

Abstract
Mutations in the cathepsin D (CTSD) gene cause an aggressive neurodegenerative disease (congenital neuronal ceroid lipofuscinosis) that leads to early death. Recent evidence suggests that presynaptic abnormalities play a major role in the pathogenesis of CTSD deficiencies. To identify the early events that lead to synaptic alterations, we investigated synaptic ultrastructure and function in pre-symptomatic CTSD knock-out (Ctsd−/−) mice. Electron microscopy revealed that there were significantly greater numbers of readily releasable synaptic vesicles present in Ctsd−/− mice than in wild-type control mice as early as postnatal day 16. The size of this synaptic vesicle pool continued to increase with disease progression in the hippocampus and thalamus of the Ctsd−/− mice. Electrophysiology revealed a markedly decreased frequency of miniature excitatory postsynaptic currents (EPSCs) with no effect on pair-pulse modulation of the evoked EPSPs in the hippocampus of Ctsd−/− mice. The reduced miniature EPSC frequency was observed before the appearance of epilepsy or any morphological sign of synaptic degeneration. Taken together, the data indicate that CTSD is required for normal synaptic function, and that a failure in synaptic trafficking or recycling may be an early and important pathological mechanism in Ctsd−/− mice; these presynaptic abnormalities may initiate synaptic degeneration in advance of subsequent neuronal loss.
Keywords: Cathepsin D, Electron microscopy, Electrophysiology, Hippocampus, Synapse, Synaptic vesicle
INTRODUCTION
Cathepsin D (CTSD) deficiency causes a devastating inherited neurological disease in humans, known as congenital neuronal ceroid-lipofuscinosis (cNCL, also called CLN10; MIM #610127) (1). Affected babies have epilepsy, extreme brain atrophy, and die from cardiac failure within the first few days of life (1-3). The disease is characterized neuropathologically by profound loss of neurons and myelin, together with pronounced glial activation throughout the neocortex (1-3). CTSD deficiency also occurs in sheep, in which it causes a congenital disease with neonatal death, and in American bulldogs, in which the disease has a more prolonged course (4, 5). In addition, models of CTSD deficiency have been created in Drosophila and mice by genetic modification (6, 7).
Ctsd knock-out (Ctsd−/−) mice, generated by gene targeting, provide an excellent tool for studying the basic pathophysiology of cNCL (7). CTSD-deficient mice appear phenotypically normal at birth, but develop a rapidly progressive neurodegenerative disease with epileptic seizures and death at postnatal day (P) 26 ± 2 (7-9). Many of the neuropathological features typical of human cNCL are recapitulated in Ctsd−/− mice, including accumulation of electron dense storage material in neurons, extensive glial activation, dysmyelination and loss of neurons (8, 10-11).
Recent data from Ctsd−/− mice, and from mice that model other forms of NCL (12-15), suggest that early synaptic alterations are characteristic of these disorders. In Ctsd−/− mice, synaptic pathology was particularly prevalent in the somatosensory cortex and the related thalamic nuclei (ventral posteromedial and ventral posterolateral nuclei, VPM/VPL); the synaptic density in these areas is also reduced at late-symptomatic stages of disease (10). Loss of synapses was accompanied by aggregation of pre-synaptic proteins, including α-synuclein and SNARE proteins (10, 16). Similar pathological alterations at the synapse have been noted in autopsy samples from CTSD-deficient patients (unpublished observation), indicating that presynaptic mechanisms play an important role in the pathogenesis of CTSD deficiencies.
In this study, we have undertaken a correlated ultrastructural and functional analysis of hippocampal synapses from Ctsd−/− mice to investigate the early events that may underlie synaptic pathology. We identified morphological and functional changes that occur in synapses of Ctsd−/− mice at pre-symptomatic time points. The most notable changes included an increase in the number of docked synaptic vesicles and a decreased frequency of miniature excitatory postsynaptic currents (mEPSCs).
MATERIALS AND METHODS
Animals
Ctsd−/− mice (7) were maintained on a mixed C57BL6J strain background at the animal facility of the Helsinki University, Biomedicum, where food and water were available ad libitum and with 12 hour/12 hour light/dark cycle. The study protocol was approved by the local Ethical Committee.
