

Abstract
The discovery of preconditioning (PC) has arguably been the single most important development in the field of ischemic biology in the past 20 years. The significance of this phenomenon transcends cardiovascular medicine, since it is ubiquitously observed in virtually every tissue of the body. This article reviews the pathophysiology and molecular basis of myocardial PC, with particular emphasis on the late phase of this cardioprotective adaptation. The article also discusses the exploitation of late PC for the development of novel gene therapy strategies aimed at inducing a permanently preconditioned cardiac phenotype (prophylactic cardioprotection). Besides its conceptual interest, deciphering the mechanism of late PC has considerable therapeutic reverberations, since transfer of the genes that underlie late PC would be expected to emulate the salubrious effects of this response of the heart to stress.
Keywords: nitric oxide synthase, protection, cyclooxygenase-2
it is a great honor to Present the 2005 Robert M. Berne Distinguished Lecture of the American Physiological Society (APS).1 The Society should be applauded for recognizing the extraordinary contributions of Dr. Berne to cardiovascular physiology.
The topic of this lecture will be the phenomenon of ischemic preconditioning (PC), which has arguably been the single most important development in the field of ischemic biology in the past 20 years. Because it involves virtually every tissue of the body, the implications of PC transcend the field of cardiovascular research and should be of interest to the APS membership at large. The lecture will focus particularly on the late phase of ischemic PC, which I believe has greater clinical relevance.
First, it is important that we establish the definition of ischemic PC. This is the phenomenon whereby brief episodes of ischemia render the heart more resistant to subsequent ischemic injury. Thus ischemic PC is an endogenous protective mechanism activated by a mild ischemic stress that makes the heart better able to cope with another similar or greater stress. In this sense, ischemic PC can be viewed as a “vaccination” of sorts against myocardial ischemia-reperfusion injury. The important conceptual implication of the discovery of PC has been the realization that the heart can sense and adapt to the environment: when exposed to a stress, it can change its phenotype in a manner that is conducive to self-preservation. This innate plasticity of the heart was not appreciated until the discovery of PC, and it has major pathophysiological and therapeutic reverberations.
To avoid confusion, it is important that ischemic PC be clearly distinguished from direct cardioprotection. Although both of these processes enhance the resistance of the heart to ischemia-reperfusion injury, they are fundamentally different. With PC, the protection persists after the therapeutic intervention has dissipated, implying the existence of a cardiac “memory” (i.e., the heart “remembers” that it has been exposed to a PC stimulus and maintains a preconditioned phenotype even after the stimulus has been withdrawn). In contrast, with direct cardioprotection the enhanced resistance to ischemia-reperfusion injury occurs only while the therapeutic intervention is being applied and requires the continued presence of this intervention; when the intervention is withdrawn, the protection disappears (i.e., the heart does not keep a “memory” of the therapeutic intervention). Thus PC is an aftereffect following exposure to a stimulus, whereas direct cardioprotection is the effect that occurs during the stimulus. These phenomena can be easily confused when the effects of pharmacological agents are examined. For example, when chronic treatment with a drug is associated with cardioprotection, this is sometimes referred to as “PC.” A drug can be said to precondition only if the protection continues after the drug has disappeared from the cardiac tissue; if the protection fades as soon as the drug is gone, then one is dealing with a direct cardioprotective effect, not a PC effect.
The phenomenon of PC was described in 1986 by Murry, Jennings, and Reimer in a landmark study (24) that has become one of the most often quoted articles in the cardiovascular literature, with at least 2,634 citations as of June 30, 2006. In this study, the authors made what at that time appeared to be a paradoxical observation. They exposed a group of open-chest dogs to a sequence of four brief ischemic episodes (5-min coronary occlusions interspersed with 5-min reperfusion periods) and then subjected them to a prolonged, more severe ischemic insult (a 40-min coronary occlusion followed by 4 days of reperfusion). Control dogs underwent the 40-min occlusion with no preceding exposure to ischemia. A priori, one would have expected that the dogs that received the four brief coronary occlusions would exhibit greater infarct size, because they had been exposed to an additional 20 min of ischemia. Surprisingly, Murry et al. found that in this group of dogs infarct size was much smaller than in controls, and that this effect was independent of differences in coronary collateral blood flow. They coined the term “ischemic PC” to describe this phenomenon, opening the floodgates for what has become one of the major themes of research in cardiovascular medicine. Following the seminal observation of Murry et al., the number of studies dealing with PC has escalated dramatically, such that in 2005 over 500 papers were published on this subject (Fig. 1). Although initially almost all studies were experimental, clinical studies soon appeared and continued to increase—a testimony to the growing level of interest among clinicians in the phenomenon of PC.
