

Abstract
Axonal pathology is a prevalent feature of Alzheimer’s disease (AD) and is thought to occur predominantly due to the accumulation of amyloid beta (Aβ). However, it remains unclear whether therapeutics geared towards reducing Aβ improves axonal deficits. We have previously used Manganese Enhanced MRI (MEMRI) to demonstrate that axonal transport deficits occur before plaque formation in the Tg2576 mouse model of AD. Here we tested whether axonal transport deficits in the Tg2576 mouse model improve in response to the Aβ42 selective lowering agent R-Flurbiprofen (R-F). We demonstrated that in young animals (before Aβ plaque formation), R-F treatment reduced Aβ42 levels and coincided with a significant improvement in axonal transport (p=0.0186) iHowever, in older animals (after plaque formation had occurred), we observed that R-F treatment did not reduce Aβ42 levels although we still observed a significant improvement in axonal transport as assessed with MEMRI (p=0.0329). We then determined that R-F treatment reduced tau hyper-phosphorylation in the older animals. These data indicate that both Aβ42 and tau comprise a role in axonal transport rate deficits in the Tg2576 models.
1. Introduction
Axonal pathology is thought to be a major contributor to Alzheimer’s Disease pathology (AD). Histological analyses of AD tissue from humans and transgenic mouse models indicate that axonal swellings likely precede synaptic deficits and could be an early indicator of AD (1–3). Previously we have reported in vivo axonal transport deficits in the Tg2576 mouse model of AD utilizing Manganese Enhanced MRI (MEMRI) (4). Using MEMRI, we demonstrated that axonal transport deficits occurred as early as 7 months of age, which is prior to Aβ plaque formation in this particular mouse model, and that the axonal transport deficits progressively worsened after plaque formation. It is important to note that the Tg2576 mouse does not exhibit neuronal loss (5–9) and that the deficits we reported were due to change in function rather than the simple loss of neurons (4). As this occurred in an animal model that exhibited increasing levels of Aβ without neuronal loss, we hypothesized that a reduction of Aβ42, the more toxic form of Aβ, should be sufficient to improve axonal transport.
R-Flurbiprofen (R-F) is an enantiomer of the non-steroidal anti-inflammatory drug (NSAID) flurbiprofen capable of reducing Aβ42). Flurbiprofen is typically produced in the racemic form and relieves pain predominantly by inhibiting cyclooxygenase-2 (COX-2), an enzyme that induces pain and inflammation. The R-F isoform has been shown to have a COX-2 independent pathway of reducing pain without the gastro-intestinal side effects associated with traditional COX-2 inhibiting NSAIDs (12–13). Currently, R-F also is thought to modulate the γ-secretase cleavage of Aβ42 to a shorter, less toxic peptide (10). In vivo application of R-F has been reported to reduce Aβ42 levels in the Tg2576 mouse model of AD at 3 months of age and at 20 months of age (10,).
The goal of this study was to determine whether R-F treatment results in improvements in axonal transport due to a reduction in Aβ42. Collectively, our data indicate that there is an association between R-F treatment and axonal transport improvements before and after plaque formation in the Tg2576 mouse model of AD through not only modulation of Aβ levels but also through a reduction in tau phosphorylation.
2. Materials and Methods
2.1 Animals
Tg2576 mice overexpressing human SwAPP695(K670N/M671L), the Swedish familial AD mutation and littermate controls were used for this study (14). Male Tg2576 mice (Tg+) were crossed with C57BL6/SJL F1 females to obtain Tg2576 overexpressing mice and littermate controls (WT). WT and Tg+ animals of both genders were treated and imaged at 7 months (pre-plaque) and 12–13 months (post-plaque) for these studies. All animal studies were conducted in accordance with the Baylor College of Medicine Institutional Animal Care and Use Committee.