Electron Microscopy
P16 and P24 Ctsd−/− mice and wild-type (wt) littermate controls were transcardially perfused with 4% paraformaldehyde. Brains were prepared for ultrastructural analysis of synapses, as previously described (17). Briefly, brains were sectioned coronally at 70 μm on a vibratome before post-fixation in 1% osmium tetroxide solution for 45 minutes. Sections were dehydrated using a descending series of alcohols and stained en-bloc using 1% uranyl acetate. Sections were mounted in Durcupan resin (Sigma, Gillingham, UK) on glass slides. VPM/VPL thalamic nuclei and the CA1 region of the hippocampus were microdissected and mounted on Durcupan resin blocks. Ultra-thin sections (~70 nm thick) were then cut and stained with lead citrate before viewing on a Philips CM12 Transmission Electron Microscope equipped with a Gatan digital camera.
Electron micrographs were analyzed and quantified using ImageJ software. One hundred randomly selected micrographs were analyzed per mouse, with the investigator blind to the age and genotype of the tissue. Only synapses the profiles of which were contained entirely within the micrograph or those crossing the top or left borders of the micrograph were counted. The vast majority (>98%) of synapses had typical excitatory, asymmetric morphology but symmetric synapses were also included in the counts when they ere observed. Pre-synaptic bouton areas were measured by the investigator manually defining the boundaries of each bouton using standard functions within the ImageJ software. Synaptic vesicles were identified by their ~50-nm size. Docked synaptic vesicles were classified as being within 100 nm of the pre-synaptic membrane directly opposing the post-synaptic density. A minimum of 50 individual synapses was assessed per mouse.
Preparation of Acute Slices
P16–P18 Ctsd−/− and wt littermate mice (P0 = day of birth) were rapidly killed by decapitation in accordance with the University of Helsinki animal welfare guidelines. Hippocampal slices (400 μm thick) were cut with a vibratome using standard methods (18). The slices were used 1 to 4 hours after cutting.
Electrophysiology
For electrophysiological recordings, the slices were placed in a submerged chamber and superfused with artificial cerebrospinal fluid containing (mM): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 15 d-glucose and 2 CaCl2; 5% CO2–95% O2, at a rate of 2–3 ml minute−1 (32°C). Whole-cell recordings were obtained from CA1 pyramidal neurons using the Axopatch 200B amplifier (Axon Instruments, Union City, CA). Cells were voltage-clamped at −70 mV with 3 to 5 MΩ pipettes filled with a solution containing (mm): 130 CsMeSO4, 10 Hepes, 0,5 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 5 N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride and 8 NaCl (285 mOsm), pH 7.2. AMPA-receptor-mediated mEPSCs (mEPSCs) were recorded from CA1 pyramidal neurons in the presence of antagonists of GABAA and voltage-dependent sodium channels (picrotoxin [100 μM] and TTX [1 μM], respectively). To measure the paired pulse facilitation, the pipette filling solution was changed (mM): 115 CsMeSO4, 10 Hepes, 10 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 5 N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride and 8 NaCl (285 mOsm), pH 7.2. A bipolar electrode was used for afferent stimulation. EPSCs were evoked by stimulation of Schaffer collateral-commissural fibers and recorded from CA1 pyramidal neurons in the presence of GABA-A and NMDAR antagonists (100 μM picrotoxin and 50 μM AP5, respectively). All compounds were from Tocris (Bristol, UK).
WinLTP (0.95b or 0.96, www.winltp.com, (19) or Axoscope 9.2 (Axon Instruments) was used for data acquisition. Offline analysis was performed using WinLTP or MiniAnalysis 6.0.3 program (Synaptosoft Inc, Decatur, GA). Spontaneous events were detected using peak detector algorithm, and all events were confirmed visually. The cumulative distributions of mEPSCs were constructed from at least 10 minutes of recording (at least 50 events) from each cell, using a bin width of 100 ms for inter-event interval and 1 pA for amplitude. The pooled data are given as mean ± SEM for the number of cells indicated. Student two-tailed t-test was used for statistical analysis. The level of significance was set as p < 0.05.
RESULTS
Synaptic Density Is Markedly Decreased in the Hippocampus and Thalamus of late-Symptomatic Ctsd−/− Mice
Our previous semiquantitative analyses revealed reduced numbers of synapses in the VPM/VPL thalamic nuclei of Ctsd−/− mice at P24 (10). We validated these findings using a more robust quantitative method, confirming a significant loss of synapses at P24 (***p < 0.001; ANOVA with Tukey’s post-hoc test; Fig. 1A). A similar analysis at pre-symptomatic age (P16) revealed a clear, but not statistically significant, reduction in synaptic density in VPM/VPL (p > 0.05; Fig. 1A), indicating that synaptic pathology was only in its early stages at P16.
