

Abstract
Bladder cancer is associated with enhanced inflammation and characterized by deregulated prostanoid metabolism. Here we examined prostaglandin E2 (PGE2) metabolism and myeloid cell subsets that infiltrate tumor tissue using two xenograft models of human bladder cancer. Human bladder tumor xenografts implanted into athymic nude mice become highly infiltrated with host CD11b myeloid cells of bone marrow origin. Fast growing SW780 bladder tumor xenografts were infiltrated with heterogeneous CD11b myeloid cell subsets including tumor-associated macrophages and myeloid-derived suppressor cells. In contrast, majority of myeloid cells in tumor tissue from slow growing bladder cancer Urothel 11 displayed more immature, homogenous phenotype and comprised mostly MHC II class-negative myeloid-derived suppressor cells. We demonstrate that human bladder tumors secrete substantial amounts of PGE2. Normal bone marrow myeloid cell progenitors cultured in the presence of a bladder tumor-conditioned medium, which is enriched for PGE2, failed to differentiate into mature APCs and acquired phenotype of the myeloid-derived suppressor cells or inflammatory macrophages with up-regulated chemokine receptor CXCR4. Collectively our data demonstrate that enhanced cancer-related inflammation and deregulated PGE2 metabolism in tumor microenvironment promote immunosuppressive pro-tumoral phenotype of myeloid cells in bladder cancer. These data also suggest that not only local tumor microenvironment but other factors such as stage of cancer disease and pace of tumor growth could markedly influence the phenotype, differentiation and immune function of myeloid cells in tumor tissue.
Keywords: Bladder cancer, Tumor microenvironment, PGE2 metabolism, Tumor-infiltrating myeloid cells, Myeloid-derived suppressor cells, Tumor-associated macrophages
1. Introduction
Bladder cancer remains the second most common genitourinary neoplasm and the fifth most common cancer in the United States. Global prevalence of bladder cancer is estimated at 1 million and is steadily increasing [1]. Despite intravesical chemotherapy and/or immunotherapy, up to 80% of patients with non-muscle-invasive bladder cancer develop recurrent tumors, of which 20%–30% evolve into more aggressive, potentially lethal tumors. Although radical cystectomy is considered gold standard for treatment of patients with localized muscle-invasive bladder cancer, nearly half of such patients develop metastases within 2 years after cystectomy and subsequently succumb to the disease.
Recent studies indicate that tumor growth is not just determined by malignant cancer cells themselves, but also by the tumor reactive stroma that is associated with increased number of fibroblasts, enhanced capillary density as well as inflammatory cells. Advanced tumors have the ability to generate local immune suppression and tolerant microenvironment through secretion of inhibitory factors and activation of negative regulatory pathways. Tumor induced immune suppression is frequently associated with cancer inflammation, which in most cases promotes the local tumor growth. One of the major events of cancer-induced inflammation is an up-regulated expression of cyclo-oxygenase 2 (COX2) in tumor resulting in enhanced production of prostaglandins including pro-tumoral PGE2 [2].
PGE2 is the most abundant prostaglandin and its expression markedly increased in human cancer including bladder, colon, lung, breast, and head and neck cancer, and is often associated with a poor prognosis [3,4]. Enhanced secretion of PGE2 by tumor cells is known to result in local immune suppression that in turn mediates malignant transformation and tumor progression [5–8]. Increased secretion of PGE2 has a major impact on intra-tumoral inflammatory cells such as inhibiting APC functions, stimulating Th2 cytokine secretion, and promoting the immunosuppressive microenvironment [9,10].
Furthermore, PGE2 can suppress cytokine and chemokine production by activated macrophages [11], regulate Th17 cell differentiation [12], and exert multiple effects on dendritic cells, including promoting survival [13], affecting antigen-presenting function [14], and regulating cytokine production [15]. PGE2 directly inhibits the activity of cytotoxic T cells through the upregulation of a CD94 and NKG2A complex and induces regulatory T cell function in vitro [16,17].
In addition, PGE2 has been shown to inhibit GM-CSF induced differentiation of myeloid antigen-presenting cells from bone marrow cell progenitors [8], induce in vitro accumulation of myeloid-derived suppressor cells and up-regulate arginase I expression [7]. Together, these observations suggest that elevated level PGE2 contributes to developing tumor-induced immune dysfunction and promoting tumor progression. In the present study we examined PGE2 metabolism and myeloid cell subsets that infiltrate tumor tissue using two xenograft models of human bladder cancer. We find that enhanced cancer-related inflammation and deregulated PGE2 catabolism in tumor microenvironment promote immunosuppressive pro-tumoral phenotype of myeloid cells in bladder cancer tissue.
2. Materials and methods
2.1. Clinical samples from bladder cancer patients
Peripheral blood samples were collected from previously untreated patients diagnosed with bladder cancer at the Department of Urology at the University of Florida (Gainesville, FL). All specimens were obtained following informed consent and approval by the institutional review board. Peripheral blood mononuclear cells (PBMC) from patients and healthy donors were separated by Lymphoprep (Accu-Prep, 1.077 g/ml, Oslo, Norway) density gradient centrifugation.