Fig. 1.

Number of publications dealing with cardiac preconditioning since the initial description of this phenomenon in 1986.
Why this extraordinary explosion of interest in PC? A major reason is that during the past 35 years, hundreds of direct cardioprotective therapies have been claimed to reduce myocardial infarct size in experimental animals. However, few of these results have been reproducible, and none has been translated into clinical therapies (5). As a result, after investing an enormous amount of time, money, and resources into the search for cardioprotective therapies, in 2006 we still do not have a drug that has been specifically approved for the reduction of infarct size in patients with acute myocardial infarction. [The reason(s) for this colossal fiasco are complex and beyond the scope of the present discussion; nevertheless, the reader is referred to an article that has examined this problem in detail (5)]. In the midst of this frustration with the failure to identify clinically effective cardioprotective therapies came the discovery of ischemic PC. It became quickly apparent that ischemic PC was quite different from direct cardioprotection. First, it was clear that this was an extremely powerful cardioprotective phenomenon. In many experimental models, ischemic PC can reduce infarct size by as much as 80–90%—a degree of infarct size limitation that is rarely, if ever, observed with direct cardioprotection. Second, and more important, ischemic PC was found to be remarkably reproducible. It has been consistently observed in every experimental model, in every laboratory, and in every species examined. In fact, it is very difficult, if not impossible, to find a study in which the protective effects of ischemic PC have not been observed. This is in stark contrast to direct cardioprotection, which is eminently nonreproducible, often being reported in some models but not in others, in some laboratories but not in others, and in some species but not in others. [The lack of consistency of studies of direct cardioprotection has been the major reason for the failure to translate these investigations into clinical therapies (5)]. Importantly, ischemic PC has been observed in humans as well (4, 39). Given these facts, the enthusiasm of the scientific community for ischemic PC is easy to understand: investigators interested in cardioprotection had finally found an intervention that was not only highly effective but also totally reproducible.
Although PC was initially described as a response of the myocardium to ischemia, it soon became apparent that a similar phenotype can be elicited by a panoply of stimuli, some of which are clinically relevant. For example, a number of pharmacological agents, including agonists of G protein-coupled receptors (GPCRs) (adenosine A1 or A3, bradykinin B2, α1-adrenergic, muscarinic M2, angiotensin AT1, endothelin, δ1-opioid, etc.), nitric oxide (NO) donors, phosphodiesterase inhibitors, and various noxious stimuli (such as endotoxin and endotoxin derivatives, various cytokines, reactive oxygen species, etc.), have all been found to elicit a PC-like phenotype (4, 39). (Indeed, it appears that almost anything that is potentially harmful to the heart can elicit a preconditioned state when applied in small quantities.) PC can also be induced by nonpharmacological stimuli, such as physical exercise [even short-term exercise for a few days is sufficient to induce a PC state (14)], heat stress, rapid pacing, and other maneuvers. The importance of these findings is that one need not use ischemia to induce a cardioprotected phenotype; stimuli that are less harmful or unpleasant (and thus more clinically relevant) can also do this.
After the initial description of PC in 1986 (24), the next major discovery came in 1993, when it was found that this phenomenon is not monolithic but rather consists of two chronologically and pathophysiologically distinct phases: an early phase, which develops very quickly (within a few minutes from the exposure to the stimulus) but is rather ephemeral, lasting only 1–2 h [this is the phenomenon originally described by Murry et al. (24)], and a late phase, which develops more slowly (requiring 6 –12 h) but lasts much longer (3– 4 days) (19, 23). The mechanisms for these two phases are completely different. The early phase is caused by rapid posttranslational modification of preexisting proteins, whereas the late phase is caused by the synthesis of new cardioprotective proteins (which explains the time course of this phenomenon). The range of protection is also different. The early phase is very effective in limiting lethal ischemia-reperfusion injury (i.e., infarction) but does not protect against reversible postischemic contractile dysfunction (myocardial “stunning”). The late phase protects against both infarction and stunning, although it is less powerful than the early phase in limiting infarct size. (The ability of either phase to prevent arrhythmias associated with ischemia-reperfusion injury remains unclear.) The two phases of ischemic PC are illustrated in Fig. 2.