2.2 Treatment
R-Flurbiprofen (R-F) obtained from Sigma, St. Louis, MO (545740) was suspended in extra virgin olive oil (vehicle) and administered by oral gavage. Animals treated in the pre-plaque and post-plaque age ranges and in the acute treatment paradigm (3 days) received 25 mg/kg/day. This dose was indicated as being effective at reducing Aβ levels previously in Tg+ mice (10). The 12–13 month old mice (post-plaque age range) in the chronic treatment paradigm (10 days) were administered a dose of 10 mg/kg/day to avoid previously reported toxicity associated with longer treatment of R-F (15).
2.3 In vivo axonal transport measurements by MEMRI
Axonal transport was measured in vivo utilizing MEMRI following minor modifications of our previously described paradigm (4).
Mn2+ Administration
MnCl2 was administered to mice within 3 hours following the final treatment with R-F. First, animals were anesthetized with 5% isoflurane, given a nasal lavage of 4 μl of 0.75 g/ml MnCl2 (Sigma, St. Louis, MO) dissolved in nanopure water, and then allowed to recover on a heating pad. 45 minutes post lavage, the mice were prepared for imaging. Anesthesia was induced with 5% isoflurane and maintained with 2% isoflurane in 100% O2. We chose this time point for imaging because the Mn2+ has not yet had the chance to travel-trans-synaptically. Animals were placed in the prone position on a Bruker mouse holder (Bruker BioSpin, Billerica, MA). Respiration was monitored with a pressure pad placed under the animal while temperature was monitored by the use of a rectal probe and maintained at 37°C via an air heating system (SA Instruments, Inc, Stony Brook, NY). The core temperature was maintained at 37°C during scanning.
MRI Images were acquired utilizing a 9.4T, Bruker Avance Biospec Spectrometer, 21 cm bore horizontal scanner with a 35 mm volume resonator (Bruker BioSpin, Billerica, MA). The imaging parameters to acquire multi-spin/multi-echo MEMRI images of the olfactory bulbs were as follows: TR = 500 ms; TE = 10.2 ms; FOV = 3.0 cm; slice thickness = 1mm; matrix = 128 × 128; NEX = 2; number of cycles = 15; each cycle took approximately 2 min 8 sec to acquire using Paravision software version 4 (Bruker BioSpin, Billerica, MA).
2.4 MRI Analysis
The region of interest (ROI) was determined as previously reported (4). Briefly, the ROI was placed on an axial slice 1 mm in front of the posterior of the olfactory bulb (OB). The ROI was vertically centered on the dorsal olfactory neuronal layer (ONL) (16). The radiologic left side of the olfactory bulb was chosen due to a chemical shift artifact on the right side as we have previously noted (17). The ROI was determined by measuring the length of the olfactory bulb, locating the midpoint of this line, then extending this midpoint out to the ONL using a 90° angle. The region closest to the midpoint within 5% error was established as the ROI for all images (16). This ROI was copied for each cycle and each ROI value normalized to the unaffected muscle of the same slice. The small region of interest collected in this study is representative of a single fascicle of axons projecting into the olfactory neuronal layer (18). Axonal transport of Mn2+ was calculated as the change in signal intensity over time (min)). For the 3 day treatment in the pre-plaque age range of animals, N = 10 and 7 for vehicle and R-F treated WT, and N = 8 and 9 for vehicle and R-F treated Tg2576 animals, respectively. For the 3 day treatment in the post-plaque age range of animals, N = 10 and 9 for vehicle and R-F treated WT animals and N = 7 and 8 for vehicle and R-F treated Tg2576 animals. For the 10 day treatment in the post-plaque age range of animals, N = 6 and 8 for vehicle and R-F treated WT animals and N = 7 and 6 for vehicle and R-F treated Tg2576 animals.
2.5 Brain Tissue Preparation
Following imaging, the brain was collected, separated into hemi-brain and olfactory bulb then flash frozen on dry ice for ELISA assays and Western blotting, respectively. Samples were homogenized with Tris-buffered Saline (TBS) extraction buffer (NaCl 137mM; Tris 20mM; EDTA 5mM; NaF 50mM; protease and phosphatase inhibitor cocktails). All chemicals unless otherwise specified were obtained from Sigma, St Louis, MO.