2.2. Isolation of CD15 and CD33 cell subsets
To isolate CD15 and CD33 cell populations from blood, we first separated peripheral mononuclear cells (PBMC) from patients diagnosed with bladder cancer or healthy donors by Lymphoprep gradient density centrifugation. The CD15+ or CD33+ cells were isolated from the PBMC by positive selection using anti-CD15 or ant-CD33 microbeads and the MACS LS columns according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA). Purity of all isolated cell populations was evaluated by flow cytometry and routinely exceeded 90%.
2.3. Bladder tumor xenograft models
SW780 human bladder transitional carcinoma cell line was obtained from ATCC (Manassas, VA). The Urothel 11 tumor cell line was established in our laboratory. Tumor cells were grown as monolayer cultures in Dulbecco's Modification of Eagle's Medium supplemented with 10% fetal bovine serum, 4.5 g/l glucose, 4 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin in a 5% CO2 humidified atmosphere in a complete culture medium. To establish subcutaneous tumors, athymic nude mice were injected into left flank with 2×106 SW780 or Urothel 11 cells. Mice were maintained under specific pathogen-free conditions throughout the study.
2.4. Isolation of CD11b cells from tumor tissue
At specified time points following the tumor instillation, mice were euthanized in a CO2 chamber and cell suspensions were prepared from solid tumors by enzymatic digestion as described before [9]. Briefly, tumors were harvested from the mice and cut into 1- to 3-mm3 pieces. The minced tissue was incubated at 37 °C in L-15 medium (BioWhittaker/Cambrex) containing FBS (HyClone), antibiotics (penicillin/streptomycin; HyClone) and collagenase cocktail. After washing in PBS, cells were resuspended in the complete medium. Viability of cells measured by trypan blue exclusion exceeded 90%.
2.5. Preparation of primary tumor-conditioned medium (TCM)
SW780 bladder tumor cell suspension was prepared as described above. Single cell suspension was incubated in DMEM/F12 medium (HyClone, Logan, UT, USA) supplemented with 5% FBS and antibiotics (penicillin/streptomycin, Hy-Clone) in culture flasks at a concentration of 2×106 cells/ml. Twenty-four hours later, supernatant (TCM) was collected, filtered, aliquoted and frozen. In all experiments where the presence of tumor-derived factors was required, 25% of SW780 TCM was added in cell cultures.
2.6. Bone marrow cell culture
Bone marrow (BM) cells were seeded into Petri dishes (Corning Inc, Lowell, MA, USA) in 20 ml DMEM/F12 culture medium containing 10 ng/ml of rGM-CSF to remove adherent cells. After 4 h of incubation, floating non-adherent cells were collected, counted, and added to six-well ultra-low attachment plate (Corning Inc, Lowell, MA, USA) at 0.5–1×106 cells/well in 5 ml complete culture medium containing 20 ng/ml of rGM-CSF and/or 25% SW-780 TCM. Fresh GM-CSF and SW780 CM were added to the wells with cells on Days 2, 4, and 6. On Day 8, cells were collected, washed with PBS and stained with indicated antibodies.
2.7. Flow cytometry
A total of 1×106 cells were re-suspended in cold PBS buffer and incubated for 5 min at 4 °C with anti-CD16/CD32 mAbs to block Fc receptors. Cells were additionally incubated for 30 min on ice in 50 μl of staining buffer with 1 μg/ml of relevant fluorochrome-conjugated or matched isotype control antibodies. Labeled cells were washed twice with cold PBS. Flow cytometry data were acquired using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and were analyzed with CXP software (Beckman Coulter, Fullerton, CA). Results were expressed as the percentage of positive cells and mean fluorescence intensity.
2.8. Quantitative RT-PCR
Total cellular RNA was prepared from tumor-infiltrated CD11b using RN-easy Plus mini kit (Qiagen) according to the manufacturer's instructions. Integrity of the RNA was analyzed in 2100 Bioanalyzer (Agilent Technologies Inc). cDNA for each RNA sample was synthesized in 20-μl reactions using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) following the manufacturer's protocol. Quantitative real-time RT-PCR analysis was performed using an Applied Biosystems Prism 7900HT Fast Real-Time PCR System according to manufacturer's specification. cDNA-specific TaqMan®Gene expression assays for 15-(NAD)PGDH and COX2 (Applied Biosystems) were used in the study. A mouse actin-beta (ACTB) was used as an endogenous control (Applied Biosystems). All samples were analyzed in triplicates, and amplification data was analyzed using Applied Biosystems Prism Sequence Detection Software (version 2.2.1). Relative quantification was calculated according to the ΔΔCt method (Applied Biosystems) using a statistical confidence of 99.9%.
2.9. PGE2 production
Analysis of PGE2 production was performed using an ELISA kit and protocol developed by Cayman Chemical (Ann Arbor, MI). Briefly, isolated cells were cultured for 24 h. Cell culture supernatants were collected, filtered and assayed for the presence of PGE2.
2.10. Statistics
The statistical significance between values was determined by the Student t-test. The data were expressed as the mean ± SD. Probability values of ≥ 0.05 were considered nonsignificant. Significant values of p ≤ 0.05 were expressed with an asterisk. The flow cytometry data shown are representative of at least three separate determinations.
