binds to d-galactose receptor, which is commonly expressed on cancer cells. After internalization to the cell, the tetramer is broken down into monomers in the lysosome; as a result, each fluorophore is separated and dequenched resulting in fluorescence activation. Three ROX molecules conjugated to a single avidin showed higher activation capacity (13-fold) compared to 0.5ROX conjugated to a single avidin (always ON) (Figure 14). When the 3-ROX conjugated probe was applied in vivo in peritoneal tumor bearing mice, tumors were detected with high sensitivity and specificity. The self-quenching system can be applied to another tumor targeting protein, galactosamine serum albumin (GSA) 135. In a similar manner as avidin, GSA binds to d-galactose receptor, is internalized and then is unfolded and broken down into small fragments in the lysosome. In an in vivo investigation, twenty ROX conjugated per GSA (GSA-20ROX) clearly visualized tumors with higher sensitivity and specificity than GSA-0.5 ROX conjugate. In this way, ROX was revealed to be an appropriate fluorophore to make use of the self-quenching mechanism.
Fig. 14.
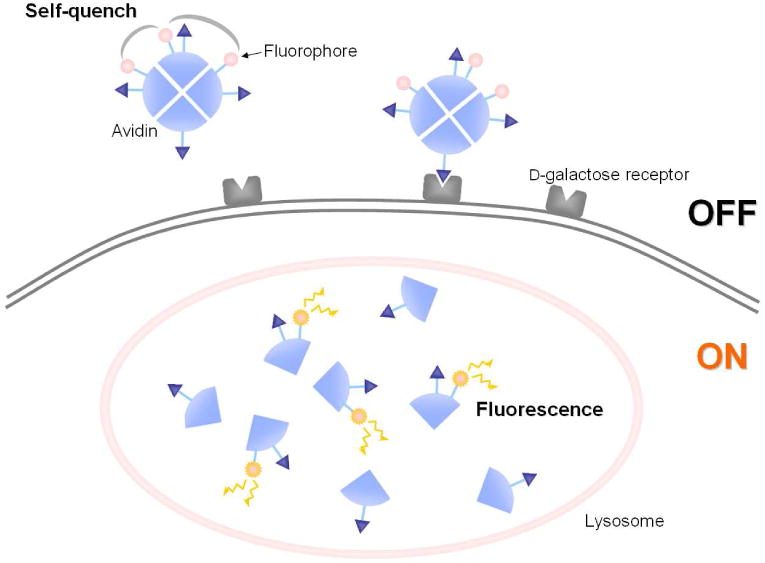
A schema for the activation mechanism of self-quenched avidin-3ROX probe targeting D-galactose receptor expressing cancer cells.
The self-quenching properties of several xanthene-based fluorophores are listed in Figure 15 136. Xanthene derivatives are known to form homo-dimers at high concentration aqueous solutions 137,138. This homo-dimer formation induces short (H-dimer) or long (J-dimer) shifts of absorbance spectra, which completely quench the emission fluorescence signal 139. The fluorophores were conjugated to the cancer targeting molecule, avidin or trastuzumab. Trastuzumab is a humanized monoclonal IgG1 antibody, which binds to human epidermal growth factor receptor type 2 (HER2). After binding to HER2, trastuzumab is gradually internalized within HER2 expressing target cells and then undergoes degradation in the lysosome 140. The fluorophore-protein conjugates showed diverse levels of fluorescence activation capacities. Also, a short shift of absorbance spectra of fluorophores (H-dimer formation) was observed (Figure 16) for the quenched probes at concentrations suitable for in vivo molecular imaging, and the quenching capacity directly related to the extent of dimerization. This means that H-dimer quenching occurs when specific fluorophores are conjugated to specific carrier proteins. R6G and TAMRA formed H-type dimers most effectively. In vitro microscopy showed fluorescence activation after cellular internalization and dissociation of the H-dimer in the target cells. After injection of avidin-TAMRA to tumor bearing mice, the peritoneal tumors were clearly visualized by fluorescence endoscopy with low background signals (Figure 17, see also Supplement video 2). Thus, H-dimer formation is shown to be a promising mechanism for activation of target-specific macromolecular optical probes for molecular imaging. These observations are consistent with our previous study using other target moieties. Several xanthene-based fluorophores were conjugated to GSA, and a series of in vitro and in vivo experiments were performed to investigate which fluorophores possess the optimal characteristics for in vivo molecular imaging. It was found that TAMRA has the optimal fluorescent properties among commonly employed in vivo molecular imaging probes28.
Fig. 15.
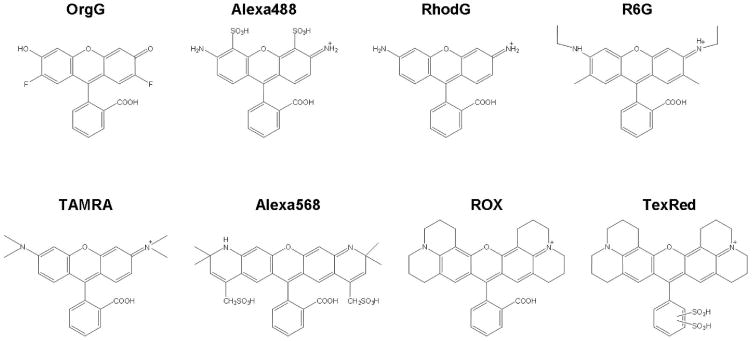
Chemical structures of xanthen-based fluorophores. Oregon Green (OrgG), Alexa Fluor488 (Alexa488), Rhodamine Green (RhodG), Rhodamine 6G (R6G), TAMRA, Alexa Fluor568 (Alexa568), ROX or Texas Red-X (TexRed)
Fig. 16.
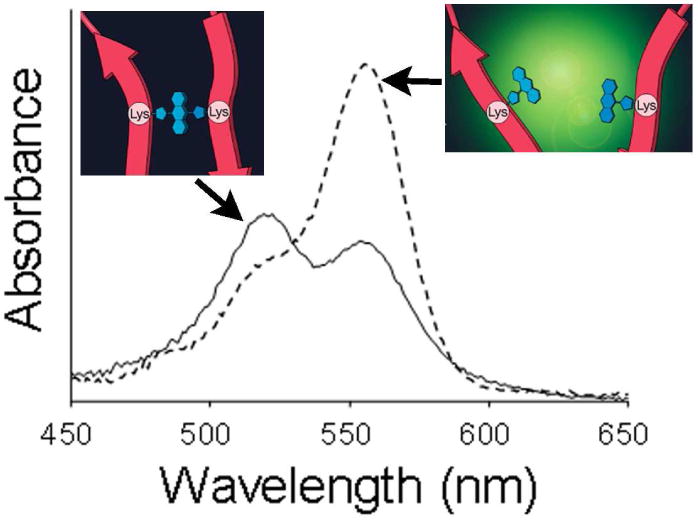
Absorbance spectrum of TAMRA conjugated avidin in PBS (solid line). The blue shifted peak (521 nm) represents the H-dimer formation of fluorophores. When the conjugate is treated with SDS to separate the fluorophores, the dimmer peak decreases and monomer peak (555 nm) increases.
Fig. 17.
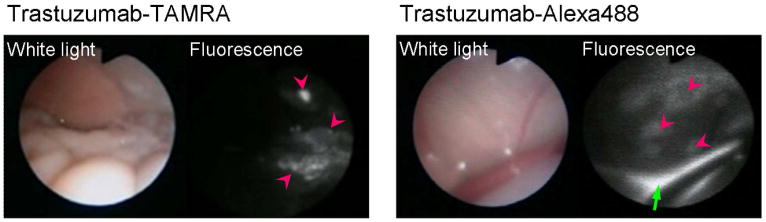
Fluorescence endoscopic images in peritoneal tumor bearing mice by avidin-TAMRA and avidin-Alexa488. The pink arrow heads show the tumor nodules. The tumors were clearly visualized with low background signal by the activatable probe, avidin-TAMRA. In contrast, the always-on probe, avidin-Alexa488 showed high background signal and high fluorescence from excess injectate in the peritoneal cavity (green arrow).