Figure 1.

Synaptic density in the ventral posteromedial and ventral posterolateral nuclei (VPM/VPL) nuclei of the thalamus and the CA1 region of hippocampus in Ctsd−/− and control mice. (A) Bar chart (mean ± SEM) showing synaptic density in the VPM/VPL nuclei quantified from electron micrographs from Ctsd−/− mice (−/−; black bars) and control mice (+/+; white bars). There is a subtle reduction in the number of synapses at postnatal day (P) 16 (pre-symptomatic; not significant p > 0.05) and a significant loss of synapses by P24 (late-symptomatic; *** p < 0.001). (B) Bar chart showing synaptic density in the CA1 region of hippocampus quantified from electron micrographs. There was no reduction in the number of synapses at P16 (pre-symptomatic; not significant p > 0.05), but there was a significant loss of synapses by P24 (late-symptomatic; ** p < 0.01). n = number of mice per genotype/age.
Because assessing synaptic function using electrophysiological methods in VPM/VPL is difficult, we extended our experiments to determine whether synapses in the hippocampus (a region ideal for electrophysiological experiments) were also affected. Synaptic density remained unchanged in the CA1 region of hippocampus at P16, but was significantly reduced at P24 (**p < 0.01; Fig. 1B). However, the magnitude of synapse loss in the hippocampus (~60% reduction) was not as large as that observed in the VPM/VPL (~90% reduction). Nevertheless these data (together with established protocols for slice preparation and electrophysiological recordings) suggest that the hippocampus provides an appropriate brain region for correlated ultrastructural and functional analyses of synapses in Ctsd−/− mice. Moreover, the data indicate that P16 represents a time point at which synaptic degeneration has not yet begun in the hippocampus, whereas P24 represents a time point when there is extensive synaptic degeneration.
Synaptic Vesicle Density Is Increased in Ctsd−/− Mice
Qualitative observations of synapses in the hippocampal CA1 region from Ctsd−/− mice suggested that there was no obvious change in the size of synaptic boutons at either pre- or late-symptomatic time points (Fig. 2A, B). Quantitative analyses confirmed these observations (Fig. 2C). However, synaptic vesicle pools appeared to be changed qualitatively in CA1 synapses at both pre- and late-symptomatic time points, i.e. denser clustering of vesicles around pre-synaptic membranes was routinely observed at P24 in Ctsd−/− mice (Fig. 2B). Quantitative analysis of total synaptic vesicle densities in CA1 showed no difference between genotypes at P16, but at P24 there was a significant increase in Ctsd−/− mice (*p < 0.05; ANOVA with Tukey’s post-hoc test; Fig. 2D). Most strikingly, however, the number of docked vesicles was significantly increased in CA1 synapses from Ctsd−/− mice, with significant changes already present at P16 (p < 0.05; Fig. 2E). The number of docked vesicles was almost double that in control mice by P24 (P < 0.05; Fig. 2E). Although not statistically significant, a similar trend was observed for the numbers of undocked vesicles in these mutant mice (Fig. 2F). These data indicate that disruption of hippocampal synaptic vesicle localization and/or recycling is an early pathological event in Ctsd−/− mice.
Figure 2.

Synaptic vesicle pools in the CA1 region of hippocampus in Ctsd−/− and control mice. (A, B) Representative electron micrographs showing synapses in the CA1 of control (A) and Ctsd−/− (B) mice at postnatal day (P) 24. Note the clustering of synaptic vesicles towards the pre-synaptic membrane in the Ctsd−/− synapses (B). (C) Bar chart (mean ± SEM) showing synaptic bouton areas in CA1 quantified from electron micrographs of Ctsd−/− (−/−; black bars) and wild-type control mice (+/+; white bars). (D) Bar chart showing synaptic vesicle densities in CA1, revealing a significant increase in Ctsd−/− mice at P24 (* p < 0.05). (E) Bar chart showing average numbers of docked synaptic vesicles per synapse in CA1, revealing a significant increase in Ctsd−/− mice at pre-symptomatic (P16) and late-symptomatic (P24) time points (* p < 0.05). (F) Bar chart showing average numbers of undocked synaptic vesicles per synapse in CA1. See Figure 1 for the number of mice in each group.