2.6 ELISA Assays
ELISA assays for Aβ40 and Aβ42 level determination were purchased from Covance and used according to manufacturer instructions (BetaMark ELISA kit x-40 or x-42). The initial homogenization was followed by the addition of sodium dodecyl sulfate (SDS) to a homogenized aliquot with a final concentration of 2% SDS. The samples were then sonicated and centrifuged at 100,000g for 1 hour. The supernatant, representing the SDS soluble fraction, was collected and saved at −80C until further use. The remaining pellet was suspended in a solution of 70% formic acid (FA), sonicated, and centrifuged at 100,000g for 1 hour. The recovered intermediate fraction, placed in neutralization buffer (1M Tris; 0.5M Na2HPO4), is referred to as the FA fraction. The SDS fraction has previously been shown to include the less aggregated forms of Aβ, whereas the FA fraction includes Aβ directly from already formed plaques. Equivalent protein fractions, determined with the Bradford method (Biorad, Hercules, CA), were loaded and the peptide concentration determined. Aβ levels were also tested in WT mice as a control for the assay, and Aβ concentrations were below detectable levels (data not shown). For mice receiving 3 days of treatment, N = 5 per group for the pre-plaque group and N = 6 for the post-plaque group. For mice receiving 10 days of treatment, N = 6 and 7 for the Tg2576 animals.
2.7 Immunoblotting
Olfactory bulbs were suspended in TBS extraction buffer, sonicated and then centrifuged at 5,000 g for 5 minutes to clear cellular debris. Protein levels were determined using the Bradford method and 10–20 μg per sample loaded onto Nupage Novex Bis-Tris Midi or Mini gels (Invitrogen, Carlsbad, CA). Samples were prepared for loading according to manufacturer recommendations. Following electrophoretic separation, proteins were transferred to nitrocellulose-backed membranes and then blocked with Blotto B (2% milk; 2% Bovine Serum Albumin and 50mM NaF in Tris buffered Saline (140mM NaCl; 20mM Tris)). Membranes were then treated with antibody overnight at 4°C (p-Tau (Ser 262) 1:500, Santa Cruz, CA), washed 3 times, placed in secondary, washed 3 times in tris-buffered saline with Tween (500mM NaCl, 20mM Tris, and 0.1% Tween20) and developed with ECL (GE Life Sciences) and ISC BioExpress film. Gels were scanned and densitometry was analyzed using Image J (19). Total tau proteins range from 46 – 62 KDa. For our data analysis, we drew a box around all bands encompassing the bands spanning from 46 – 62 KDa to calculate the total tau levels. We then used this value as a baseline level to normalize the p-Tau 262 band. N = 4 and 5 for vehicle and R-F treated WT samples and N = 7 and 8 for vehicle and R-F treated Tg2576 samples, respectively.
2.8 Statistical analyses
All results are reported as mean ± SEM. In this study, vehicle treated Tg+ served as controls and were expected to have decreased axonal transport compared to WT groups as we have previously reported (4). A priori planned comparisons were performed with two-tailed student’s t tests between: vehicle WT and vehicle Tg+ groups, vehicle Tg+ and R-F Tg+ groups, and vehicle WT and R-F WT groups for MEMRI and immunoblotting data. For the ELISA assays, Mann-Whitney-Wilcoxon U tests between vehicle Tg+ and R-F Tg+ were conducted due to the non-Gaussian distribution of the values. All statistics were calculated and prepared using SPSS16 (SPSS Inc., Chicago, IL).
3. Results
3.1 R-F treatment in pre-plaque Tg+ mice results in improved axonal transport and reduction of Aβ levels
Axonal transport in Tg+ mice acutely treated with R-F at the pre-plaque age of 7 months was measured with MEMRI (Figure 1A–D). In agreement with our prior studies (4), our data indicate that axonal transport in the vehicle Tg+ group decreased significantly in comparison to the vehicle WT group (**p=0.003, Figure 1E). The R-F Tg+ group treatment resulted in improved axonal transport compared to vehicle Tg+(*p=0.02, Figure 1E). The data indicate an improvement in axonal transport with R-F treatment. Further comparison of vehicle WT and R-F WT groups results in equivalent axonal transport (p=0.73, Figure 1E). These results demonstrate that improvement of the axonal transport deficit is possible with R-F treatment in the Tg2576 mouse model of AD.