Since NIR probes are of great interest for in vivo imaging, the self-quenching properties of NIR fluorophores were also investigated. The cyanine-based NIR fluorophores, AlexaFluor 680 (Alexa680) or Cy5.5 were conjugated to trastuzumab 141. These two fluorophores have identical absorbance spectrums and fluorescence emission spectrums. Seven fluorophores were conjugated to each trastuzumab molecule to make the following self quenching probes, Tra-Cy5.5(SQ) and Tra-Alexa680(SQ). For comparison, always-ON probes, Tra-Cy5.5(ON) and Tra-Alexa680(ON), were prepared with one fluorophore per trastuzumab. The quenching capacities were 9, 2, 8 and 2-fold for Tra-Cy5.5(SQ), Tra-Cy5.5(ON), Tra-Alexa680(SQ) and Tra-Alexa680(ON), respectively. In vitro microscopy demonstrated that the fluorescent signals of Tra-Cy5.5(SQ) and Tra-Alexa680(SQ) are quenched while the antibody-conjugates are outside the HER2 positive cell but they are activated after internalization into the cell (Figure 18). In contrast, with always-ON probes, bright surface fluorescence was detected. When these probes were applied in vivo, Tra-Alexa680(SQ) showed a high tumor-to-background ratio and only the target HER2 positive tumor was detected. In contrast, Tra-Cy5.5(SQ) caused high non-specific binding, and the HER2 negative tumor demonstrated even higher signal than the HER2 positive tumor. The always-ON probes, as expected, showed high background signals arising from the blood pool. Thus, the self-quenched Alexa680 probe demonstrated target specific activation. As predicted from the chemical structures, the lipophilicity of Cy5.5 was higher than Alexa680. This characteristic may have led to higher non-specific protein binding, although other factors (e.g. charge, molecular weight) might also affect its binding, and pharmacokinetics. This system was validated by comparing it with a similarly labeled humanized anti-CD25 antibody, daclizumab, to demonstrate that this method is widely applicable to a broad range of humanized antibodies.
Fig. 18.
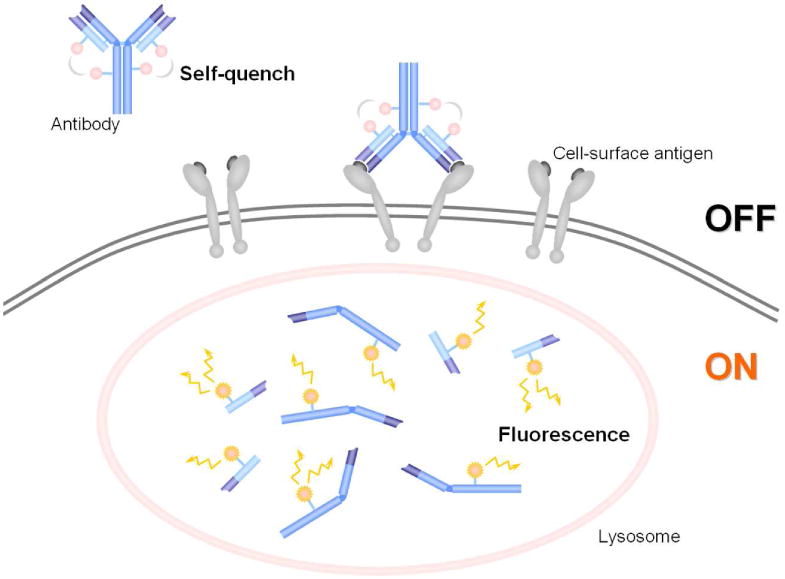
A schema for the concept of a self-quenching activation system with avidin-fluorophore or antibody-fluorophore conjugates. The fluorescence is self-quenched outside of the cell. When it binds to the target and is internalized, it is catabolized within the lysosome and dequenched. Thus, fluorescence is activated only inside the target cells.
Currently indocyanine green (ICG) is the only NIR fluorophore that has been approved by the FDA for clinical use. The absorption peak is at 780 nm and the emission peak is at 820 nm, which is longer than the emission wavelength of Cy5.5, which means better penetration and lower autofluorescence. The ICG-conjugated (Figure 19) antibody, however, loses its fluorescence, a property that has not until recently been understood. Therefore, ICG has not been used in imunohistochemistry in vitro 142,143. However, ICG-conjugated monoclonal antibodies can be used as target cell-specific activatable probes, during in vivo molecular imaging 74. ICG was conjugated to daclizumab, panitumumab (human anti-HER1 monoclonal antibody), and trastuzumab at 1:1 or 1:5 antibody-ICG ratios. The quenching capacities were 43, 6, 58, 44, and 10 for Dac-ICG(1:5), Dac-ICG(1:1), Pan-ICG(1:5), Tra-ICG(1:5) and Tra-ICG(1:1), respectively. Surprisingly, even the 1:1 conjugate showed fluorescence quenching. In addition, the quenching capacity of 1:5 conjugates was extremely high, although self quenching usually leaves a weak residual fluorescence emission. The mechanism has not been entirely elucidated yet, but non-covalent interaction between ICG and hydrophobic amino acid sequences on the IgG might cause loss of fluorescence from the ICG. In vitro microscopy demonstrated fluorescence activation only within target cells, and surface fluorescent signal was undetectable (Figure 20). In in vivo studies, target tumor was specifically visualized with ICG-conjugated daclizumab with a high tumor to background ratio. The fluorescence intensity of the tumor increased only in the target ATAC4 (IL2-Rα+) tumor, and it was higher for Dac-ICG(1:5) than Dac-ICG(1:1). The background and the non-target tumor fluorescence was low for both Dac-ICG(1:1), and Dac-ICG(1:5). When the HER1 targeting Pan-ICG(1:5) and HER2 targeting Tra-ICG(1:5) were injected into mice bearing HER1+ and HER2+ tumors, HER1+ tumors (MDA-MB468 and A431) were only visualized by Pan-ICG(1:5) and not by Tra-ICG(1:5). On the other hand, the HER2+ tumor (3T3/HER2+) was imaged only by Tra-ICG(1:5). Thus, ICG-IgG is an activatable probe for in vivo molecular imaging that can be conjugated with highly specific monoclonal antibodies. It is possible to detect and characterize tumors in vivo using ICG conjugated to antibodies (Figure 21).
Fig. 19.
A chemical schema of the amine-reactive ingocyanine green probe.
Fig. 20.
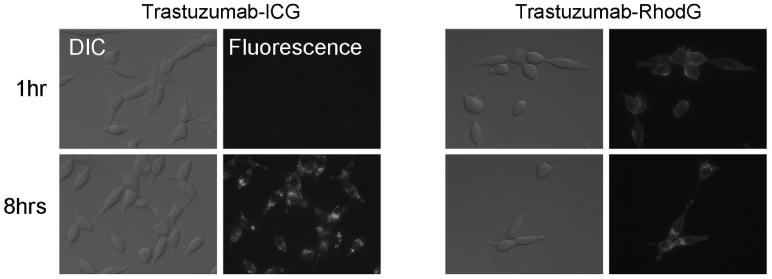
Fluorescence microscopy and differential interference contrast (DIC) images with 3T3/HER2 cells. For trastuzumab-ICG, the fluorescent signal was detected after internalization into the cells by 8hr incubation. The fluorescence signal was not detected by 1hr incubation while the conjugates were outside the cells binding to the surface receptors. In contrast, the always-ON fluorescence probe, trastuzumab-Rhodamine Green (RhodG) showed cell surface fluorescence by 1hr incubation. Many fluorescent dots were observed after the antibody internalization by 8hr incubation as well as the surface fluorescence.
Fig. 21.