Fig. 2.
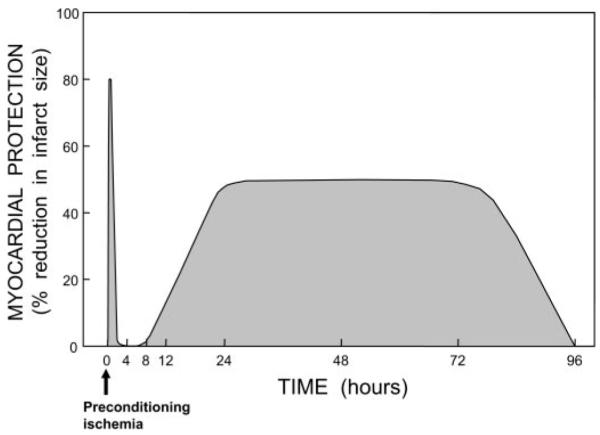
Schematic illustration of the 2 phases of ischemic preconditioning (PC). Myocardial protection (e.g., % reduction in infarct size) is represented on the y-axis, and time is represented on the x-axis. After the application of a PC stimulus (in this case, ischemia), there is the rapid development of a preconditioned state that confers powerful protection (up to 80–90% reduction in infarct size in some models). However, the protection is ephemeral, dissipating after 1–2 h. This is followed by the slower development of a late phase of protection, which becomes fully manifested at 24 h after the PC stimulus and persists for ~72 h. Thus the duration of the late phase is 30–50 times longer than that of the early phase of PC.
We reckoned that because the late phase has a broader range of protection, and because its duration is 30–40 times greater than that of the early phase, it is likely to have greater clinical relevance. Accordingly, the remainder of this talk will focus on the late phase of PC. The fundamental question to be addressed here pertains to the mechanism of the late phase of PC, because if this issue is not resolved, therapeutic exploitation of this phenomenon will be difficult if not impossible. Figure 3 summarizes our current understanding of the mechanism of late PC. As shown in this figure, a mild, sublethal ischemic stress causes the release of chemical signals that are referred to as “triggers” of late PC. These signals can be viewed as “alarm bells” that go off in the presence of a threat, telling the heart that there is trouble looming on the horizon and that it needs to get ready for the impending threat. Numerous triggers have been identified for the late phase of ischemic PC; among them, NO [generated by endothelial NO synthase (eNOS)], reactive oxygen species, opioid agonists, and adenosine play a prominent role. The release of these chemicals causes the activation of a complex (and yet incompletely understood) signal transduction cascade that includes the ∊-isoform of protein kinase C (PKC), the Src/Lck isoforms of tyrosine kinases, Janus-activated kinases 1 and 2 (JAK1/2), and probably other kinases as well, resulting in the activation of cytoplasmic and normally dormant stress-responsive transcription factors, such as NF-κB, STAT1 and STAT3, and most likely many other transcriptional regulators. This process culminates in the upregulation of cardioprotective genes, leading to the synthesis of new proteins that mediate the protection afforded by late PC. The first gene to be identified as a mediator of late PC was the inducible isoform of NOS (iNOS) (7, 12); subsequently, other genes have been discovered, including cyclooxygenase (COX)-2, heme oxygenase (HO)-1, and antioxidant enzymes such as extracellular SOD (ecSOD), aldose reductase, manganese SOD, etc. (3, 4, 6, 15). Heat stress proteins have been suggested to contribute to late PC, but this remains to be confirmed. Thus late PC is a genetic reprogramming of the heart that is elicited by the exposure to potentially threatening stimuli and is mediated by the activation of stress-responsive and protective genes.
Fig. 3.