Figure 1.

A–D. Representative images of manganese transport in the olfactory bulb Outlined square depicts ROI. A & C. 2 and 32 min scans of vehicle Tg+. (B & D) the 2 and 32 min scans of R-F Tg+. E. Averaged axonal transport reported as percent control indicate significant reduction in vehicle Tg+ compared to vehicle WT (**p<0.003). R-F Tg+ axonal transport significantly increased compared to vehicle Tg+ (*p=0.02). F. Representative MEMRI data of WT and Tg2576 animals treated with sucrose. Note the lower rate of increase in signal in the Tg2576 mice. G. Representative MEMRI data of WT and Tg2576 animals treated with R-F. Note the increased rate of signal in the RF-treated Tg2576 mice.
We then determined whether the improvements in axonal transport are due to a reduction in Aβ42 levels. Brain samples acquired during the pre-plaque age range were tested for changes in Aβ42 levels with R-F treatment and in Aβ40 to determine whether measured changes are specific to Aβ42 or Aβ overall. The data demonstrated that Aβ40 levels are unchanged in vehicle Tg+ compared to R-F Tg+ in the SDS fraction (Figure 2A) at 7 months of age. Comparison of the vehicle Tg+ and R-F Tg+ groups showed a 50% decrease in Aβ42 levels with treatment in the SDS fraction (*p=0.04, Figure 2B). This result indicates that R-F treatment specifically reduced Aβ42 levels at an age range before plaque formation occurs in this mouse model. These data are in agreement with a study from a different group in which R-F treatment resulted in Aβ42 decreases in young Tg2576 mice(10).
Figure 2.

A. Aβ40 concentrations show the accumulation of Aβ40 in the SDS fraction and that treatment did not result in altered levels between the two groups tested. B. Aβ42 levels in the SDS fraction are increased in the vehicle Tg+ group, while treatment reduces these levels in the R-F Tg+ group.(*p=0.04 U=5.000) C. Ratio of Aβ42/Aβ40 levels in SDS fraction were significantly shifted towards the less amyloidogenic Aβ40 in R-F samples (*p=0.03 U=2).
The levels of both Aβ species in the FA samples from both vehicle Tg+ and R-F Tg+ treated groups did not significantly differ from each other (Aβ40 p=0.08; Aβ42 p=1.00, Aβ42/Aβ40 p=0.10, Figure 2A, B, & C). The FA fraction did not have an increase in Aβ levels indicating a minimal amount of FA soluble Aβ accumulated pre-plaque. However, the Aβ42/Aβ40 ratios were also calculated as this ratio is increasingly being considered as a clinical indicator of AD progression (20). Our data demonstrate that there is a shift in the Aβ42/Aβ40 ratio in the SDS fraction (Aβ42/Aβ40 *p=0.03, Figure 2C).
3.2 Acute R-F treatment in post-plaque Tg+ mice improves axonal transport without significantly reducing Aβ levels
R-F treatment of Tg+ mice in the post-plaque age range (12 months) was conducted to determine whether reducing Aβ42 improves axonal transport at advanced stages of Aβ accumulation. Tg+ and WT mice received an acute treatment of R-F or vehicle, and axonal transport was measured with MEMRI following the final dose. Comparison between vehicle WT and vehicle Tg+ groups shows a significant and expected decrease in axonal transport of vehicle Tg+ group (**p=0.01, Figure 3A). R-F Tg+ compared to Vehicle Tg+ showed a significant 50% increase in axonal transport (*p=0.03, Figure 3A). Vehicle WT and R-F WT axonal transport were comparable to each other (p=0.68, Figure 3A). These data demonstrate that in vivo treatment with R-F improves axonal transport after Aβ plaque accumulation has occurred.