3. Results
3.1. Bladder tumors are infiltrated by CD11b myeloid cells
To characterize myeloid cell subsets that infiltrate the bladder cancer tissue we used two bladder tumor xenograft models. To establish subcutaneous tumors, athymic nude mice were injected with fast-growing SW780 bladder tumor cells (2×106/mouse) or slow-growing Urothel 11 bladder tumor cells (2×106/mouse). Animals bearing SW780 tumors were sacrificed three weeks after tumor cell inoculation and those bearing Urothel 11 tumors after three months. To characterize phenotype of tumor-infiltrating cells, tumor tissues were disaggregated with enzymatic cocktail, and single cell suspensions were stained with fluorochrome-conjugated antibodies and analyzed by multi-color flow cytometry.
Results indicate that tissues from bladder cancer xenografts (SW780 and Urothel 1) were infiltrated by a large number of CD11b+ myeloid cells (Fig. 1). We first analyzed myeloid cell subsets presented in the fast growing SW780 tumors (Fig. 2A). Based on expression of F4/80 and CD11c markers, two major cell subpopulations among SW780 tumor-infiltrating myeloid cells can be identified. About two third of the F4/80-positive tumor-infiltrating myeloid cells did not express CD11c marker, whereas the other third co-expressed both F8/40 and CD11c markers (Fig. 2A). Further analysis of those two cell subsets revealed that CD11c+F480+ cells exhibited the phenotype of activated antigen-presenting cells with the high level of MHC class II and CD86 expression on their surface, whereas most of F4/80+CD11c– cells were negative for those markers. F4/80+CD11c– and F4/80+CD11c+ cells differed in expression of chemokine receptor CXCR4. We observed that only F4/80+CD11c+ cells were able to express CXCR4 marker but not F4/80+CD11c– cells. This suggests that F4/80+CD11c+ that also co-expresses MHC class II, CD86 and CXCR4, represents a subset of activated mature myeloid cells that selectively present in fast growing SW780 xenograft but not in slow growing Urothel 11 tumor (Fig. 2B). Furthermore, almost exclusively Ly-6C+F4/80+ and F4/80+ MHC class II-negative myeloid cell subsets were detected in Urothel 11 tumors. Overall, these results clearly indicate that bladder cancer xenografts are highly infiltrated with a heterogeneous population of myeloid cells in which immunosuppressive MHC class II negative Ly-6C+F4/80+ cell subset is predominant.
Fig. 1.
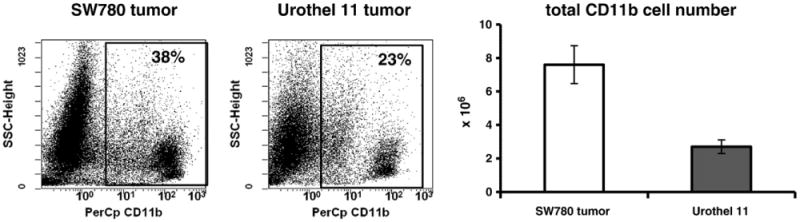
Human bladder cancer xenografts are infiltrated with CD11b myeloid cells. The SW780 or Urothel 11 tumor cells were subcutaneously inoculated into athymic nude mice. Tumor-bearing mice were euthanized at three (SW780) and eleven (Urothel 11) weeks after tumor cell inoculation. Tumor cell suspensions were prepared from individual mice by enzymatic disaggregation of minced tumor as described in Materials and methods and stained with PerCp-conjugated anti-CD11b monoclonal Abs. Percentage of tumor-infiltrated CD11b cells was estimated by flow cytometry. 7-AAD positive cells were excluded from analysis. Results of one out of two similar experiments are shown.
Fig. 2.
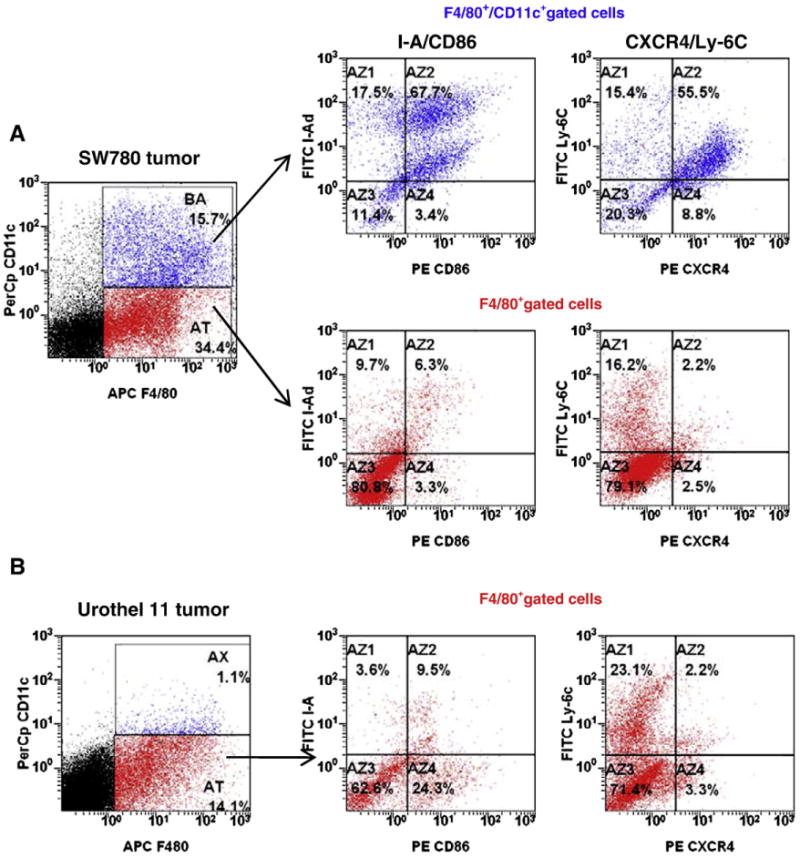
Bladder tumor-infiltrating myeloid cells are heterogeneous. The SW780 cells or Urothel 11 cells were s.c. inoculated into nude mice. Tumor cell suspensions were prepared from individual mice by enzymatic digestion of minced tumor and stained with myeloid subset-specific fluorochrome-conjugated Abs. The presence of myeloid cell subsets in the tumor tissues was assessed by flow cytometry. The F4/80+CD11c+ cells and F4/80+CD11c– cells were gated and further individually analyzed. The results of one out of two similar experiments are shown.