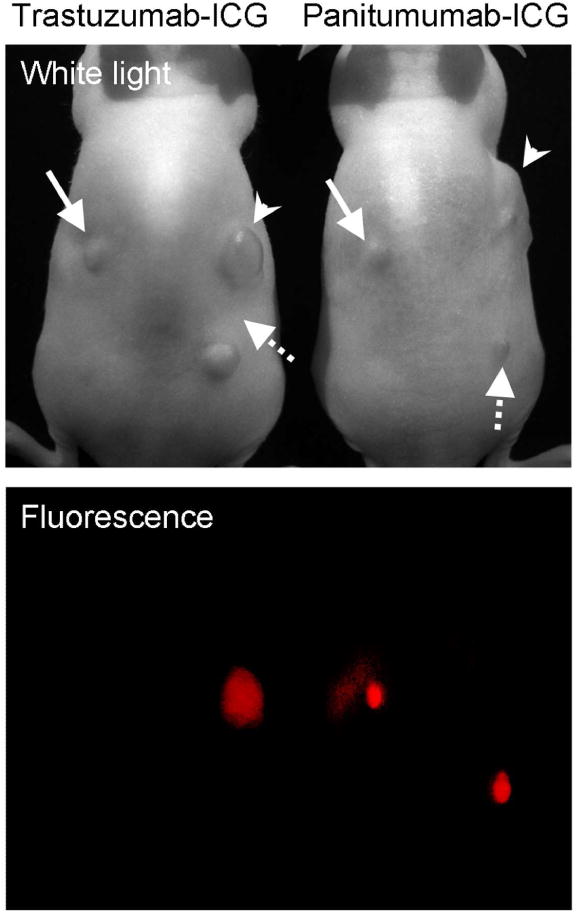
In vivo fluorescence images 4 days after the trastuzumab-ICG or panitumumab-ICG injection to the HER1 positive and HER2 positive tumor bearing mice. Arrow head; 3T3/HER2 tumors (HER2 positive), solid arrow; MDA-MB468 tumors (HER1 positive), dashed arrow; A431 (HER1 positive) tumors. Only the target specific tumor was detected by both conjugates.
5.2.2.2.2 Fluorophore-quencher activation in the targeted cells
Similar to enzyme-specific activatable probes, another method to accomplish target cell-specific fluorescence switching is to utilize fluorophore-quencher combinations. We found that TAMRA (fluorophore) and QSY7 (quencher) combination work well for in vivo molecular imaging by combination with targeting proteins 144. Both TAMRA and QSY7 were conjugated to avidin or trastuzumab to synthesize Av-TM-Q7 or Traz-TM-Q7. Both probes have large quenching capacities, 40-fold for Av-TM-Q7 and 13-fold for Traz-TM-Q7. As expected, activation took place intracellularly by releasing the quencher from TAMRA during lysosomal degradation of the carrier molecule. The tumors in the peritoneal cavity were clearly imaged by Av-TM-Q7. Traz-TM-Q7 visualized lung tumors after intravenous injection with fairly low background signal. The tumor to normal lung tissue ratios of Traz-TM-Q7 was 22.4 ± 4.3, and the ratio was 5.7 ± 1.8 for non-quencher conjugated always-ON probe, Traz-TM. Thus, activatable probes based on a fluorophore-quencher pair conjugated to a targeting ligand are promising for target specific molecular imaging. The mechanism of this activation was thought to be caused by FRET between TAMRA and QSY7. However, since altered absorbance profiles of both TAMRA and QSY7 have been demonstrated, also it is suggested that part of this activation is derived from hetero H-dimer formation with these two different molecules.
5.2.3. Macromolecular targeted agents conjugated to activatable small molecule probes
Macromolecular contrast agents provide a number of advantages over low molecular weight agents. The multiple binding sites afforded by macromolecules allow multiple binding ligands and/or signaling molecules to be bound to the agent. When a targeting moiety and an activatable probe are coupled to a single molecule, the conjugate can be designed to yield a signal only under specific conditions, such as only after binding to a targeted cell type. Since the targeting moiety is independent from the signaling moiety, this design is advantageous compared to conventional strategies because; 1. The targeting moieties on the macromolecule can be flexibly selected to adapt to specific cell surface targets of interest 2. The pharmacokinetics of the probe can be easily altered by changing the size of macromolecule. 3. Appropriate activation mechanisms including oxidation-reduction, or proton concentration (pH) (both potentially reversible), and enzymatic cleavage (usually irreversible) can be used.
Using this concept, Urano and Kobayashi et al. designed a highly specific in vivo cancer imaging probe that incorporates an “activatable” fluorescent imaging probe within the macromolecule. This agent is activated, after cellular internalization, by changes in pH which occur during lysosomal degradation of the probe 36. A library of pH-activatable probes based on the BODIPY fluorophore and the PeT mechanism were created (see Figure 8h), each activating at a different pH, and these were conjugated to a monoclonal antibody targeting a cancer-specific cell surface receptor. These pH-activatable probe-antibody conjugates were almost non-fluorescent under neutral pH conditions, however, after selective uptake by tumor cells the conjugate was transported to the lysosome to undergo degradation at pH 4-5 (Figure 22a). Upon activation the probe became highly fluorescent. As proof of concept, ex and in vivo imaging of human HER2-positive lung tumors in mice were performed with a humanized antibody against HER2, trastuzumab, conjugated with a BODIPY-based pH-activatable small molecular fluorophore. The probes did not yield fluorescence signal on the surface of HER2-positive cells but internalized and activated the signal only in the lysosome in vitro (Figure 22b), and were highly specific for tumors and had only minimal background signal in vivo (Figure 22c).
Fig. 22.
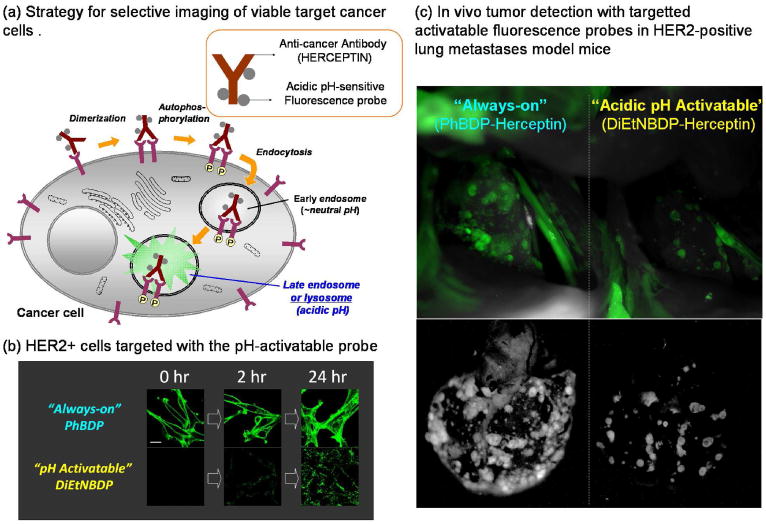
a. A schematic strategy for selective imaging of viable target cancer cells with acidic pH-sensitive small molecular probe conjugated with cell surface molecule targeted monoclonal antibodies. b. Confocal microscope images obtained just after the addition of the probes, and at 2, and 24 hours post-addition of “always on” and “pH-activatable” BDP-conjugated trastuzumab against HER2. c. In vivo tumor detection with targeted activatable fluorescence probes in HER2-positive lung metastases model mice. The pH-activatable probe produces a fluorescence signal only from tumors in the lung. However, the control “always on” probe produces a fluorescence signal not only from tumors, but also from the background normal lung and heart.
As mentioned previously, in vivo tumor imaging with another cancer-targeting molecule, a galactosyl serum albumin (GSA), which is known to bind to a galactose-binding lectin on the cell surface and then be internalized, demonstrated successful imaging with high target to background ratios. The pH-activatable probe-GSA conjugate, was given to living mice with peritoneally disseminated ovarian cancer expressing galactose-binding lectin. By using fluorescence micro-endoscopy, tiny tumor sites, invisible to the naked eye, became clearly visible, demonstrating this method might be useful in situations such as laparoscopic surgery by providing the surgeon with a roadmap of the resectable lesions.
Another advantage of this probe is its reversibility. Because, the probes lose signal when they leak out of the cell and into the more neutral conditions of the extracellular environment, cellular viability can be assessed since this is an energy consuming process. Thus, viable cancer cells can be distinguished from dead or apoptotic cells permitting a better delineation of the relevant target population of cells. Thus, activatable probes conjugated to macromolecular targeting agents have specific advantages over low molecular weight probes.