Figure 3.

A. Vehicle Tg+ group axonal transport was significantly reduced compared to vehicle WT (**p=0.01). Treatment with R-F resulted in improved axonal transport between vehicle Tg+ and R-F Tg+ groups (*p=0.03). Transport reported as % Control. Vehicle WT (100.0% ± 16.17); Vehicle Tg+ (28.30% ± 17.20); R-F WT (112.9 ± 26.17); R-F Tg+ (79.48 ± 13.29). B. ELISA Assays for Aβ40 are not significantly reduced as a result of treatment. C. Aβ42 levels did not show the expected reduction with R-F treatment in either the SDS or the FA fraction with an acute treatment post-plaque. D. Aβ42/Aβ40 ratio did not vary between vehicle and R-F Tg+ groups.
We then tested whether improved axonal transport in the post-plaque age range correlates with a reduction in Aβ42 levels. Our results show that in the post-plaque age range, overall Aβ levels increase in both the SDS and FA fractions compared to the Aβ levels at the pre-plaque stage. Figure 3B shows the Aβ40 levels in the SDS and FA fractions remain unchanged with R-F treatment in the post-plaque age range. The measurement of Aβ42 in the post-plaque age range also shows minimal differences between vehicle Tg+ and R-F Tg+ and the ratio between Aβ42/Aβ40 is unaltered between the vehicle Tg+ and R-F Tg+ (Figure 3C & D). This result contrasts with the Aβ42 reduction observed at the pre-plaque age range reported by the Eriksen group but is similar to non-significant decrease reported by Kukar et al (10,15). However, because Kukar et al reported benefits with longer treatments at this age range, we tested whether an increase in treatment duration may be required to detect significant changes in the Aβ42 levels (15).
3.3 Chronic R-F treatment in post-plaque Tg+ mice improves axonal transport without significantly reducing Aβ levels
In the chronic treatment study mice at 12 months of age were treated with R-F for 10 days at the reduced dose of 10mg/kg/day. This treatment and dose was modeled after the treatment administered by Kukar and colleagues that showed reduced Aβ42 levels at 20 months of age (15). We measured axonal transport with MEMRI in the chronic post-plaque treatment group and continued to observe a significant decrease in the vehicle Tg+ group compared to vehicle WT (***p=0.001, Figure 4A). We observed that 10 days of treatment also instilled a recovery in axonal transport in the R-F Tg+ group compared to vehicle Tg+ group (**p=0.01, Figure 4A), while vehicle WT and R-F WT showed a slight yet statistically insignificant decrease (p=0.16).
Figure 4.

A. Averaged axonal transport rates reported as % Control indicate a significant reduction in vehicle Tg+ compared to vehicle WT (***p=0.001). The R-F Tg+ group axonal transport rates significantly increased compared to vehicle Tg+ (**p=0.01). Vehicle WT (100.0% ± 10.88); Vehicle Tg+ (27.28% ± 10.16); R-F WT (80.49% ± 7.902); R-F Tg+ (98.98% ± 19.75. B. Treatment did not visibly reduce R-F Tg+ Aβ40 levels compared to vehicle Tg+ C. Aβ42 ELISA assays show trends towards a decrease in both SDS and FA fractions. SDS p=0.21 U=14; FA p=0.40 U=20. D. Aβ42/Aβ40 ratio shifted significantly towards increased levels of Aβ40 in the SDS fraction (*p=0.0200 U=6).
As expected, we found that Aβ40 levels remained unaltered with R-F treatment in either the SDS or FA fractions (SDS p=0.73; FA p=1.000, Figure 4B). We observed a 60% reduction in Aβ42 in the SDS fraction in the R-F treated Tg+ group compared to the vehicle treated Tg+ group, although the p-value remained short of statistical significance (p=0.21, Figure 4C). In the FA fraction Aβ42 is only slightly reduced in the R-F Tg+ group compared to vehicle Tg+ Aβ42 levels (p=0.40, Figure 4C). However, the longer treatment significantly shifted the ratio of Aβ towards the less amyloidogenic Aβ40 peptide compared to Aβ42 (Aβ42/Aβ40 *p=0.02, Figure 4D). At this dose, R-F appeared to reduce Aβ42 although to a lesser extent, after plaque formation occurred. These data are also consistent with previous studies in Tg+ mice at this age point (15).