To determine whether the changes observed in synaptic vesicle densities in CA1 were a common feature of synaptic pathology in Ctsd−/− mice, we quantified synaptic vesicle densities in VPM/VPL at P16 and P24 (Fig. 3A, B). The number of docked synaptic vesicles and the total number of vesicles in VPM/VPL were significantly increased (**p < 0.01; ***p < 0.001; Fig. 3C, D) at P24, but not at P16.
Figure 3.

Synaptic vesicle pools in synapses from ventral posteromedial and ventral posterolateral thalamic nuclei (VPM/VPL) nuclei of Ctsd−/− and control mice. (A, B) Representative electron micrographs showing synapses in the VPM/VPL of control (A) and Ctsd−/− (B) mice at postnatal day (P) 24. Note the clustering of synaptic vesicles towards the pre-synaptic membrane in the Ctsd−/− synapses (B). (C) Bar chart (mean ± SEM) showing synaptic vesicle densities in the VPM/VPL, revealing a significant increase in Ctsd−/− mice (−/−; black bars) at P24 compared to control mice (+/+; white bars; *** p < 0.001). (D) Bar chart showing average numbers of docked synaptic vesicles per synapse in the VPM/VPL, revealing a significant increase in Ctsd−/− mice at P24 (** p < 0.01). See Figure 1 for the number of mice in each group.
mEPSC Frequency Is Decreased in Ctsd−/− Mice
To determine the functional effects of the increased numbers of docked synaptic vesicles in the hippocampal CA1 region of Ctsd−/− mice, pharmacologically isolated AMPA mEPSCs were recorded from hippocampal pyramidal cells in the CA1 subfield (Fig. 4A-D). At P16-P18 there was a lower mEPSC frequency in Ctsd−/− mice vs. controls. The mEPSC inter-event interval in the mutants was about twice as long as in wild-type mice (wt: 2.74 ± 0.31 seconds; Ctsd−/−: 5.89 ± 1.17 seconds; p < 0.05; Fig. 4E). There was, however, no difference between genotypes in the AMPA mEPSC amplitude (wt: 14.04 ± 0.80 pA; Ctsd−/−: 14.48 ± 0.99 pA; p > 0.05; Fig. 4F).
Figure 4.

(A-D) Characteristics of miniature excitatory postsynaptic currents (EPSCs) recorded from hippocampal CA1 pyramidal neurons of wild-type (A, B) and Ctsd−/− mice (C, D) at postnatal day (P) 16-17. Asterisks in example recordings (A, C) indicate regions presented with the expanded time scale (B, D). (E) Cumulative distribution and averaged values of mEPSC inter-event interval, * p < 0.05. (F) The cumulative distribution and averaged values of mEPSC amplitude.
Release Probability Is Unaltered in Ctsd−/− Mice
Because an increased number of docked synaptic vesicles may be associated with increased neurotransmitter release (20), we next measured paired-pulse facilitation (PPF) in the CA3-CA1 synapse because it is inversely correlated with a change in the initial release probability (Pr) (20, 22, 23). To assess PPF we used inter-pulse intervals of 20 ms (50 Hz), 50 ms (20 Hz) and 150 ms (6, 7 Hz). We found no difference in PPF between the genotypes at any of the inter-pulse intervals used (pair-pulse ratio, 20-ms-interval: wt 2.54 ± 0.08, Ctsd−/− 2.61 ± 0.19; 50-ms-interval: wt 2.13 ± 0.14, Ctsd−/− 2.17 ± 0.22; 150-ms-interval: wt 1.50 ± 0.05, Ctsd−/− 1.27 ± 0.19; p > 0.05; Fig. 5), indicating that the release probability is unaltered in Ctsd−/− mice.
Figure 5.