5.2.4 Pretargeting activation/quenching strategy
A pretargeting activation/quenching strategy is another unique method for the target specific activation. Hama et al.133 and Ogawa et al.145 reported two successful pretargeting activation/quenching systems for target-specific fluorescence imaging. In the strategy described by Hama, biotinylated target-specific monoclonal antibody was first injected, and then after clearance of non-bound antibody, a second injection was made with a non-fluorescing neutravidin-BODIPY conjugate, which was activated upon binding to biotin. This strategy led to the successful visualization of HER1-positive ovarian cancer in the peritoneal cavity with high tumor to background ratios. Another approach was to first target the specific antigen with the injection of a monoclonal antibody conjugated with an “always on” fluorophore and biotin molecules, and then after sufficient accumulation of the fluorescing antibody at the target, another injection was made with a neutravidin-quencher conjugate to minimize the background signal from unbound antibody. Unbound antibody was bound to the neutravidin-quencher reducing signal and resulting in faster clearance via the liver. By using this pretargeting quenching/chasing method, Ogawa et al successfully visualized the HER2-positive breast cancers with minimal background signals.
6. Additional signaling mechanisms using fluorescent probes
For the previously described activation strategies, fluorescence was induced by excitation light leading to emission light. Although fluorescence intensity is the most straightforward way to obtain signal, fluorophores can be also detected by observing other emission properties such as fluorescence decay (lifetime). Moreover light can be induced with acoustic energy. These technologies are still under development for use in medical imaging, however, these agents have great potential to overcome some limitations of conventional optical imaging including the reduction of background signal derived from autofluorescence or the enhancement of tissues that are deep to the skin. In this section, we review these emerging technologies as potential adjuncts to fluorescence imaging.
6.1. Fluorescence lifetime imaging
Fluorescence lifetime imaging is the production of, spatially resolved images which reflect the time required for an excited fluorophore to transition to its ground state (typically in nanoseconds). This lifetime value can be measured through the rate of fluorescence decay, which is proportional to the number of energy dissipation pathways made available to the fluorophore by its environment. Methods to dissipate energy include: fluorescence quantum yield, quenching, internal conversion, photolysis, FRET, etc. This provides a useful additional source of contrast as lifetime is largely independent of fluorophore concentration, light scattering, excitation laser power, and light path length 146,147, and other artifacts which prevent reliable interpretation of fluorescence intensity. If a probe's lifetime is distinct from tissue autofluorescence, fluorescence lifetime imaging provides a simple means of removing autofluorescence from the image.
Fluorescence lifetime is measured through both time domain and frequency domain techniques. Time domain measurements are performed through time correlated single photon counting, and through a series of nanosecond excitation pulses and exposures. This method is more sensitive than frequency domain techniques but has a lower temporal resolution (∼1 per second). Frequency domain measurements are done through the application of an excitation light of a known frequency; demodulating the resulting emission light and determining lifetime from the phase shift 148. This technique has a much higher temporal resolution (∼ 10 per second), allowing a near real time monitoring. Commercial time domain cameras have become available in recent years broadening the applicability of this technique to provide new insights into biology. The majority of work done with fluorescence lifetime has been done with microscopy (confocal, multi-photon, and wide-field microscopes) and is often referred to as fluorescence lifetime imaging microscopy (FLIM). The principle use of FLIM has been imaging of FRET studies. The transfer of energy from the donor to the acceptor in FRET decreases donor lifetime, while increasing the lifetime of the acceptor. FRET-FLIM is advantageous because the presence of FRET can be determined independently of concentration, FRET-FLIM is less susceptible to tissue scattering, the technique offers improved quantification149,150, and allows for the elimination of the potential artifacts of photoconversion of FPs151 and spectral bleedthrough from donor to acceptor152. A recently developed assay technique utilizes multi-photon FRET-FLIM to quantify integrin-GFP, effector-mRFP association, and the role this may play in drug development. Wide field FRET-FLIM revealed intracellular interactions between the transforming oncogene RhoC and RhoGDIγ, a GDP dissociation inhibitor, utilizing enhanced cyan fluorescent protein (donor) and yellow fluorescence protein (acceptor) as a FRET pair, a scenario in which FRET intensity imaging did not produce a clear result153. Beyond FRET imaging, lifetime can be influenced by pH154,155, viscosity156, and oxygen content 157.
Small animal and clinical fluorescence lifetime imaging systems lag behind FLIM in development, but are increasingly becoming more widespread. To image through greater than 1 to 2 mm of tissue requires an accurate photon migration model, which the community only recently appears to be settling upon 158,159. Much work is being done to bring the advantages of FLIM to larger biological systems 159-161.
6.2. Photo-acoustic/Acoustic-optical
Photoacoustic (synonymous with optoacoustic) imaging for biological applications first appeared in the 1970s 162, but only recently have photoacoustic fluorescent probes been explored as imaging agents 163,164. In photoacoustic imaging a pulsatile excitation laser (or radiowave) is applied to a tissue, the differential absorbance of which results in thermal expansion and the propagation of pressure waves, characteristic of each absorbing analyte, to an ultrasonic detector. Several centimeters of tissue can be stimulated at a particular absorption peak and resolved, as acoustic waves demonstrate 2-3 orders of magnitude less scattering in tissue than light165. The limited numbers of fluorescent probes designed for photoacoustic imaging have been in the NIR range, not only to increase penetration depth, but also to avoid endogenous chromophores with high absorbances (most notably hemoglobin). Recently, photoacoustic techniques were able to distinguish HER2(+) and HER2(-) cell lines in vitro utilizing Herceptin-AlexaFluor750 conjugates. This study furthermore demonstrated increasing photoacoustic signal with increasing conjugation ratios, despite decreasing fluorescence at the higher ratios due to quenching166. Alexa Flour 750, ICG 163, Cy5.5167, inorganic dyes (gold nanoparticles168 and nanoshells169), and fluorescent proteins170 have been imaged successfully with photoacoustic techniques.
Supplementary Material
Supporting Information Available:
Supplemental video 1: Videos demonstrating in vivo (video S1) real-time multicolor fluorescence lymphatic imaging. Each draining lymph node can be identified by its distinct color.
Supplemental video 2: Fluorescence-guided laparoscopy of a disseminated ovarian cancer implants model. A tiny implanted tumor, which can hardly be pointed out with the conventional white light endoscope (left window), was clearly detected using the fluorescence guide injected with galactosyl serum albumin-based D-galactose receptor targeted activatable probe on the green fluorescence view (right window). This information is available free of charge via the Internet at http://pubs.acs.org/.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Biographies

Hisataka Kobayashi, MD, PhD
Dr. Hisataka Kobayashi is the Chief of the Basic/Preclinical Development Section in the Molecular Imaging Program at the National Cancer Institute of the National Institutes of Health in Bethesda, MD. Dr. Kobayashi is awarded MD and PhD (Immunology/Medicine) from the Kyoto University in Japan. He joined as a post-doc fellow in the Nuclear Medicine Department at the Clinical Center of the National Institutes of Health in 1995 and moved to the Molecular Imaging Program at NCI in 2004. His interest is in developing the novel molecular imaging agents and technology especially for targeting cancers.

Mikako Ogawa, PhD
Dr. Mikako Ogawa is a postdoctoral fellow in the Molecular Imaging Program at the Center for Cancer Research of the National Cancer Institute in Bethesda, MD. Dr. Ogawa is awarded PhD (Pharmaceutical Sciences) from the Kyoto University in Japan. She joined to the Molecular Imaging Program at NCI in 2007. Her interest is in developing the novel molecular imaging agents.

Raphael Alford
Raphael Alford is in pursuit of his MD degree from Case Western Reserve University School of Medicine. He is a graduate of Duke University, where he received a dual degree in Biomedical and Electrical Engineering. He is interested in pursuing a career in the development and application of molecular imaging technologies.

Peter L. Choyke, M.D.
Dr. Peter Choyke is the Chief of the Molecular Imaging Program at the Center for Cancer Research of the National Cancer Institute in Bethesda, Maryland. Dr. Choyke is a graduate of Jefferson Medical School and received training in Diagnostic Radiology at Yale University and the University of Pennsylvania. He joined the Diagnostic Radiology Department at the Clinical Center of the National Institutes of Health in 1988 and formed the Molecular Imaging Program at NCI in 2004. His interest is in accelerating the treatment of cancer by using novel molecular imaging agents which target specific features of cancers.