3.4 R-F Treatment in post-plaque Tg+ mice results in reduced tau hyper-phosphorylation
Because of the insignificant reduction in Aβ42 in the 12 months old Tg2576 animals, we decided to address whether an additional factor associated with AD and axonal transport in this mouse model is affected with R-F treatment. We tested whether tau hyperphosphorylation at the serine 262 (S262) site is perturbed. The S262 site is located in the microtubule binding region of tau (21). Hyperphosphorylation of the S262 site impairs tau binding and microtubule stabilization, and in the Tg+ mouse the hyperphosphorylated tau S262 surrounds Aβ plaques (22).
Olfactory bulbs from the chronic treatment groups of 10 mg/kg/day at the post-plaque disease stage were used for immunoblotting. Vehicle Tg+ compared to vehicle WTexhibited a significant increase in tau hyperphosphorylation (*p=0.02, Figure 5). This result demonstrates that Tg+ mice have an abnormal increase in tau phosphorylation at a key phosphorylation site for axonal transport, consistent with previous reports of tau phosphorylation in this mouse model (22).
Figure 5.
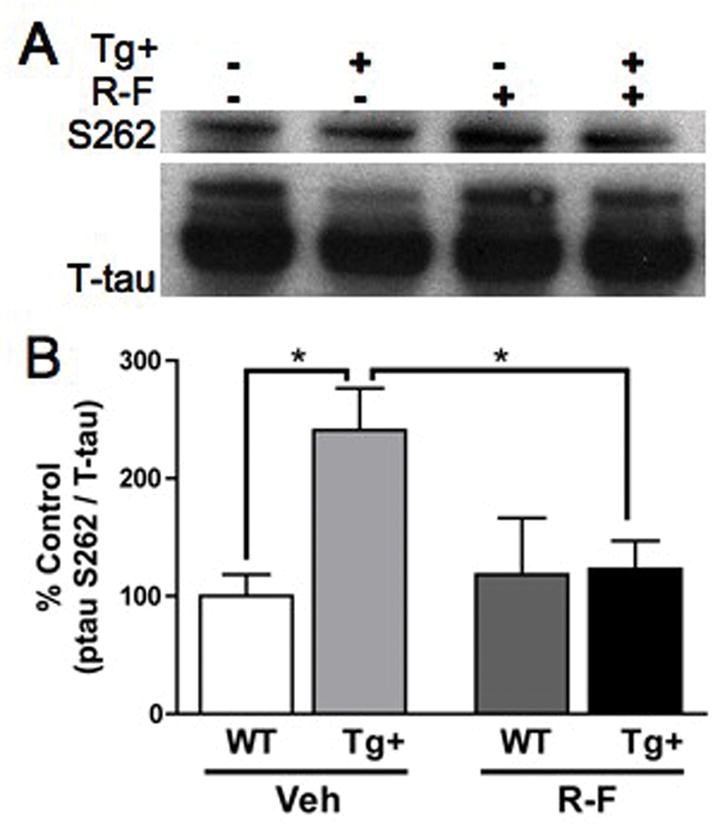
A. Representative immunoblot images with pS262 and total tau. B. Densitometry analysis of pS262 normalized for levels of total tau shows that pS262 more than doubles in the vehicle Tg+ group post-plaque compared to vehicle WT (*p=0.02). Chronic treatment with R-F eliminates the pS262 hyperphosphorylation seen in the vehicle Tg+ (*p=0.01). Vehicle WT (100.0 ± 18.30) Vehicle Tg+ (240.9% ± 35.47); R-F WT (118.5% ± 48.00) R-F Tg+ (123.2% ± 23.80).