3.2. Evaluation of PGE2 metabolism in the bladder cancer tissue and tumor-infiltrating myeloid cells
Cancer-related inflammation is frequently associated with enhanced PGE2 metabolism. Increased PGE2 level has a major impact on both intra-tumoral immune and inflammatory cells favoring Th2 cytokine milieu, inhibiting APC differentiation, and promoting immunosuppressive microenvironment [5,7,9,18,19]. Of note, up-regulated expression of the PGE2-forming enzyme COX-2 has previously been reported in bladder cancer [20,21].
Here we wanted to evaluate PGE2 production by bladder cancer cells as well as by myeloid cells. To evaluate the levels of PGE2 secreted by epithelial bladder cancer cells we have compared PGE2 production by bladder and kidney human cancer cell lines. To this end, SW780, Urothel 11 (bladder) as well as Caki 1 and A498 (kidney) tumor cell lines were grown as monolayer cultures in a complete culture medium. When cultures reached ∼70% of confluence, cells were detached, counted and transferred in 6-well plates (1×106 cells per well). After culturing for 24 h cell-free culture supernatants were collected and assayed for the presence of PGE2. Fig. 3 (left panel) shows that bladder cancer cells secrete substantial amounts of PGE2 which are higher than the amounts of PGE2 produced by kidney cancer cells.
Fig. 3.

Evaluation of PGE2 production by bladder tumor cells. Human bladder tumor cell lines (SW780, Urothel 11) and human renal cell carcinoma cell lines (Caki 1, A498) were grown as monolayer cultures in complete culture medium. When cultures reached ∼70% of confluence, cells were detached, counted and transferred in 6-well plates (1×106 cells per well). After culturing for 24 h cell-free culture supernatants were collected. The concentration of PGE2 in cell culture supernatants was measured by ELISA (left panel). The CD15 and CD14 cells were isolated individually from PBMC of four bladder patients or healthy donors with magnetic beads as described in Material and methods. PGE2 production by circulating CD15 and CD14 myeloid cell subsets was measured in cell culture supernatants by ELISA (right panel). Average ± SD is shown.
To examine the ability of blood circulating myeloid cells from bladder cancer patients to produce PGE2 we compared PGE2 levels in circulating PBMC-derived myeloid cell subsets obtained from patients suffering from bladder cancer and healthy volunteers. Data presented in Fig. 3 (right panel) demonstrate that overall circulating myeloid cells produce relatively low amounts of PGE2 (500–1200 pg/ml). However, granulocyte-type CD15+ myeloid cells from cancer patients secreted twice as much PGE2 in comparison to similar cells derived from healthy donors.
It is well established that tumors are frequently infiltrated with inflammatory myeloid cells. To better evaluate contribution of bladder tumor-infiltrated inflammatory cells to the enhanced production of PGE2 by bladder tumor cells, we utilized the bladder cancer xenograft model. SW-780 tumor cells were implanted into athymic nude mice and three weeks later tumor-bearing mice were sacrificed in order to get tumor tissue and tumor-infiltrated myeloid cells.
We compared COX-2 expression in cultured SW-780 tumor cell line and in SW-780 tumor ex-vivo excised from tumor-bearing mice. As shown in Fig. 4A, the expression of COX-2 in ex-vivo excised tumors was 4-fold higher compared to the tumor cell line. These data suggest that the presence of host cells in tumor microenvironment results in the markedly increased expression of COX-2 in tumor.
Fig. 4.