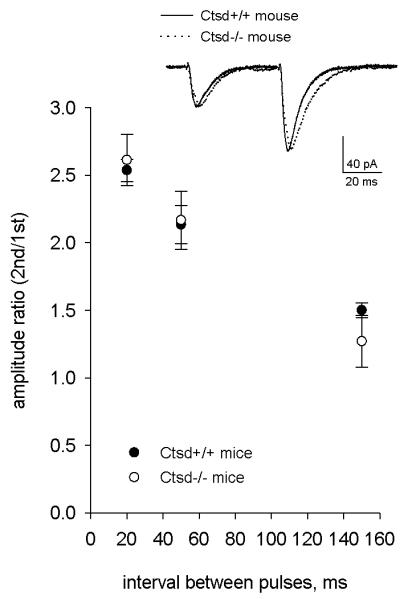
Characteristics of synaptic responses evoked by stimulation of Shaffer collateral and recorded from hippocampal CA1 pyramidal neurons of wild-type and Ctsd−/− mice. Mean values of amplitude paired-pulse ratio recorded with 20-, 50- and 150-ms-interval. Inset: example response at 50-ms-interval (averages of 10 sweeps, normalized by the amplitude of the first peak.
DISCUSSION
Readily Releasable Synaptic Vesicle Pool Size Increases with Disease Progression in Ctsd−/− Mice
Our most important ultrastructural finding in the synapses of Ctsd−/− mice was a marked increase in vesicle pool size compared to controls. Indeed, the readily releasable synaptic vesicle pool size was already significantly increased (~1.2-fold) in hippocampal neurons of Ctsd−/− mice at P16, which represents a pre-symptomatic stage of the disease. If there were a significant loss of synapses at P16, an increased number of synaptic vesicles could reflect a compensatory mechanism as a response to synaptic loss. However, at this stage, synaptic loss was not yet evident in CA1 of these affected mice. By P24, the size of the readily releasable vesicle pool had increased markedly, not only in the CA1 (~1.7-fold) but also in the thalamus (~1.5-fold). In addition, the total number of synaptic vesicles per bouton was significantly increased in both of these brain regions, suggesting that the synaptic vesicle pool undergoes abnormal changes throughout the brain of Ctsd−/− mice. Inasmuch as the changes in the vesicle pool size occurred in advance of synaptic degeneration in the hippocampus of Ctsd−/− mice, the data raise the possibility that alterations in synaptic vesicle recycling may contribute to the initiation and progression of synaptic degeneration in these mice. These findings are in line with our previously reported alterations in the distribution of presynaptic proteins in Ctsd−/− mice (10).
Electrophysiological Data Indicate Early Dysfunction of Synapses in Ctsd−/− Mice
The synaptic vesicle pool size and, particularly, the number of readily releasable synaptic vesicles (so called “docked” vesicles) affect the efficacy of synaptic transmission (20). The increased number of docked vesicles may lead to an increased neurotransmitter release and to a consequent increase in postsynaptic currents (20, 22-23). Somewhat surprisingly, our observations contradict this principle, i.e. despite the increased number of docked vesicles in Ctsd−/− mice, the paired-pulse data suggested no change in the Pr-. Rather, the increased number of readily releasable vesicles was associated with a lower frequency of AMPA-mediated mEPSCs. The lower mEPSC frequency might be explained by the presence of synaptic degeneration and loss, which would not necessarily alter the overall Pr in these synapses. Although we found no morphological evidence for synaptic loss in the CA1 of Ctsd−/− mice at P16, it is reasonable to suggest that the reduced mEPSC frequency was likely caused by changes in the functional parameters of synapses before they underwent morphologically visible degeneration. In previous electrophysiological analyses, spontaneous burst discharges of an epileptiform nature were recorded from the hippocampus of Ctsd−/− mice at late-symptomatic ages (P24) (8, 21), but were not observed at P16 (21). This suggests that the synaptic dysfunction reported in the present study precedes the onset of epilepsy and synaptic degeneration in Ctsd−/−mice.
Comparison with Other Forms of NCL
The NCLs are a genetically heterogenous group of fatal neurodegenerative diseases, sharing common clinical symptoms and pathological events (24, 25). The existence of several diverse mouse models of NCL allows comparison of the basic mechanisms underlying the diseases (24-25). In contrast to our findings in Ctsd−/− mice, the size of the readily releasable synaptic vesicle pool has been shown to progressively decline in cultured neurons of palmityl-protein thioesterase 1 (Ppt1) knock-out mice, a model of infantile NCL disease (12). This reduction was associated with decreased frequency of spontaneous mEPSCs (12). Whether the differences observed between this study and ours reflect different roles of PPT1 and CTSD in regulating synaptic vesicle dynamics, or whether other factors affected the results (e.g. comparing in vitro changes with in vivo pathology) remains unclear. The major neuropathological changes in Ppt1 knock-out mice closely resemble those in Ctsd−/− mice, with synaptic pathology occurring in a regionally selective manner, preceding the onset of neuronal loss in the thalamus and cortex (10, 15). Similar pathological changes are evident in autopsy material of infantile NCL patients with PPT1 deficiency and it has been suggested that the lack of PPT1 activity leads to altered synaptic vesicle trafficking at nerve terminals (14). Thus, whatever the basis for these events, it is becoming apparent that the presynaptic compartment is targeted early in the pathogenesis of the NCLs. Although the precise nature of the early events underlying synaptic pathology may differ between the forms of NCL, invariably these changes lead to synapse degeneration and subsequent neuronal loss.