Schematic representation of the cellular mechanisms underlying late PC. A nonlethal cellular stress (e.g., reversible ischemia, heat stress, ventricular pacing, or exercise) causes release of chemical signals [nitric oxide (NO), reactive oxygen species (ROS), adenosine, and possibly opioid receptor agonists] that serve as triggers for the development of late PC. These substances activate a complex signal transduction cascade that includes protein kinase C (PKC; specifically, the ∊-isoform), protein tyrosine kinases (specifically, Src and/or Lck), and probably other as yet unknown kinases. A similar activation of PKC and downstream kinases can be elicited pharmacologically by a wide variety of agents, including naturally occurring—and often noxious—substances (e.g., endotoxin, interleukin-1, TNF-α, TNF-β, leukemia inhibitor factor, or ROS), as well as clinically applicable drugs (NO donors, adenosine A1 or A3 receptor agonists, endotoxin derivatives, or δ1-opioid receptor agonists). The recruitment of PKC and distal kinases leads to activation of NF-κB and almost certainly other transcription factors, resulting in increased transcription of multiple cardioprotective genes and synthesis of multiple cardioprotective proteins that serve as comediators of protection 2–4 days after the PC stimulus. The mediators of late PC identified thus far include inducible nitric oxide synthase (iNOS), cyclooxygenase (COX)-2, heme oxygenase (HO)-1, aldose reductase, extracellular (ec)SOD, and Mn SOD. Among the products of COX-2, PGE2 and/or prostacyclin (PGI2) appear to be the most likely effectors of COX-2-dependent protection. Increased synthesis of heat stress proteins (HSPs) is unlikely to be a mechanism of late PC, although the role of posttranslational modification of preexisting HSPs remains to be determined. In addition, the occurrence of cardioprotection on days 2–4 requires the activity of protein tyrosine kinases and possibly p38 MAPKs, potentially because iNOS and other mediators need to undergo posttranslational modulation to confer protection against ischemia. Opening of ATP-sensitive K+ (KATP) channels is also essential for the protection against infarction (but not against stunning) to become manifest. The exact interrelationships among iNOS, COX-2, HO-1, aldose reductase, ecSOD, Mn SOD, and KATP channels are unclear, although recent evidence suggests that COX-2 is downstream of iNOS (i.e., COX-2 is activated by NO) and NO induces HO-1. eNOS, endothelial NOS.
As mentioned above, iNOS was the first protein to be identified as a mediator of late PC (7, 12). Approximately 10 years ago, we postulated (3) that iNOS could participate in late PC because this enzyme is known to be induced by stress and because NO possesses a host of cardioprotective actions that would be expected to be beneficial during myocardial ischemia-reperfusion. Indeed, the literature is replete with studies documenting the salubrious actions of NO during myocardial ischemia-reperfusion injury (3). For example, as illustrated in Fig. 4, in the decade 1990–2000 a total of 91 full-length articles addressed the role of NO in modulating myocardial ischemia-reperfusion injury. Of these, 66 concluded that NO is beneficial and only 11 that NO is detrimental (there were, however, methodological problems with many of these 11 “negative” studies). The precise mechanism whereby NO induces cardioprotection is beyond the scope of this lecture; the reader is referred to a recent review on this topic (15). Briefly, plausible mechanisms whereby NO enhances resistance to cell death following ischemia-reperfusion include inhibition of calcium influx, antagonism of β-adrenergic stimulation, reduction in myocardial oxygen demands, opening of sarcolemmal and/or mitochondrial ATP-sensitive K+ (KATP) channels, activation of COX-2 leading to the synthesis of cardioprotective prostanoids, and possibly direct antioxidant actions, such as inhibition of the effects of superoxide anion (O2•–) and peroxynitrite (ONOO–).
Fig. 4.
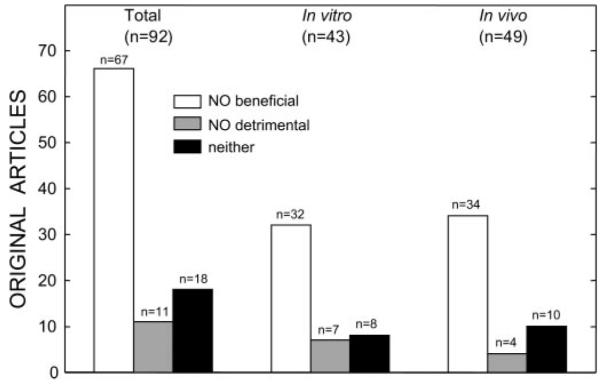
Studies of the role of NO in modulating myocardial ischemia-reperfusion injury. The figure illustrates the studies published in the decade 1990–2000. Of the 92 full-length original manuscripts, 67 concluded that NO is beneficial and only 11 that NO is detrimental (however, several of these “negative” studies have methodological problems). A similar distribution was observed when studies were subdivided according to whether the experiments were performed in vitro (43 studies) or in vivo (49 studies); in both cases, the preponderance of the evidence supports a beneficial role of NO in myocardial ischemia-reperfusion injury. Therefore, the protective effects of NO are not dependent on the use of in vivo or in vitro models. Reprinted from Ref. 3 (Copyright 2001), with permission from Elsevier.