Analysis of COX-2 and 15-PGDH gene expression in tumor-infiltrating CD11b cells. (A): SW780 tumor cells (1×106) were injected into athymic nude mice. At day 21 after injection mice were sacrificed, excised tumors were digested with enzymatic cocktail and single cell population was obtained. The expression of COX-2 in tumor cell line and cells from ex-vivo excised SW-780 tumors was analyzed by quantitative RT-PCR. Results of one out of two similar experiments performed with a mixture of cells obtained from three individual mice are displayed. (B and C): CD11b cells were isolated from tumor tissue and spleens of SW780 tumor-bearing mice using magnetic beads. Total RNA was isolated from purified pooled CD11b cells with RNeasy reagent (Qiagen). Quantitative real-time RT-PCR analysis was performed using an Applied Biosystems Prism 7900HT Fast Real-Time PCR System. Samples were run in triplicates, and amplification data were analyzed using Applied Biosystems Prism Sequence Detection Software. Results of one out of two similar experiments performed with a mixture of tumor CD11b cells obtained from three individual mice are displayed.
We examined the possible role of inflammatory tumor-infiltrating myeloid cells in enhanced COX2 expression in the bladder tumor. Tumor-infiltrating CD11b-positive myeloid cells were isolated from SW780 tumor-bearing mice using magnetic beads and expression of COX-2 as well as 15-PGDH was measured using real-time RT-PCR. Results indicate that bladder cancer tissue-infiltrating CD11b myeloid cells express markedly higher levels of COX-2 gene (Fig. 4A and B) and significantly less 15-PGDH gene (Fig. 4C). Together, these results show that tumor-infiltrating CD11b cells express COX2 and are involved in the regulation of PGE2 level in bladder tumor tissue.
3.3. Bladder tumor-conditioned medium inhibits in vitro differentiation of bone marrow-derived antigen-presenting cells and promote accumulation of Ly-6C+F4/80+ monocytic myeloid-derived suppressor cells and Ly-6C+-F4/80+ macrophages
Previous studies have demonstrated that the local microenvironment and cytokine milieu in the tumor block immunological functions of newly recruited mononuclear phagocytes, such as maturation of dendritic cell, antigen presentation and cytotoxicity toward tumors, and divert them toward specialized immunosuppressive tumor-associated macrophages (TAM) or myeloid-derived suppressor cells (reviewed in [22–25]). Recently we showed that addition of CT-26 colon tumor-conditioned medium (TCM) to murine bone marrow (BM) cell cultures skews the GM-CSF-driven generation of Th1-oriented F4/80+CD11c+ myeloid APCs to M2-oriented arginase-expressing macrophage-like cells, enabling them to produce substantial amounts of IL-10, VEGF and MIP-2 [18]. We tested whether the culturing of BM cells in the presence of the SW-780 human bladder tumor-derived conditioned medium inhibits the in vitro GM-CSF-driven development of myeloid CD11c+ DC. Fig. 5 shows that the addition of bladder TCM to the cell culture with GM-CSF profoundly decreased the expression of CD11c (from 93% down to 10%) but not the F4/80 marker (Fig. 5A), as compared to control cell culture where only GM-CSF was added. Moreover, TCM markedly reduced the expression of the MHC class II molecule (Fig. 5B).
Fig. 5.
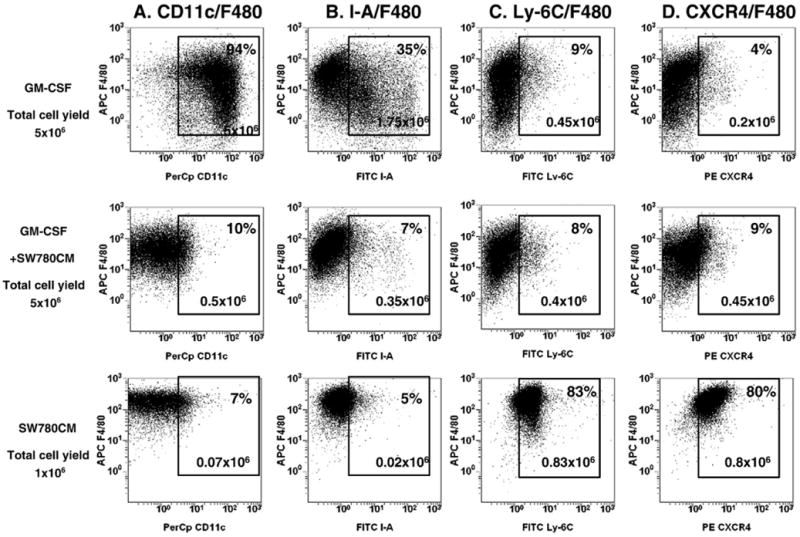
SW780 bladder tumor-conditioned medium inhibits GM-CSF-driven differentiation of bone marrow-derived myeloid antigen-presenting cells and promotes accumulation of myeloid cells with immunosuppressive phenotype. BM cells from naive BALB/c mice were cultured in the presence of mouse rGM-CSF (20 ng/ml) and/or SW780 conditioned medium (25% of total volume) in ultra-low attachment plates. On day eight cells were collected, washed, stained with CD11c and F4/80 (A), F4/80 and I-A (B), F4/80 and Ly-6C (C) and F4/80 and CXCR4 (D) antibodies and analyzed by flow cytometry. Results of one representative experiment out of three are shown.