Decreased frequency of spontaneous mEPSCs at an early age has only been reported in a few mouse models. These models have genetic defects affecting elementary functions of the brain and/or causing severe neurological diseases, such as neocortical maturation and autism (26), frontotemporal dementia (27), and Huntington disease (28). The early appearance of functional alterations in the synapses of Ctsd−/− mice suggests that CTSD has a fundamental role in maintenance and perhaps even development of synapses and neuronal networks. This possibility is supported by our previous findings showing that the somatosensory thalamocortical system is a focus of major pathological alterations in these mice (10). The abnormal accumulation of synaptic vesicles in Ctsd−/− brains indicates that the function of CTSD may be related to recycling or trafficking of presynaptic vesicles.
Relationship to Other “Synaptopathies”
In the most common forms of age-related dementia, such as Alzheimer disease and Parkinson disease, the role of synaptic loss in the neurodegenerative process has been recognized since the early 1990s (29, 30). Subsequently, there has been growing evidence of early synaptic vulnerability in a number of different neurodegenerative conditions affecting both central and peripheral nervous systems, including Alzheimer and Huntington disease, prion diseases, and spinal muscular atrophy (31-36). In particular, disruption of synaptic functions may precede synaptic degeneration and loss in many of these diseases (31-34), whereas the exact functional mechanisms may vary from one disease to another.
Summary
We have quantitatively assessed synaptic pathology in the thalamus and hippocampus of Ctsd−/− mice at an ultrastructural and functional level. We observed a significant increase in the number of docked vesicles at individual synapses occurring in advance of synaptic degeneration and neuron loss. These structural changes were accompanied by a markedly decreased frequency of mEPSCs, but no effects on evoked excitatory post-synaptic potentials. Taken together, the present data provide the first direct evidence for functional and structural changes at the synapse in pre-symptomatic Ctsd−/− mice and strongly suggest that a failure in the presynaptic function initiates synaptic degeneration, which leads to neuronal degeneration in these mice. These findings further emphasize the role of CTSD in maintaining synaptic integrity in vivo.
Acknowledgments
Support: Academy of Finland (JT), BDSRA and the Wellcome Trust (THG and JDC), Batten Disease Family Association and The Natalie Fund (JDC), Academy of Finland and Sigrid Juselius Foundation (SM and TT).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Siintola E, Partanen S, Strömme P, et al. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–45. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- 2.Garborg I, Torvik A, Hals J, et al. Congenital neuronal ceroid lipofuscinosis. A case report. Acta Pathol Microbiol Immunol Scand A. 1987;95:119–25. doi: 10.1111/j.1699-0463.1987.tb00017_95a.x. [DOI] [PubMed] [Google Scholar]
- 3.Fritchie K, Siintola E, Armao D, et al. Novel mutation and the first prenatal screening of cathepsin D deficiency (CLN10) Acta Neuropathol. 2009;117:201–8. doi: 10.1007/s00401-008-0426-7. [DOI] [PubMed] [Google Scholar]
- 4.Tyynelä J, Sohar I, Sleat DE, et al. A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J. 2000;19:2786–92. doi: 10.1093/emboj/19.12.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Awano T, Katz ML, O’Brien DP, et al. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006;87:341–8. doi: 10.1016/j.ymgme.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Myllykangas L, Tyynelä J, Page-McCaw A, et al. Cathepsin D-deficient Drosophila recapitulate the key features of neuronal ceroid lipofuscinoses. Neurobiol Dis. 2005;19:194–9. doi: 10.1016/j.nbd.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 7.Saftig P, Hetman M, Schmahl W, et al. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995;14:3599–608. doi: 10.1002/j.1460-2075.1995.tb00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koike M, Nakanishi H, Saftig P, et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haapanen A, Ramadan UA, Autti T, et al. In vivo MRI reveals the dynamics of pathological changes in the brains of cathepsin D-deficient mice and correlates changes in manganese-enhanced MRI with microglial activation. Magn Reson Imaging. 2007;25:1024–31. doi: 10.1016/j.mri.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Partanen S, Haapanen A, Kielar C, et al. Synaptic changes in the thalamocortical system of cathepsin D-deficient mice: a model of human congenital neuronal ceroid-lipofuscinosis. J Neuropathol Exp Neurol. 2008;67:16–29. doi: 10.1097/nen.0b013e31815f3899. [DOI] [PubMed] [Google Scholar]