R-F Tg+ compared to vehicle Tg+ demonstrates a significant reduction in tau hyperphosphorylation at S262 (*p=0.01, Figure 5). These data indicate that R-F treatment benefits extend beyond lowering Aβ42. This presents an additional and unexpected effect of R-F treatment after plaque formation has occurred in the Tg2576 mouse.
4. Discussion
In this study we have shown that treatment with R-F improves in vivo axonal transport in the olfactory system of the Tg2576 mouse model of AD as assessed with MEMRI. R-F treatment concurrently shifted Aβ peptide levels towards the less amyloidogenic Aβ40 peptide at all age points. Additionally, we observed a reduction in soluble levels of Aβ42 in Tg+ mice in the pre-plaque age range. We interpret these results to mean that drug therapies known to reduce Aβ42 appear to be accompanied by improvements in axonal transport in the Tg2576 mouse model of AD and that MEMRI can be utilized to characterize improvements in axonal pathology.
In addition to lowering Aβ levels, R-F is known to have minimal COX-2 inhibitory effects (13), affects nuclear factor-kappaB signaling (23), affects glutamate release (24) and activates c-Jun N terminal kinase (25). However, these particular functions of R-F do not explain a direct path to improvements in axonal transport. Tau hyperphosphorylation precedes the development of neurofibrillary tangles in AD (26–27) and impairs the ability of tau to bind to and stabilize microtubules resulting in microtubule impairments (28–29). Because axonal transport deficits in disease are often associated with changes in molecular motor proteins or microtubule associated protein dysfunction, in particular tau hyperphosphorylation, we determined whether R-F also influenced tau-hyperphosphorylation. Our data demonstrate that treatment with R-F exerts effects through an additional pathway that results in reduced tau hyperphosphorylation in the Tg2576 mouse following formation of plaques. These data can partially explain the mechanism of the observed improvement in axonal transport.
An additional possible explanation of the deficits and improvements we observed in this model include perturbations in Ca2+ handling in the Tg2576 mouse model. However, it has been previously shown that Ca2+ influx actually increases with age in this model (30). This would not explain the initial deficit we observed previously and in the current study in the untreated animals. Additionally, we are not assessing absolute values in our measurements but rather the rates of increase.
In this work, whereas R-F has previously been regarded as primarily an Aβ42 lowering agent, our results indicate that there are additional pathways impacted by R-F treatment in the Tg2576 mouse model that impart improvements in axonal transport. These data also indicate that both Aβ42 and tau comprise a role in axonal transport deficits in the Tg2576 model of AD.
Acknowledgments
This work was supported by NIH/NIA R01AG029977 (RGP); and NIH/NHLBI T32 HL07676 (Dr. Susan Hamilton). The authors thank Dr. F. Serrano and Dr. C.A. Massaad and all members of the Pautler lab for helpful comments throughout this study.
References
- 1.Hiruma H, Katakura T, Takahashi S, Ichikawa T, Kawakami T. Glutamate and amyloid beta-protein rapidly inhibit fast axonal transport in cultured rat hippocampal neurons by different mechanisms. J Neurosci. 2003;23:8967–8977. doi: 10.1523/JNEUROSCI.23-26-08967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 3.Muresan V, Muresan Z. Is abnormal axonal transport a cause, a contributing factor or a consequence of the neuronal pathology in Alzheimer’s disease? Future Neurol. 2009;4:761–773. doi: 10.2217/fnl.09.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith KD, Kallhoff V, Zheng H, Pautler RG. In vivo axonal transport rates decrease in a mouse model of Alzheimer’s disease. Neuroimage. 2007;35:1401–1408. doi: 10.1016/j.neuroimage.2007.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Duff K, ECkman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid B42 in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 7.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 8.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 9.Stein TD, Johnson JA. Lack of Neurodegeneration in Transgenic Mice Overexpressing Mutant Amyloid Precursor Protein is Associated with Increased Levels of Transthyretin and the Activation of Cell Survival Pathways. J Neurosci. 2002;22(17):7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest. 2003;112:440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem. 2003;278:31831–31837. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]