It is accepted that tumor-derived factors promote the accumulation of immature myeloid cells or MSDCs [26]. To address this issue, we examined whether human bladder SW-780 cell line derived conditioned media could impact markers of MDSCs, such as Ly-6G (granulocytic MDSC) or Ly-6C (monocytic MDSC). We find that the culturing of BM cells in the presence of SW-780 TCM alone induces expression of monocyte/macrophage marker F4/80 and Ly-6C (Fig. 5C), a marker commonly associated with mononuclear myeloid-derived suppressor cells. There was no detectable expression of Ly-6G marker on the surface of BM cells after 8 days of culture in the presence of TCM alone or together with GM-CSF. In addition, bladder TCM alone promoted the expression of the SDF-1 chemokine receptor CXCR4 (Fig. 5D). Addition of TCM prepared from Urothel 11 tumors, instead of SW780, elicited similar results (data not shown). Taken together, these results demonstrate that bladder tumors are able to inhibit GM-CSF-driven in vitro differentiation of BM myeloid cell progenitors into mature APCs and promote expansion of myeloid-derived suppressor cells and macrophages. Furthermore, this phenotype of myeloid cells cultured in the presence of tumor-derived factors highly resembles the phenotype of myeloid cells isolated from bladder tumor tissues.
4. Discussion
Multiple immunosuppressive cell types and inflammatory mediators are mobilized into tumor tissue in response to inflammation similar to that seen in chronic inflammatory responses and tissue repair. A significant portion of inflammatory cells in tumor tissue is represented by tumor-recruited CD11b cells that play multiple roles in cancer progression, contributing to local immunosuppression, tumor neovasculogenesis and development of metastasis [9,26–28].
Tumor-infiltrated myeloid CD11b cells comprised heterogeneous subpopulations that include tumor-associated macrophages, immature dendritic cells and myeloid-derived suppressor cells (MDSC). Induction of MDSCs is considered an important immune-evading strategy used by tumors [29,30]. Numerous studies have shown that tumor growth is accompanied by the increased presence of MDSCs in peripheral immune organs, blood and in tumor tissue. In murine tumor models, MDSC co-expresses CD11b and Gr-1 markers [31,32] and consists of two major subsets: monocytic Ly6G– MO-MDSCs and granulocytic Ly6G+ PMN-MDSCs [33,34]. MDSCs migrate from peripheral lymphoid organs or from bone marrow into tumor site and differentiate mostly into F4/80-positive tumor-associated macrophages [35]. However, limited available data and poor understanding of relationship between different myeloid cell populations within the tumor site limit our understanding of the biology of tumor progression and the development of targeted therapeutics [36].
In the present study we characterized the phenotype of myeloid cell infiltrated bladder tissue and evaluated the production of PGE2 in tumor tissue. We have demonstrated that bladder tumors are highly infiltrated with a heterogeneous population of myeloid cells where the MHC class II negative Ly-6C+F4/80+ cells were the most prominent subset. These results are in agreement with previous studies showing that the tumor-infiltrating myeloid compartment is highly heterogeneous, with coexistence of distinct subsets of mononuclear phagocytes [36,37]. Emerging evidence demonstrates that at the murine mammary tumor site, Ly6Chi monocytes exclusively give a rise to distinct subsets of inflammatory macrophages [38]. Our results indicate that human bladder tumor implanted into athymic nude mice become profoundly infiltrated with Ly-6C+CD11b myeloid-derived suppressor cells and Ly-6C-negative F4/80-positive tumor-associated macrophages. Accumulation of Ly6C+F/40+CD11c– myeloid-derived suppressor cells could be reproduced in in vitro experiments when bone marrow myeloid progenitors were cultured in the presence of bladder TCM alone (Fig. 5). Addition of GM-CSF to the bone marrow cultures together with bladder tumor-conditioned medium promoted differentiation of Ly6C+F/40+ MDSCs into MHC class II-negative Ly6C– F4/80+ macrophages.
Bladder tumor-infiltrated myeloid cell subsets also co-express CXCR4. It has been reported that CXCR4 involved in the spread and progression of a variety of different tumors such as myelogenous leukemia [38,39] and breast carcinoma [40]. CXCR4 activation by its ligand CXCL12 induces migration and/or survival of the neoplastic cells, including tumor cells from neuroblastoma [41], colorectal [42], prostate [43], melanoma [44] and ovarian cancers [45]. It has been demonstrated that CXCR4-positive myeloid cells participate in tumor angiogenesis [27]. Thus, the CXCR4 positive myeloid cells can also contribute to tumor progression. Promising results in preclinical tumor models indicate that CXCR4 antagonists may have antitumor activity in patients with various malignancies [46,47]. Overall, the bladder tumor tissue accumulates or forms the different myeloid cell populations that are known to promote growth of tumor cells.
Striking inhibitory effects of human bladder tumor supernatants (SW780, Urothel 11) on in vitro differentiation of murine bone marrow-derived APCs are likely associated with high PGE2 concentrations in bladder tumor supernatants. These data are consistent with previously published reports that indicate that high PGE2 levels directly inhibit APC differentiation from bone marrow myeloid cell progenitors trough PGE2-specific EP2 and EP4 receptors. PGE2 is not a species-specific lipid, and therefore in human xenograft tumor model PGE2 of human origin could easily bind to the PGE2-specific EP2/EP4 receptors on myeloid cells of murine origin and exert its inhibitory effects.