To determine whether iNOS mediates the late phase of ischemic PC, we undertook a study in mice (12). First, we asked whether iNOS is upregulated by ischemic PC. We found that 24 h after exposure of the heart to an ischemic PC protocol (6 cycles of 4-min occlusion and 4-min reperfusion) there was a significant, albeit modest, increase in the total content of iNOS in the ischemic-reperfused region (+40%). The small magnitude of this increase is important because iNOS is known to be toxic when expressed at very high levels; thus a mild upregulation of iNOS is beneficial, whereas a severe upregulation can be detrimental. The increased protein expression was associated with increased activity (as determined by the l-citrulline assay), documenting that the protein is functionally competent. Of course, just because iNOS is upregulated 24 h after ischemic PC does not mean that the protein is involved in the genesis of this phenomenon. Myriad proteins are upregulated after ischemic PC, and most of them have nothing to do with the genesis of the protection (i.e., they represent epiphenomena). To determine whether the upregulation of iNOS was causally related to protection, we performed the second phase of the study in which wild-type (WT) or iNOS–/– mice were subjected to myocardial infarction (30-min coronary occlusion followed by 24 h of reperfusion) (12). We found that under control (unstressed) conditions, infarct size was similar in WT and iNOS–/– mice, implying that iNOS does not modulate myocardial ischemia-reperfusion injury in the absence of stress (this was not surprising, as myocardial iNOS is barely detectable under control conditions). Subjecting mice to a sham PC protocol (open-chest state for 1 h with no coronary occlusion) had no detectable effect on infarct size in WT or iNOS–/– mice. When WT mice were subjected to ischemic PC 24 h earlier (six cycles of 4-min occlusion and 4-min reperfusion), infarct size was markedly reduced compared with sham-operated controls, indicating a robust late PC effect. However, when iNOS–/– mice were subjected to the same ischemic PC protocol, infarct size was similar to that observed in sham-operated mice. Thus, in the absence of iNOS, the late phase of ischemic PC did not develop (12). To determine whether this was due to an inherent inability of the myocardium of iNOS–/– mice to be preconditioned, we also studied the early phase of ischemic PC. WT and iNOS–/– mice were subjected to six cycles of 4-min occlusion and 4-min reperfusion and then, 10 min later, were subjected to a 30-min coronary occlusion followed by 24-h reperfusion. Both WT and iNOS–/– mice exhibited a pronounced reduction in infarct size, of comparable magnitude, demonstrating that iNOS–/– myocardium can indeed be protected and that iNOS is not required for the early phase of ischemic PC. Collectively, these results provided conclusive molecular genetic evidence that iNOS is necessary for the protective effects of late PC to occur (3).
The cellular type that expresses iNOS is important in determining its effects on ischemia-reperfusion injury. In subsequent studies, we found that, after ischemic PC, iNOS is upregulated in cardiac myocytes, as demonstrated both by in situ hybridization at 3 h after the PC stimulus and by iNOS immunohistochemistry at 24 h after the PC stimulus (35). In contrast, in the setting of an infarction (permanent coronary occlusion), we found (using both in situ hybridization and immunohistochemistry) that iNOS was expressed in inflammatory cells (at 48–66 h after occlusion), but not in surviving myocytes. We propose that the expression of iNOS in cells such as myocytes that do not produce large quantities of O2•– is protective, whereas its expression in cells that generate large quantities of O2•– (e.g., macrophages, neutrophils) is detrimental, because of the formation of ONOO–. Another important difference between the upregulation of iNOS in ischemic PC and that observed in other pathophysiological situations is the magnitude of protein expression. As mentioned above, the increase in cardiac iNOS following ischemic PC is mild (~40%), whereas after application of endotoxin (lipopolysaccharide), it is massive (>30-fold) (35).
Returning to our pathophysiological scheme, it is now apparent that NO plays a pivotal role both as a trigger of late PC, at the beginning of the cascade, and as a mediator of late PC, at the end of the cascade (6) (Fig. 3). This was somewhat unexpected, as there is no a priori reason why the same molecule should serve as a trigger as well as a mediator. Nevertheless, these observations emphasize the fundamental role of NO in late PC: that is, NO plays a dual role in the late phase of this phenomenon, acting initially as the trigger and subsequently as the mediator of late PC.