Yasuteru Urano, PhD
Dr. Yasuteru Urano is the associate professor of Graduate School of Pharmaceutical Sciences, the University of Tokyo. Dr. Urano is awarded PhD (Pharmaceutical Sciences) from the University of Tokyo in 1995. He joined Graduate School of Pharmaceutical Sciences, the University of Tokyo as a JSPS post-doc fellow in 1995, and was promoted to assistant professor in 1997 and to associate professor in 2005. His interest is in developing novel small molecular-based photo-functional tools such as fluorescence probes, photosensitizers, and caged compounds for biological researches and molecular imaging and treatment of cancers.
References
- 1.Weissleder R, Mahmood U. Radiology. 2001;219:316. doi: 10.1148/radiology.219.2.r01ma19316. [DOI] [PubMed] [Google Scholar]
- 2.Thakur M, Lentle BC. Radiology. 2005;236:753. doi: 10.1148/radiol.2363051160. [DOI] [PubMed] [Google Scholar]
- 3.Schaeffter T. Prog Drug Res. 2005;62:15. doi: 10.1007/3-7643-7426-8_2. [DOI] [PubMed] [Google Scholar]
- 4.Boswell CA, Brechbiel MW. Nucl Med Biol. 2007;34:757. doi: 10.1016/j.nucmedbio.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funovics M, Weissleder R, Tung CH. Anal Bioanal Chem. 2003;377:956. doi: 10.1007/s00216-003-2199-0. [DOI] [PubMed] [Google Scholar]
- 6.Weissleder R, Tung CH, Mahmood U, Bogdanov A., Jr Nat Biotechnol. 1999;17:375. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 7.Ntziachristos V, Yodh AG, Schnall M, Chance B. Proc Natl Acad Sci U S A. 2000;97:2767. doi: 10.1073/pnas.040570597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corlu A, Choe R, Durduran T, Rosen MA, Schweiger M, Arridge SR, Schnall MD, Yodh AG. Opt Express. 2007;15:6696. doi: 10.1364/oe.15.006696. [DOI] [PubMed] [Google Scholar]
- 9.Hawrysz DJ, Sevick-Muraca EM. Neoplasia. 2000;2:388. doi: 10.1038/sj.neo.7900118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi H, Hama Y, Koyama Y, Barrett T, Regino CA, Urano Y, Choyke PL. Nano Lett. 2007;7:1711. doi: 10.1021/nl0707003. [DOI] [PubMed] [Google Scholar]
- 11.Hama Y, Koyama Y, Urano Y, Choyke PL, Kobayashi H. J Invest Dermatol. 2007;127:2351. doi: 10.1038/sj.jid.5700892. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman RM. Nat Rev Cancer. 2005;5:796. doi: 10.1038/nrc1717. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman RM, Yang M. Nat Protoc. 2006;1:1429. doi: 10.1038/nprot.2006.223. [DOI] [PubMed] [Google Scholar]
- 14.Marten K, Bremer C, Khazaie K, Sameni M, Sloane B, Tung CH, Weissleder R. Gastroenterology. 2002;122:406. doi: 10.1053/gast.2002.30990. [DOI] [PubMed] [Google Scholar]
- 15.Hama Y, Urano Y, Koyama Y, Kamiya M, Bernardo M, Paik RS, Krishna MC, Choyke PL, Kobayashi H. Neoplasia. 2006;8:607. doi: 10.1593/neo.06268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheth RA, Upadhyay R, Stangenberg L, Sheth R, Weissleder R, Mahmood U. Gynecol Oncol. 2009;112:616. doi: 10.1016/j.ygyno.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blumenthal D. FDA consumer. U.S Government Printing Office; Washington, DC: 1990. [Google Scholar]
- 18.Butner RW, McPherson AR. Ann Ophthalmol. 1983;15:1084. [PubMed] [Google Scholar]
- 19.Hope-Ross M, Yannuzzi LA, Gragoudas ES, Guyer DR, Slakter JS, Sorenson JA, Krupsky S, Orlock DA, Puliafito CA. Ophthalmology. 1994;101:529. doi: 10.1016/s0161-6420(94)31303-0. [DOI] [PubMed] [Google Scholar]
- 20.Janssen YMW. Lab Invest. 1993;69:261. [PubMed] [Google Scholar]
- 21.Smith K. The Science of Photobiology. Plenum Press; New York: 1977. [Google Scholar]
- 22.Ntziachristos V, Ripoll J, Weissleder R. Opt Lett. 2002;27:333. doi: 10.1364/ol.27.000333. [DOI] [PubMed] [Google Scholar]
- 23.Ntziachristos V, Bremer C, Weissleder R. Eur Radiol. 2003;13:195. doi: 10.1007/s00330-002-1524-x. [DOI] [PubMed] [Google Scholar]
- 24.Chan WC, Nie S. Science. 1998;281:2016. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 25.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Derfus AM, Chan WCW, Bhatia SN. Nano Lett. 2004;4:11. doi: 10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hama Y, Urano Y, Koyama Y, Bernardo M, Choyke PL, Kobayashi H. Bioconjug Chem. 2006;17:1426. doi: 10.1021/bc0601626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longmire MR, Ogawa M, Hama Y, Kosaka N, Regino CA, Choyke PL, Kobayashi H. Bioconjug Chem. 2008;19:1735. doi: 10.1021/bc800140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bremer C, Bredow S, Mahmood U, Weissleder R, Tung CH. Radiology. 2001;221:523. doi: 10.1148/radiol.2212010368. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi T, Urano Y, Kamiya M, Ueno T, Kojima H, Nagano T. J Am Chem Soc. 2007;129:6696. doi: 10.1021/ja070376d. [DOI] [PubMed] [Google Scholar]
- 31.Kamiya M, Kobayashi H, Hama Y, Koyama Y, Bernardo M, Nagano T, Choyke PL, Urano Y. J Am Chem Soc. 2007;129:3918. doi: 10.1021/ja067710a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pham W, Choi Y, Weissleder R, Tung CH. Bioconjug Chem. 2004;15:1403. doi: 10.1021/bc049924s. [DOI] [PubMed] [Google Scholar]
- 33.Law B, Curino A, Bugge TH, Weissleder R, Tung CH. Chem Biol. 2004;11:99. doi: 10.1016/j.chembiol.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 34.Pham W, Weissleder R, Tung CH. Angew Chem Int Ed Engl. 2002;41:3659. doi: 10.1002/1521-3773(20021004)41:19<3659::AID-ANIE3659>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 35.Koide Y, Urano Y, Kenmoku S, Kojima H, Nagano T. J Am Chem Soc. 2007;129:10324. doi: 10.1021/ja073220m. [DOI] [PubMed] [Google Scholar]
- 36.Urano Y, Asanuma D, Hama Y, Koyama Y, Barrett T, Kamiya M, Nagano T, Watanabe T, Hasegawa A, Choyke PL, Kobayashi H. Nat Med. 2009;15:104. doi: 10.1038/nm.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medvedeva N, Martin VV, Weis AL, Likhtenshten GI. J Photochem Photobiol A Chem. 2004;163:45. [Google Scholar]
- 38.Heim R, Cubitt AB, Tsien RY. Nature. 1995;373:663. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 39.Shu X, Royant A, Lin MZ, Aguilera TA, Lev-Ram V, Steinbach PA, Tsien RY. Science. 2009;324:804. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kishimoto H, Zhao M, Hayashi K, Urata Y, Tanaka N, Fujiwara T, Penman S, Hoffman RM. Proc Natl Acad Sci U S A. 2009;106:14514. doi: 10.1073/pnas.0906388106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ando R, Mizuno H, Miyawaki A. Science. 2004;306:1370. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- 42.Alivisatos AP. Science. 1996;271:933. [Google Scholar]