Interestingly that enhanced tumor progression in mice bearing SW780 tumors was accompanied with enhanced influx of myeloid cells in tumor tissue and relatively “mature”, more differentiated phenotype (F4/80+CD11c+CXCR4+ and F4/80+CD11c+CD86+MHCII+) of tumor-infiltrating myeloid cell subsets. In contrast, progression of “slow growing” Urothel 11 tumors was associated with less differentiated tumor-infiltrating myeloid cells that lack expression of CD11c, CD86 and CXCR4. It is possible that more differentiated and “educated” in tumor microenvironment myeloid cells are more efficient in promotion of tumor progression through mechanism(s) that might include stimulation of tumor angiogenesis, stimulation of tumor cell proliferation and evasion of immune surveillance.
One such mechanism could be represented by enhanced metabolism of PGE2 in tumor microenvironment. It is established that in addition to tumor-derived PGE2, tumor-infiltrating myeloid cells also express COX2 and are able to produce PGE2. Concerted secretion of PGE2 by epithelial cancer cells, inflammatory tumor-infiltrating myeloid cells and possibly by cancer-associated fibroblasts promotes increased PGE2 levels in bladder tumor microenvironment. Moreover, down-regulated expression of PGE2-degrading enzyme 15-PGDH in bladder cancer cells [48] and in tumor-infiltrating myeloid cells (current study) further maintains elevated PGE2 levels in bladder tumor tissue. As a result, high PGE2 levels in bladder tumor micro-environment lead to poor differentiation of recruited myeloid cells and accumulation of immunosuppressive myeloid cells in tumor tissue.
Collectively, these and previously published data strongly suggest that enhanced and disturbed metabolism of PGE2 in some cancers such as bladder cancer could contribute to inhibition of differentiation of recruited myeloid cells and promote accumulation of myeloid-derived suppressor cells within tumor tissue thus promoting tumor escape from immune recognition and destruction. Correction of PGE2 metabolism in bladder tumor microenvironment could provide an effective way to improve differentiation of myeloid cells within tumor bed and reduce numbers of tumor-infiltrating immunosuppressive myeloid cells. It is important to note that bladder cancer is an appealing candidate for novel modalities such as gene therapy or immunotherapy due to its accessibility through transurethral noninvasive drug delivery or inspection. One of the promising potential approaches could be the overexpression of PGE2-inactivating enzyme 15-PGDH in bladder cancer tissue which could provide a new therapeutic tool for treatment of this deadly disease.
Acknowledgments
This work has been supported by grant from James & Esther King Florida Biomedical Research Program to S.K.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:30–42. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, Dubois R. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–93. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nat Rev Cancer. 2005;5:713–25. doi: 10.1038/nrc1697. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–22. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stolina M, Sharma S, Lin Y, Dohadwala M, Gardner B, Luo J, et al. Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J Immunol. 2000;164:361–70. doi: 10.4049/jimmunol.164.1.361. [DOI] [PubMed] [Google Scholar]
- 6.Sombroek CC, Stam AGM, Masterson AJ, Lougheed SM, Schakel MJAG, Meijer CJLM, et al. Prostanoids play a major role in the primary tumor-induced inhibition of dendritic cell differentiation. J Immunol. 2002;168:4333–43. doi: 10.4049/jimmunol.168.9.4333. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez PC, Hernandez CP, Quiceno D David, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–9. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 9.Eruslanov E, Kaliberov S, Daurkin I, Kaliberova L, Buchsbaum D, Vieweg J, et al. Altered expression of 15-hydroxyprostaglandin dehydrogenase in tumor-infiltrated CD11b myeloid cells: a mechanism for immune evasion in cancer. J Immunol. 2009;182:7548–57. doi: 10.4049/jimmunol.0802358. [DOI] [PubMed] [Google Scholar]
- 10.Daurkin I, Eruslanov E, Vieweg J, Kusmartsev S. Generation of antigen-presenting cells from tumor-infiltrated CD11b myeloid cells with DNA demethylating agent 5-aza-2′-deoxycytidine. Cancer Immunol Immunother. 2010;59:697–706. doi: 10.1007/s00262-009-0786-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takayama K, García-Cardena G, Sukhova G, Comander J, Gimbrone M, Libby JP. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J Biol Chem. 2002;277:44147–54. doi: 10.1074/jbc.M204810200. [DOI] [PubMed] [Google Scholar]
- 12.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, et al. Prostaglandin E2–EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15:633–40. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 13.Vassiliou E, Sharma V, Jing H, Sheibanie F, Ganea D. Prostaglandin E2 promotes the survival of bone marrow-derived dendritic cells. J Immunol. 2004;173:6955–64. doi: 10.4049/jimmunol.173.11.6955. [DOI] [PubMed] [Google Scholar]
- 14.Sharma S, Stolina M, Yang SC, Baratelli F, Lin JF, Atianzar K, et al. Tumor cyclo-oxygenase 2-dependent suppression of dendritic cell function. Clin Cancer Res. 2003;9:961–8. [PubMed] [Google Scholar]