When we discovered the role of iNOS in late PC, we assumed (in retrospect naively) that the problem of identifying the mediator of late PC had been solved. It soon became apparent, however, that late PC is not mediated by just one protein, but rather by a battery of cardioprotective proteins, all of which are necessary for the cardioprotected phenotype to appear (3, 4, 8). Thus late PC should be viewed as a polygenic adaptation of the heart to stress that is underlain by the concerted activation of a number of cardioprotective genes that act in a coordinated fashion to confer resistance to ischemia-reperfusion injury.
The second protein (after iNOS) to be identified as a mediator of late PC was COX-2. It is known that COX-2 is frequently coinduced together with iNOS in response to stress. Furthermore, it has been known for many years that prostanoids [such as prostacyclin (PGI2) and PGE2] are protective during myocardial ischemia-reperfusion (8). The exact mechanism for this beneficial effect is unclear, but it probably involves a combination of actions, including antagonism of adenylyl cyclase, opening of KATP channels, inhibition of calcium influx, antiadrenergic actions, and possibly attenuation of neutrophil infiltration, some of which are reminiscent of those that underlie the protective actions of NO (8). On the basis of these facts, we postulated that COX-2 may be involved as a mediator of late PC together with iNOS. To test this hypothesis, we utilized a conscious rabbit model of late PC and first asked the question of whether ischemic PC upregulates COX-2 expression (28). We found that after a standard ischemic PC protocol (6 cycles of 4-min occlusion and 4-min reperfusion), there was robust upregulation of COX-2 protein content in the ischemic-reperfused region 24 h later (28). This was associated with an increased content of the by-products of COX-2 activity, mainly PGE2 and 6-keto-PGF1α (the stable by-product of PGI2). As in the case of iNOS, however, the mere observation that COX-2 is upregulated does not signify that this protein participates in the genesis of the protection. To establish whether the increased activity and expression of COX-2 are causally related to the protection of late PC, we subjected rabbits to a 30-min coronary occlusion followed by 72 h of reperfusion in the presence or absence of ischemic PC 24 h earlier. Treated rabbits received a selective COX-2 inhibitor (either NS-398 or celecoxib) on day 2, just before the 30-min occlusion, in order to inhibit COX-2 activity during the development of infarction. We reasoned that if the increased production of COX-2 metabolites plays a necessary role in the protection of late PC, pretreatment with COX-2 inhibitors ought to abolish the protection; conversely, if the increased activity of COX-2 is not responsible for protection, the administration of COX-2 inhibitors should have no effect. First, we documented that the doses of NS-398 and celecoxib that we used were able to prevent the increase in the myocardial levels of PGE2 and 6-keto-PGF1α, i.e., that they completely inhibited COX-2 activity 24 h after ischemic PC. Next, we tested the effect of these doses on protection (infarct size). We found that after pretreatment with either NS-398 or celecoxib the reduction in infarct size observed during late PC in control rabbits was completely abrogated, indicating that COX-2 activity is necessary (28). These studies were the first to demonstrate that COX-2 is an obligatory mediator of the cardioprotective effects of late PC.
The recognition that COX-2 is a necessary mediator of the salubrious effects of late PC (8) came as a surprise, as this protein was commonly thought to exert nefarious effects on the cardiovascular system, largely because of its postulated involvement in atherosclerosis. Even if the role of COX-2 in vascular lesions is detrimental, however, our results demonstrate that this is not the case when it comes to myocardial ischemia-reperfusion injury. Our results (28) impelled a reassessment of current views regarding COX-2, revealing a here-tofore unappreciated role of this enzyme as a fundamental cardioprotectant. Indeed, the demonstration that COX-2 is a major cardioprotective protein has been an important paradigm shift in cardiovascular biology. It should be noted that this discovery (28) occurred several years before the controversy on the cardiovascular effects of COX-2 inhibitors erupted. Although most of the publicity has focused on the prothrombotic effects of COX-2 inhibitors, we suggest that a fundamental reason for their adverse cardiovascular profile is that they deprive the heart of its ability to shift to a preconditioned (protected) phenotype in response to stress (26).