- 43.Burda C, Chen X, Narayanan R, El-Sayed MA. Chem Rev. 2005;105:1025. doi: 10.1021/cr030063a. [DOI] [PubMed] [Google Scholar]
- 44.Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nat Biotech. 2003;21:47. doi: 10.1038/nbt767. [DOI] [PubMed] [Google Scholar]
- 45.Giepmans BNG, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH. Nat Meth. 2005;2:743. doi: 10.1038/nmeth791. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Yan, So Mk, Loening AM, Yao H, Gambhir SS, Rao J. Angew Chem Int Ed Engl. 2006;45:4936. doi: 10.1002/anie.200601197. [DOI] [PubMed] [Google Scholar]
- 47.Yao Hequan, Zhang Y, Xiao F, Xia Z, Rao J. Angew Chem Int Ed Engl. 2007;46:4346. doi: 10.1002/anie.200700280. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Yu WW, Zhang J, Aldana J, Peng X, Xiao M. Phys Rev B. 2003;68:125318. [Google Scholar]
- 49.Ohlson M, Sorensson J, Haraldsson B. Am J Physiol Renal Physiol. 2001;280:F396. doi: 10.1152/ajprenal.2001.280.3.F396. [DOI] [PubMed] [Google Scholar]
- 50.Zimmer JP, Kim SW, Ohnishi S, Tanaka E, Frangioni JV, Bawendi MG. J Am Chem Soc. 2006;128:2526. doi: 10.1021/ja0579816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SW, Zimmer JP, Ohnishi S, Tracy JB, Frangioni JV, Bawendi MG. J Am Chem Soc. 2005;127:10526. doi: 10.1021/ja0434331. [DOI] [PubMed] [Google Scholar]
- 52.Longmire M, Choyke PL, Kobayashi H. Nanomedicine. 2008;3:703. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tung JW, Heydari K, Tirouvanziam R, Sahaf B, Parks DR, Herzenberg LA. Clin Lab Med. 2007;27:453. doi: 10.1016/j.cll.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baumgarth N, Roederer M. J Immunol Methods. 2000;243:77. doi: 10.1016/s0022-1759(00)00229-5. [DOI] [PubMed] [Google Scholar]
- 55.De Rosa SC, Roederer M. Clin Lab Med. 2001;21:697. [PubMed] [Google Scholar]
- 56.Devous MD, Sr, Payne JK, Lowe JL. J Nucl Med. 1992;33:1919. [PubMed] [Google Scholar]
- 57.Du Y, Frey EC. IEEE Nuclear Science Symposium Conference Record. 2007:4213. [Google Scholar]
- 58.Yang M, Luiken G, Baranov E, Hoffman RM. Biotechniques. 2005;39:170. doi: 10.2144/05392BM02. [DOI] [PubMed] [Google Scholar]
- 59.Hoffman RM, Yang M. Nat Protoc. 2006;1:928. doi: 10.1038/nprot.2006.119. [DOI] [PubMed] [Google Scholar]
- 60.Kogure T, Karasawa S, Araki T, Saito K, Kinjo M, Miyawaki A. Nat Biotechnol. 2006;24:577. doi: 10.1038/nbt1207. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi H, Koyama Y, Barrett T, Hama Y, Regino CA, Shin IS, Jang BS, Le N, Paik CH, Choyke PL, Urano Y. ACS Nano. 2007;1:258. doi: 10.1021/nn700062z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kobayashi H, Koyama Y, Barrett T, Hamai Y, Choyke PL. Progress in Biomedical Optics and Imaging - Proceedings of SPIE. SPIE; San Jose, CA: 2008. p. 6867. [Google Scholar]
- 63.Koyama Y, Barrett T, Hama Y, Ravizzini G, Choyke PL, Kobayashi H. Neoplasia. 2007;9:1021. doi: 10.1593/neo.07787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haugland RP. The Handbook—A Guide to Fluorescent Probes and Labeling Technologies. 10th. Molecular Probes; Eugene, OR: 2005. [Google Scholar]
- 65.Osterman H, Schutz-Geschwender A. Seeing beyond the visible with IRDye Infrared Dyes. Licor Biosciences; Lincoln, NE: 2007. [Google Scholar]
- 66.Kovar JL, Simpson MA, Schutz-Geschwender A, Olive DM. Anal Biochem. 2007;367:1. doi: 10.1016/j.ab.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 67.Buschmann V, Weston KD, Sauer M. Bioconjug Chem. 2003;14:195. doi: 10.1021/bc025600x. [DOI] [PubMed] [Google Scholar]
- 68.Lin Y, Weissleder R, Tung CH. Bioconjug Chem. 2002;13:605. doi: 10.1021/bc0155723. [DOI] [PubMed] [Google Scholar]
- 69.Ye Y, Bloch S, Kao J, Achilefu S. Bioconjug Chem. 2005;16:51. doi: 10.1021/bc049790i. [DOI] [PubMed] [Google Scholar]
- 70.Longmire M, Kosaka N, Ogawa M, Choyke PL, Kobayashi H. Cancer Sci. 2009;100:1099. doi: 10.1111/j.1349-7006.2009.01133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hama Y, Koyama Y, Urano Y, Choyke PL, Kobayashi H. Breast Cancer Res Treat. 2007;103:23. doi: 10.1007/s10549-006-9347-0. [DOI] [PubMed] [Google Scholar]
- 72.Kosaka N, Ogawa M, Sato N, Choyke PL, Kobayashi H. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hama Y, Koyama Y, Choyke PL, Kobayashi H. J Biomed Opt. 2007;12:034016. doi: 10.1117/1.2745306. [DOI] [PubMed] [Google Scholar]
- 74.Ogawa M, Kosaka N, Choyke PL, Kobayashi H. Cancer Res. 2009;69:1268. doi: 10.1158/0008-5472.CAN-08-3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barrett T, Koyama Y, Hama Y, Ravizzini G, In SS, Jang BS, Paik CH, Urano Y, Choyke PL, Kobayashi H. Clin Cancer Res. 2007;13:6639. doi: 10.1158/1078-0432.CCR-07-1119. [DOI] [PubMed] [Google Scholar]
- 76.Kosaka N, Ogawa M, Longmire MR, Choyke PL, Kobayashi H. J Biomed Opt. 2009;14:014023. doi: 10.1117/1.3083449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Nature. 1997;388:882. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 78.Rotman B, Papermaster BW. Proc Natl Acad Sci U S A. 1966;55:134. doi: 10.1073/pnas.55.1.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leytus SP, Melhado LL, Mangel WF. Biochem J. 1983;209:299. doi: 10.1042/bj2090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kenmoku S, Urano Y, Kojima H, Nagano T. J Am Chem Soc. 2007;129:7313. doi: 10.1021/ja068740g. [DOI] [PubMed] [Google Scholar]
- 81.Miura T, Urano Y, Tanaka K, Nagano T, Ohkubo K, Fukuzumi S. J Am Chem Soc. 2003;125:8666. doi: 10.1021/ja035282s. [DOI] [PubMed] [Google Scholar]
- 82.Ueno T, Urano Y, Setsukinai K, Takakusa H, Kojima H, Kikuchi K, Ohkubo K, Fukuzumi S, Nagano T. J Am Chem Soc. 2004;126:14079. doi: 10.1021/ja048241k. [DOI] [PubMed] [Google Scholar]
- 83.Umezawa N, Tanaka K, Urano Y, Kikuchi K, Higuchi T, Nagano T. Angew Chem Int Ed Engl. 1999;38:2899. doi: 10.1002/(sici)1521-3773(19991004)38:19<2899::aid-anie2899>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 84.Tanaka K, Miura T, Umezawa N, Urano Y, Kikuchi K, Higuchi T, Nagano T. J Am Chem Soc. 2001;123:2530. doi: 10.1021/ja0035708. [DOI] [PubMed] [Google Scholar]
- 85.Kojima H, Urano Y, Kikuchi K, Higuchi T, Hirata Y, Nagano T. Angew Chem Int Ed Engl. 1999;38:3209. doi: 10.1002/(sici)1521-3773(19991102)38:21<3209::aid-anie3209>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 86.Gabe Y, Urano Y, Kikuchi K, Kojima H, Nagano T. J Am Chem Soc. 2004;126:3357. doi: 10.1021/ja037944j. [DOI] [PubMed] [Google Scholar]