- 12.Geisslinger G, Muth-Selbach U, Coste O, Vetter G, Schrodter A, Schaible HG, Brune K, Tegeder I. Inhibition of noxious stimulus-induced spinal prostaglandin E2 release by flurbiprofen enantiomers. a microdialysis study. Journal of Neurochemistry. 2000;74:2094–2100. doi: 10.1046/j.1471-4159.2000.0742094.x. [DOI] [PubMed] [Google Scholar]
- 13.Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001;15:2057–2072. doi: 10.1096/fj.01-0390rev. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 15.Kukar T, Prescott S, Eriksen JL, Holloway V, Murphy MP, Koo EH, Golde TE, Nicolle MM. Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 2007;8:54. doi: 10.1186/1471-2202-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. 2. Academic Press; San Diego, Calif.: London: 2001. [Google Scholar]
- 17.Sbarbati A, Calderan L, Nicolato E, Marzola P, Lunati E, Donatella B, Bernardi P, Osculati F. Magnetic resonance imaging of the rat Harderian gland. J Anat. 2002;201(3):231–238. doi: 10.1046/j.1469-7580.2002.00086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akins MR, Greer CA. Cytoskeletal organization of the developing mouse olfactory nerve layer. J Comp Neurol. 2006;494:358–367. doi: 10.1002/cne.20814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Image J Reference Rasband, W S, ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2009. http://rsb.info.nih.gov/ij/ [Google Scholar]
- 20.Hansson O, Zetterberg H, Buchhave P, Andreasson U, Londos E, Minthon L, Blennow K. Prediction of Alzheimer’s disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;23:316–320. doi: 10.1159/000100926. [DOI] [PubMed] [Google Scholar]
- 21.Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117:5721–5729. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 22.Puig B, Gomez-Isla T, Ribe E, Cuadrado M, Torrejon-Escribano B, Dalfo E, Ferrer I. Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding betaA plaques in APP Tg2576 mice. Neuropathol Appl Neurobiol. 2004;30:491–502. doi: 10.1111/j.1365-2990.2004.00569.x. [DOI] [PubMed] [Google Scholar]
- 23.Morihara T, Chu T, Ubeda O, Beech W, Cole GM. Selective inhibition of Abeta42 production by NSAID R-enantiomers. J Neurochem. 2002;83:1009–1012. doi: 10.1046/j.1471-4159.2002.01195.x. [DOI] [PubMed] [Google Scholar]
- 24.Arbez N, Gautheron V, Brugg B, Mariani J, Rovira C. Beta-amyloid(1–42) induces a reduction in the parallel fiber responses of Purkinje cells: possible involvement of pro-inflammatory processes. Exp Gerontol. 2007;42:951–962. doi: 10.1016/j.exger.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Grosch S, Tegeder I, Schilling K, Maier TJ, Niederberger E, Geisslinger G. Activation of c-Jun-N-terminal-kinase is crucial for the induction of a cell cycle arrest in human colon carcinoma cells caused by flurbiprofen enantiomers. FASEB J. 2003:02-0919fje. doi: 10.1096/fj.02-0919fje. [DOI] [PubMed] [Google Scholar]
- 26.Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM. Accumulation of abnormally phosphorylated [tau] precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Research. 1989;477:90–99. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- 27.Bancher C, Grundke-Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM. Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Research. 1991;539:11–18. doi: 10.1016/0006-8993(91)90681-k. [DOI] [PubMed] [Google Scholar]
- 28.Terwel D, Dewachter I, Van Leuven F. Axonal transport, tau protein, and neurodegeneration in Alzheimer’s disease. Neuromolecular Med. 2002;2:151–165. doi: 10.1385/NMM:2:2:151. [DOI] [PubMed] [Google Scholar]
- 29.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–108530. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Xie CW. Calcium-regulated signaling pathways: role in amyloid beta-induced synaptic dysfunction. Neuromol Med. 2004;6(1):53–64. doi: 10.1385/NMM:6:1:053. [DOI] [PubMed] [Google Scholar]