- 11.Mutka AL, Haapanen A, Käkelä R, et al. Murine cathepsin D deficiency is associated with dysmyelination/myelin disruption and accumulation of cholesteryl esters in the brain. J Neurochem. 2010;112:193–203. doi: 10.1111/j.1471-4159.2009.06440.x. [DOI] [PubMed] [Google Scholar]
- 12.Virmani T, Gupta P, Liu X, et al. Progressively reduced synaptic vesicle pool size in cultured neurons derived from neuronal ceroid lipofuscinosis-1 knockout mice. Neurobiol Dis. 2005;20:314–23. doi: 10.1016/j.nbd.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–9. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- 14.Kim SJ, Zhang Z, Sarkar C, et al. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Invest. 2008;118:3075–86. doi: 10.1172/JCI33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kielar C, Wishart TM, Palmer A, et al. Molecular correlates of axonal and synaptic pathology in mouse models of Batten disease. Hum Mol Genet. 2009;18:4066–80. doi: 10.1093/hmg/ddp355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen V, Lindfors M, Ng J, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2:5. doi: 10.1186/1756-6606-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillingwater TH, Ingham CA, Parry KE, et al. Delayed synaptic degeneration in the CNS of Wlds mice after cortical lesion. Brain. 2006;129:1546–56. doi: 10.1093/brain/awl101. [DOI] [PubMed] [Google Scholar]
- 18.Lauri SE, Vesikansa A, Segerstråle M, et al. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron. 2006;50:415–29. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–56. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 20.Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu T, Hayashi Y, Yamasaki R, et al. Proteolytic degradation of glutamate decarboxylase mediates disinhibition of hippocampal CA3 pyramidal cells in cathepsin D-deficient mice. J Neurochem. 2005;94:680–90. doi: 10.1111/j.1471-4159.2005.03250.x. [DOI] [PubMed] [Google Scholar]
- 22.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 23.Dobrunz LE. Release probability is regulated by the size of the readily releasable vesicle pool at excitatory synapses in hippocampus. Int J Dev Neurosci. 2002;20:225–36. doi: 10.1016/s0736-5748(02)00015-1. [DOI] [PubMed] [Google Scholar]
- 24.Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009;1793:697–709. doi: 10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Kohlschütter A, Schulz A. Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 2009;31:499–502. doi: 10.1016/j.braindev.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Yashiro K, Riday TT, Condon KH, et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci. 2009;12:777–83. doi: 10.1038/nn.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belly A, Bodon G, Blot B, et al. CHMP2B mutants linked to frontotemporal dementia impair maturation of dendritic spines. J Cell Sci. 2010;1:2943–5. doi: 10.1242/jcs.068817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cepeda C, Hurst RS, Calvert CR, et al. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J Neurosci. 2003;23:961–9. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 30.Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–15. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- 31.Chapman PF, White GL, Jones MW, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–6. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 32.Jeffrey M, Halliday WG, Bell J, et al. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol Appl Neurobiol. 2000;26:41–54. doi: 10.1046/j.1365-2990.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- 33.Klapstein GJ, Fisher RS, Zanjani H, et al. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J Neurophysiol. 2001;86:2667–77. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- 34.Coleman P, Federoff H, Kurlan R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology. 2004;63:1155–62. doi: 10.1212/01.wnl.0000140626.48118.0a. [DOI] [PubMed] [Google Scholar]
- 35.DiProspero NA, Chen EY, Charles V, et al. Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. 2004;33:517–33. doi: 10.1007/s11068-004-0514-8. [DOI] [PubMed] [Google Scholar]
- 36.Murray LM, Comley LH, Thomson D, et al. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–62. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]