- 87.Kojima H, Hirotani M, Nakatsubo N, Kikuchi K, Urano Y, Higuchi T, Hirata Y, Nagano T. Anal Chem. 2001;73:1967. doi: 10.1021/ac001136i. [DOI] [PubMed] [Google Scholar]
- 88.Sasaki E, Kojima H, Nishimatsu H, Urano Y, Kikuchi K, Hirata Y, Nagano T. J Am Chem Soc. 2005;127:3684. doi: 10.1021/ja042967z. [DOI] [PubMed] [Google Scholar]
- 89.Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. J Biol Chem. 2003;278:3170. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 90.Ueno T, Urano Y, Kojima H, Nagano T. J Am Chem Soc. 2006;128:10640. doi: 10.1021/ja061972v. [DOI] [PubMed] [Google Scholar]
- 91.Izumi S, Urano Y, Hanaoka K, Terai T, Nagano T. J Am Chem Soc. 2009;131:10189. doi: 10.1021/ja902511p. [DOI] [PubMed] [Google Scholar]
- 92.Fujikawa Y, Urano Y, Komatsu T, Hanaoka K, Kojima H, Terai T, Inoue H, Nagano T. J Am Chem Soc. 2008;130:14533. doi: 10.1021/ja802423n. [DOI] [PubMed] [Google Scholar]
- 93.Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. J Am Chem Soc. 2005;127:4888. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- 94.Kamiya M, Urano Y, Ebata N, Yamamoto M, Kosuge J, Nagano T. Angew Chem Int Ed Engl. 2005;44:5439. doi: 10.1002/anie.200501542. [DOI] [PubMed] [Google Scholar]
- 95.Vogel SS, Thaler C, Koushik SV. Sci STKE. 2006;2006:re2. doi: 10.1126/stke.3312006re2. [DOI] [PubMed] [Google Scholar]
- 96.Li IT, Pham E, Truong K. Biotechnol Lett. 2006;28:1971. doi: 10.1007/s10529-006-9193-5. [DOI] [PubMed] [Google Scholar]
- 97.Massey M, Algar WR, Krull UJ. Anal Chim Acta. 2006;568:181. doi: 10.1016/j.aca.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 98.Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Proc Natl Acad Sci U S A. 2006;103:13872. doi: 10.1073/pnas.0601455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fujimoto K, Shimizu H, Inouye M. J Org Chem. 2004;69:3271. doi: 10.1021/jo049824f. [DOI] [PubMed] [Google Scholar]
- 100.Chen Y, Yang CJ, Wu Y, Conlon P, Kim Y, Lin H, Tan W. Chembiochem. 2008;9:355. doi: 10.1002/cbic.200700542. [DOI] [PubMed] [Google Scholar]
- 101.Yang CJ, Jockusch S, Vicens M, Turro NJ, Tan W. Proc Natl Acad Sci U S A. 2005;102:17278. doi: 10.1073/pnas.0508821102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Winnik FM. Chem Rev. 1993;93:587. [Google Scholar]
- 103.Sharma V, Wang Q, Lawrence DS. Biochem Biophys Acta. 2008;1784:94. doi: 10.1016/j.bbapap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lawrence DS, Wang Q. Chembiochem. 2007;8:373. doi: 10.1002/cbic.200600473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Torchilin VP. Eur J Pharm Sci. 2000;11 2:S81. doi: 10.1016/s0928-0987(00)00166-4. [DOI] [PubMed] [Google Scholar]
- 106.Baban DF, Seymour LW. Adv Drug Deliv Rev. 1998;34:109. doi: 10.1016/s0169-409x(98)00003-9. [DOI] [PubMed] [Google Scholar]
- 107.Obermajer N, Doljak B, Kos J. Expert Opin Biol Ther. 2006;6:1295. doi: 10.1517/14712598.6.12.1295. [DOI] [PubMed] [Google Scholar]
- 108.Le Gall C, Bonnelye E, Clezardin P. Curr Opin Support Palliat Care. 2008;2:218. doi: 10.1097/SPC.0b013e32830baea9. [DOI] [PubMed] [Google Scholar]
- 109.Haque A, Banik NL, Ray SK. C N S Neurol Disord Drug Targets. 2008;7:270. doi: 10.2174/187152708784936653. [DOI] [PubMed] [Google Scholar]
- 110.Benes P, Vetvicka V, Fusek M. Crit Rev Oncol Hematol. 2008;68:12. doi: 10.1016/j.critrevonc.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Conus S, Simon HU. Biochem Pharmacol. 2008;76:1374. doi: 10.1016/j.bcp.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 112.Tung CH, Bredow S, Mahmood U, Weissleder R. Bioconjug Chem. 1999;10:892. doi: 10.1021/bc990052h. [DOI] [PubMed] [Google Scholar]
- 113.Bremer C, Tung CH, Bogdanov A, Jr, Weissleder R. Radiology. 2002;222:814. doi: 10.1148/radiol.2223010812. [DOI] [PubMed] [Google Scholar]
- 114.Jaffer FA, Kim DE, Quinti L, Tung CH, Aikawa E, Pande AN, Kohler RH, Shi GP, Libby P, Weissleder R. Circulation. 2007;115:2292. doi: 10.1161/CIRCULATIONAHA.106.660340. [DOI] [PubMed] [Google Scholar]
- 115.Kozloff KM, Quinti L, Patntirapong S, Hauschka PV, Tung CH, Weissleder R, Mahmood U. Bone. 2009;44:190. doi: 10.1016/j.bone.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jezierska A, Motyl T. Med Sci Monit. 2009;15:RA32. [PubMed] [Google Scholar]
- 117.Koutroulis I, Zarros A, Theocharis S. Expert Opin Ther Targets. 2008;12:1577. doi: 10.1517/14728220802560307. [DOI] [PubMed] [Google Scholar]
- 118.Bremer C, Bredow S, Mahmood U, Weissleder R, Tung CH. Radiology. 2001;221:523. doi: 10.1148/radiol.2212010368. [DOI] [PubMed] [Google Scholar]
- 119.Bremer C, Tung CH, Weissleder R. Nat Med. 2001;7:743. doi: 10.1038/89126. [DOI] [PubMed] [Google Scholar]
- 120.Deguchi JO, Aikawa M, Tung CH, Aikawa E, Kim DE, Ntziachristos V, Weissleder R, Libby P. Circulation. 2006;114:55. doi: 10.1161/CIRCULATIONAHA.106.619056. [DOI] [PubMed] [Google Scholar]
- 121.Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Proc Natl Acad Sci U S A. 2004;101:17867. doi: 10.1073/pnas.0408191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Farber SA, Pack M, Ho SY, Johnson ID, Wagner DS, Dosch R, Mullins MC, Hendrickson HS, Hendrickson EK, Halpern ME. Science. 2001;292:1385. doi: 10.1126/science.1060418. [DOI] [PubMed] [Google Scholar]
- 123.Bullok K, Piwnica-Worms D. J Med Chem. 2005;48:5404. doi: 10.1021/jm050008p. [DOI] [PubMed] [Google Scholar]
- 124.Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Nat Chem Biol. 2007;3:668. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- 125.Hsu AR, Hou LC, Veeravagu A, Greve JM, Vogel H, Tse V, Chen X. Mol Imag Biol. 2006;8:315. doi: 10.1007/s11307-006-0059-y. [DOI] [PubMed] [Google Scholar]
- 126.Chen X, Conti PS, Moats RA. Cancer Res. 2004;64:8009. doi: 10.1158/0008-5472.CAN-04-1956. [DOI] [PubMed] [Google Scholar]
- 127.Diagaradjane P, Orenstein-Cardona JM, Colon-Casasnovas NE, Deorukhkar A, Shentu S, Kuno N, Schwartz DL, Gelovani JG, Krishnan S. Clin Cancer Res. 2008;14:731. doi: 10.1158/1078-0432.CCR-07-1958. [DOI] [PubMed] [Google Scholar]
- 128.Ke S, Wen X, Gurfinkel M, Charnsangavej C, Wallace S, Sevick-Muraca EM, Li C. Cancer Res. 2003;63:7870. [PubMed] [Google Scholar]
- 129.Ballou B, Fisher GW, Deng JS, Hakala TR, Srivastava M, Farkas DL. Cancer Detect Prev. 1998;22:251. doi: 10.1046/j.1525-1500.1998.0oa25.x. [DOI] [PubMed] [Google Scholar]