The discovery that both iNOS and COX-2 mediate late PC (Fig. 3) led to the obvious question of whether these two proteins act in concert or independently. To address this question, we conducted a series of studies in mice and rabbits, using both pharmacological and genetic inhibition of COX-2 (29, 37). When COX-2–/– mice were subjected to ischemic PC, there was, 24 h later, an increase in iNOS protein expression and activity, as expected (37). When iNOS–/– mice were subjected to ischemic PC, COX-2 protein expression was increased 24 h later, as expected; however, the increase in COX-2 protein content was not accompanied by increased myocardial content of COX-2 by-products (PGF2 and 6-keto-PGF1α), indicating that COX-2 was not active. Thus the absence of COX-2 did not affect iNOS expression or activity following ischemic PC; in contrast, the absence of iNOS prevented the activity of the induced COX-2 protein following ischemic PC (37). We also found that iNOS and COX-2 coprecipitated 24 h after ischemic PC in homogenates of preconditioned myocardium (37), indicating a physical interaction between these two proteins. Although a recent study has confirmed the coprecipitation of iNOS and COX-2 in nonmyocytes (16), it was the study by Xuan et al. (37) that provided the first evidence for this phenomenon. We have obtained similar results in conscious rabbits, in which pharmacological inhibition of COX-2 had no effect on iNOS activity whereas pharmacological inhibition of iNOS with 1400W blocked the increased COX-2 activity (29). In that study, administration of the soluble guanylate cyclase inhibitor ODQ did not block COX-2 activity, indicating that NO activates COX-2 via cGMP-independent mechanisms (29). Together, these studies (29, 37) demonstrate that, in late PC, COX-2 activity requires iNOS-derived NO whereas iNOS activity is independent of COX-2-derived prostanoids, implying that COX-2 is located downstream of iNOS in the protective pathway of late PC. Thus these two proteins act in series, with iNOS being the protein that drives COX-2 activity.
Figure 5 summarizes the mechanism of late PC. As can be seen, the initial PC ischemia activates at least two signaling pathways that act in parallel: the PKC∊-Src/Lck-NF-κB pathway and the JAK1/JAK2-STAT1/STAT3 pathway. These pathways lead to nuclear translocation of NF-κB and STAT1/STAT3, which bind to the cognate promoter sequences of the iNOS and COX-2 genes, resulting in increased synthesis of the respective proteins. iNOS produces NO, which is in itself cardioprotective but also acts by activating COX-2, leading to the synthesis of the cytoprotective prostanoids PGI2 and PGE2. The combined (and possibly synergistic) actions of NO and PGI2/PGE2 result in myocardial protection.
Fig. 5.

Schematic representation of the mechanism of late PC. The PC stimulus activates at least 2 signaling pathways: the PKC∊-Src/Lck-NF-κB pathway and the Janus-activated kinase (JAK)1/JAK2-STAT1/STAT3 pathway. PKC∊ activates the Src and Lck tyrosine kinases, leading to tyrosine phosphorylation of IκBα (the inhibitor of NF-κB); in addition, PKC∊ directly activates IKKα/IKKβ, leading to inhibition of IκBα. NF-κB translocates to the nucleus, where it binds to its cognate sequence on the promoter region of target genes, including iNOS and COX-2. The PC ischemia also activates JAK1 and JAK2, which then tyrosine phosphorylate and activate STAT1 and STAT3. In addition to tyrosine phosphorylation, full activation of STAT1 and STAT3 also requires serine phosphorylation via a PKC∊-Raf-1-MEK1/2-p44/42 MAPK signaling pathway. The activated STAT1/STAT3 heterodimer translocates to the nucleus, where it binds to the promoter of target genes. The combinatorial actions of NF-κB and STAT1/STAT3, and almost certainly other transcription factors, result in transcriptional activation of iNOS, COX-2, HO-1, and ecSOD, leading to the synthesis of the respective proteins. (A multitude of transcription factors are likely to be mobilized during late PC and to lead to the recruitment of cardioprotective genes, acting in concert.) iNOS produces NO, which directly protects the myocardium; in addition, NO activates COX-2, leading to the synthesis of cardioprotective prostanoids, mainly PGI2 and PGE2. It follows from this that the activity of newly synthesized COX-2 protein requires iNOS-dependent NO generation whereas the activity of iNOS does not require COX-2-dependent prostanoid generation; thus COX-2 is downstream of iNOS in the pathophysiological cascade of late PC. In addition, NO can also induce HO-1. Thus iNOS-derived NO can protect the myocardium from recurrent ischemia via direct actions, via activation of COX-2-dependent synthesis of cardioprotective prostanoids, and via generation of HO-1 by-products (CO and biliverdin). Among the products of COX-2, PGE2 and/or PGI2 appear to be the most likely effectors of cytoprotection. A similar upregulation of COX-2 can be elicited pharmacologically by δ-opioid receptor agonists but not by adenosine A1 or A3 receptor agonists.