- 15.Khayrullina T, Yen JH, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J Immunol. 2008;181:721–35. doi: 10.4049/jimmunol.181.1.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma S, Yang SH, Zhu L, Reckamp K, Gardner B, Baratelli B, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005;65:5211–20. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- 17.Zeddou M, Greimers R, Valensart N, Nayjib B, Tasken K, Boniver J, et al. Prostaglandin E2 induces the expression of functional inhibitory CD94/NKG2A receptors in human CD8+ T lymphocytes by a cAMP dependent protein kinase A type I pathway. Biochem Pharmacol. 2005;70:714–24. doi: 10.1016/j.bcp.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 18.Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal advance: tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages cells by altering intracellular PGE2 catabolism in myeloid cells. J Leukoc Biol. 2010;88:839–48. doi: 10.1189/jlb.1209821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Yamagata N, Yadav R, Brandon S, Courtney RL, Morrow JD, et al. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111:727–35. doi: 10.1172/JCI16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wülfing C, Eltze E, von Struensee D, Wülfing P, Hertle L, Piechota H. Cyclooxygenase-2 expression in bladder cancer: correlation with poor outcome after chemotherapy. Eur Urol. 2004;45:46–52. doi: 10.1016/j.eururo.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 21.Wild P, Kunz-Schughart A, Stoehr R, Burger M, Blaszyk H, Simon R, et al. High-throughput tissue microarray analysis of COX2 expression in urinary bladder cancer. Int J Oncol. 2005;27:385–91. [PubMed] [Google Scholar]
- 22.Pollard J. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 23.Mantovani A, Schioppa T, Porta C, Allavena P, Sica A Antonio. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–22. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 24.Kusmartsev S, Gabriovich D. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Rev Metastasis. 2006;25:323–31. doi: 10.1007/s10555-006-9002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talmadge J, Donkor M, Scholar E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev. 2007;26:373–400. doi: 10.1007/s10555-007-9072-0. [DOI] [PubMed] [Google Scholar]
- 26.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Du R, Lu K, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–20. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagaraj S, Gabrilovich D. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–44. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 31.Kusmartsev S, Li Y, Chen SH. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 co-stimulation. J Immunol. 2000;165:779–85. doi: 10.4049/jimmunol.165.2.779. [DOI] [PubMed] [Google Scholar]
- 32.Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162:5728–37. [PMC free article] [PubMed] [Google Scholar]
- 33.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 34.Youn J, Nagaraj S, Collazo M, Gabrilovich D. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kusmartsev S, Gabrilovich D. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880–91. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 36.Corzo C, Condamine T, Lu L, Cotter M, Youn J, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–53. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Movahedi K, Laoui D, Gyseman SC, Baeten M, Van den Bossche J, Mack M, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C (high) monocytes. Cancer Res. 2010;70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 38.Burger J, Burger M, Kipps T. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94:3658–67. [PubMed] [Google Scholar]
- 39.Mohle R, Bautz F, Rafii S, Moore M, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. 1998;91:4523–30. [PubMed] [Google Scholar]
- 40.Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan M, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 41.Geminder H, Sagi-Assif O, Goldberg L, Meshel T, Rechavi G, Witz I, et al. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J Immunol. 2001;167:4747–57. doi: 10.4049/jimmunol.167.8.4747. [DOI] [PubMed] [Google Scholar]
- 42.Zeelenberg I, Ruuls-Van Stalle L, Roos E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res. 2003;63:3833–9. [PubMed] [Google Scholar]
- 43.Sakamoto N, Porvasnik S, Kusmartsev S, Eruslanov E, Cao W, Urbanek C, et al. Effects of CXCR4 antagonist CTCE-9908 on prostate tumor growth. Prostate. 2009;69:1460–9. doi: 10.1002/pros.21008. [DOI] [PubMed] [Google Scholar]
- 44.Scala S, Ottaiano A, Ascierto P, Cavalli M, Simeone E, Giuliano P, et al. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clin Cancer Res. 2005;11:1835–41. doi: 10.1158/1078-0432.CCR-04-1887. [DOI] [PubMed] [Google Scholar]
- 45.Scotton C, Wilson L, Scott K, Stamp G, Wilbanks D, Fricker S, et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002;62:5930–8. [PubMed] [Google Scholar]
- 46.Devine S, Flomenberg N, Vesole D, Liesveld J, Weisdorf D, Badel K, et al. Rapid mobilization of CD34+ cells following administration of the CXCR4 antagonist AMD3100 to patients with multiple myeloma and non-Hodgkin's lymphoma. J Clin Oncol. 2004;22:1095–102. doi: 10.1200/JCO.2004.07.131. [DOI] [PubMed] [Google Scholar]
- 47.Tamamura H, Hori A, Kanzaki N, Hiramatsu K, Mizumoto M, Nakashima H, et al. T140 analogs as CXCR4 antagonists identified as antimetastatic agents in the treatment of breast cancer. FEBS Lett. 2003;550:79–83. doi: 10.1016/s0014-5793(03)00824-x. [DOI] [PubMed] [Google Scholar]
- 48.Tseng-Rogenski S, Gee J, Ignatoski KW, Kunju LP, Bucheit A, Kintner HJ, et al. Loss of 15-hydroxyprostaglandin dehydrogenase expression contributes to bladder cancer progression. Am J Pathol. 2010;176:1462–8. doi: 10.2353/ajpath.2010.090875. [DOI] [PMC free article] [PubMed] [Google Scholar]