- 130.Gee MS, Upadhyay R, Bergquist H, Alencar H, Reynolds F, Maricevich M, Weissleder R, Josephson L, Mahmood U. Radiology. 2008;248:925. doi: 10.1148/radiol.2482071496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hilger I, Leistner Y, Berndt A, Fritsche C, Haas KM, Kosmehl H, Kaiser WA. Eur Radiol. 2004;14:1124. doi: 10.1007/s00330-004-2257-9. [DOI] [PubMed] [Google Scholar]
- 132.Wu AM, Senter PD. Nat Biotechnol. 2005;23:1137. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 133.Hama Y, Urano Y, Koyama Y, Choyke PL, Kobayashi H. Cancer Res. 2007;67:3809. doi: 10.1158/0008-5472.CAN-06-3794. [DOI] [PubMed] [Google Scholar]
- 134.Hama Y, Urano Y, Koyama Y, Kamiya M, Bernardo M, Paik RS, Shin IS, Paik CH, Choyke PL, Kobayashi H. Cancer Res. 2007;67:2791. doi: 10.1158/0008-5472.CAN-06-3315. [DOI] [PubMed] [Google Scholar]
- 135.Hama Y, Urano Y, Koyama Y, Gunn AJ, Choyke PL, Kobayashi H. Clin Cancer Res. 2007;13:6335. doi: 10.1158/1078-0432.CCR-07-1004. [DOI] [PubMed] [Google Scholar]
- 136.Ogawa M, Kosaka N, Choyke PL, Kobayashi H. ACS Chem Biol. 2009;4:535–46. doi: 10.1021/cb900089j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lopez Arbeloa I, Ruiz Ojeda P. Chem Phys Lett. 1982;87:556. [Google Scholar]
- 138.Kemnitz K, Yoshihara K. J Phys Chem. 1991;95:6095. [Google Scholar]
- 139.Valdes-Aguilera O, Neckers DC. Acc Chem Res. 1989;22:171. [Google Scholar]
- 140.Harari D, Yarden Y. Oncogene. 2000;19:6102. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 141.Ogawa M, Regino CA, Choyke PL, Kobayashi H. Mol Cancer Ther. 2009;8:232. doi: 10.1158/1535-7163.MCT-08-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tadatsu Y, Muguruma N, Ito S, Tadatsu M, Kusaka Y, Okamoto K, Imoto Y, Taue H, Sano S, Nagao Y. J Med Invest. 2006;53:52. doi: 10.2152/jmi.53.52. [DOI] [PubMed] [Google Scholar]
- 143.Muguruma N, Ito S, Hayashi S, Taoka S, Kakehashi H, Li K, Shibamura S, Takesako K. J Gastroenterol. 1998;33:467. doi: 10.1007/s005350050116. [DOI] [PubMed] [Google Scholar]
- 144.Ogawa M, Kosaka N, Longmire MR, Urano Y, Choyke PL, Kobayashi H. Mol Pharm. 2009;6:386. doi: 10.1021/mp800115t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ogawa M, Kosaka N, Choyke PL, Kobayashi H. Bioconjug Chem. 2009;20:147. doi: 10.1021/bc8003765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chang CW, Sud D, Mycek MA. Methods Cell Biol. 2007;81:495. doi: 10.1016/S0091-679X(06)81024-1. [DOI] [PubMed] [Google Scholar]
- 147.van Munster EB, Gadella TW. Adv Biochem Eng Biotechnol. 2005;95:143. doi: 10.1007/b102213. [DOI] [PubMed] [Google Scholar]
- 148.Peter CS, Robert MC. Rev Sci Instrum. 1997;68:4107. [Google Scholar]
- 149.Padilla-Parra S, Audugé N, Coppey-Moisan M, Tramier M. Biophys J. 2008;95:2976. doi: 10.1529/biophysj.108.131276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Spriet C, Trinel D, Riquet F, Vandenbunder B, Usson Y, Heliot L. Cytometry A. 2008;73A:745. doi: 10.1002/cyto.a.20581. [DOI] [PubMed] [Google Scholar]
- 151.Valentin G, Verheggen C, Piolot T, Neel H, Coppey-Moisan M, Bertrand E. Nat Meth. 2005;2:801. doi: 10.1038/nmeth1105-801. [DOI] [PubMed] [Google Scholar]
- 152.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd. Kluwer Academic; Plenum, New York: 1999. [Google Scholar]
- 153.Zhong W, Wu M, Chang CW, Merrick KA, Merajver SD, Mycek MA. Opt Express. 2007;15:18220. doi: 10.1364/oe.15.018220. [DOI] [PubMed] [Google Scholar]
- 154.Hanson KM, Behne MJ, Barry NP, Mauro TM, Gratton E, Clegg RM. Biophys J. 2002;83:1682. doi: 10.1016/S0006-3495(02)73936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hille C, Berg M, Bressel L, Munzke D, Primus P, Lohmannsroben HG, Dosche C. Anal Bioanal Chem. 2008;391:1871. doi: 10.1007/s00216-008-2147-0. [DOI] [PubMed] [Google Scholar]
- 156.Kuimova MK, Yahioglu G, Levitt JA, Suhling K. J Am Chem Soc. 2008;130:6672. doi: 10.1021/ja800570d. [DOI] [PubMed] [Google Scholar]
- 157.Borst JW, Hink MA, van Hoek A, Visser AJ. J Fluoresc. 2005;15:153. doi: 10.1007/s10895-005-2523-5. [DOI] [PubMed] [Google Scholar]
- 158.Vishwanath K, Pogue B, Mycek MA. Phys Med Biol. 2002;47:3387. doi: 10.1088/0031-9155/47/18/308. [DOI] [PubMed] [Google Scholar]
- 159.Hassan M, Riley J, Chernomordik V, Smith P, Pursley R, Lee SB, Capala J, Gandjbakhche AH. Mol Imaging. 2007;6:229. [PMC free article] [PubMed] [Google Scholar]
- 160.Hall DJ, Sunar U, Farshchi-Heydari S, Han SH. Appl Opt. 2009;48:D74. doi: 10.1364/ao.48.000d74. [DOI] [PubMed] [Google Scholar]
- 161.Bloch S, Lesage F, McIntosh L, Gandjbakhche A, Liang K, Achilefu S. J Biomed Opt. 2005;10:054003. doi: 10.1117/1.2070148. [DOI] [PubMed] [Google Scholar]
- 162.Rosencwaig A. Science. 1973;181:657. doi: 10.1126/science.181.4100.657. [DOI] [PubMed] [Google Scholar]
- 163.Kruger RA, Kiser WL, Reinecke DR, Kruger GA, Miller KD. Mol Imaging. 2003;2:113. doi: 10.1162/15353500200303109. [DOI] [PubMed] [Google Scholar]
- 164.Razansky D, Vinegoni C, Ntziachristos V. Opt Lett. 2007;32:2891. doi: 10.1364/ol.32.002891. [DOI] [PubMed] [Google Scholar]
- 165.Wang LV. Med Phys. 2008;35:5758. doi: 10.1118/1.3013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bhattacharyya S, Wang S, Reinecke D, Kiser W, Kruger RA, DeGrado TR. Bioconjug Chem. 2008;19:1186. doi: 10.1021/bc700482u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Razansky D, Baeten J, Ntziachristos V. Med Phys. 2009;36:939. doi: 10.1118/1.3077120. [DOI] [PubMed] [Google Scholar]
- 168.Eghtedari M, Oraevsky A, Copland JA, Kotov NA, Conjusteau A, Motamedi M. Nano Lett. 2007;7:1914. doi: 10.1021/nl070557d. [DOI] [PubMed] [Google Scholar]
- 169.Wang Y, Xie X, Wang X, Ku G, Gill KL, O'Neal DP, Stoica G, Wang LV. Nano Lett. 2004;4:1689. [Google Scholar]
- 170.Razansky D, Vinegoni C, Ntziachristos V. Proceedings of the SPIE, Volume 7177; SPIE: San Jose, CA. 2009:7177. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available:
Supplemental video 1: Videos demonstrating in vivo (video S1) real-time multicolor fluorescence lymphatic imaging. Each draining lymph node can be identified by its distinct color.
Supplemental video 2: Fluorescence-guided laparoscopy of a disseminated ovarian cancer implants model. A tiny implanted tumor, which can hardly be pointed out with the conventional white light endoscope (left window), was clearly detected using the fluorescence guide injected with galactosyl serum albumin-based D-galactose receptor targeted activatable probe on the green fluorescence view (right window). This information is available free of charge via the Internet at http://pubs.acs.org/.