Before moving to the topic of gene therapy, it is important to note that virtually all of the studies of PC described above were performed in relatively young, healthy animals. Whether the results of these studies apply to animals (or humans) that are old or have risk factors for cardiovascular disease (e.g., hypercholesterolemia, diabetes, hypertension) is an important issue that needs to be thoroughly investigated. Considerable evidence indicates that hypercholesterolemia abrogates both early (33) and late (30, 31) PC, although the effects on early PC remain controversial (17). In addition, it appears that early and late PC are either attenuated or absent in the presence of diabetes (10, 18, 32). No information is available regarding the impact of hypertension on PC. The effect of aging on PC is unclear. Some studies (1, 11) but not others (25) support the concept that the early PC response is absent in old animals. Aging abolishes the late phase of ischemia-induced PC in mice (36) but not the late phase of opioid-induced late PC in rats (27). Because most patients with ischemic heart disease are old and/or have risk factors, defining the impact of these conditions on PC and its mechanism is of the utmost importance.
Our motivation to study the mechanism of late PC stemmed not only from the intellectual drive to unravel the molecular basis of this powerful adaptation to stress but, most importantly, from a desire to exploit this phenomenon for therapeutic purposes. From the outset 15 years ago, it seemed clear to us that if we could understand how PC makes the heart resistant to ischemia, it should then be possible to induce this phenomenon in patients at risk for myocardial infarction. One could argue that protection of ischemic myocardium in patients can be achieved by using direct cardioprotective strategies (rather than PC); however, there are several inherent limitations in this approach. Direct cardioprotection in patients can usually be implemented only at the time of reperfusion (when the patient presents to the hospital). Unfortunately, therapies started at reperfusion have limited efficacy, particularly because, in most cases, reperfusion cannot be implemented until 4–5 h after the onset of symptoms. By that time, it is likely that a significant part (perhaps most) of the damage has already occurred. Administration of an infarct-sparing drug 4–5 h after the onset of myocardial infarction is too late, and this is the predicament in which most patients find themselves. On the other hand, it is usually impossible to pretreat patients because the onset of an acute infarction is unpredictable. Although one could administer infarct-sparing agents chronically in anticipation of an infarct, this would be an exceptionally cumbersome and expensive approach to infarct size reduction. Similarly, chronic treatment with a PC-mimetic agent would be quite impractical. Consequently, we reasoned that the most efficacious strategy to limit infarct size may be to induce a chronically protected (defensive) cardiac phenotype analogous to that associated with late PC, by transferring the genes responsible for late PC. We refer to this approach as prophylactic cardioprotection.
In my mind, the most important implication of understanding the mechanism of late PC is that this knowledge offers a template for implementing prophylactic cardioprotection in patients with gene therapy. The rationale is clear. As discussed above, late PC is a genetic reprogramming of the heart that confers a protected (defensive) phenotype and is underlain by the upregulation of specific cardioprotective genes, some of which have already been identified. Based on this premise, it is plausible to postulate that long-term expression of the proteins that mediate the anti-ischemic effects of late PC should protect the heart whenever ischemia occurs and throughout the entire ischemia-reperfusion sequence. The important points of this hypothesis are that the protection afforded by gene therapy would be present at any time when ischemia strikes (which is unpredictable) and would be operative from the very onset of ischemia (unlike direct cardioprotection, which usually is implemented at the onset of reperfusion). Thus prophylactic gene therapy has the potential for limiting the size of the infarct whenever it occurs and for combating the damage inflicted by both ischemia and reperfusion, not just that inflicted by reperfusion.
As a first step toward the development of prophylactic gene therapy, we sought to determine whether it is possible to induce a chronically protected cardiac phenotype by transferring to the myocardium the genes responsible for the salubrious actions of late PC. We have developed viral vectors encoding several of the genes identified as mediators of late PC, specifically, iNOS, COX-2, HO-1, and ecSOD. We started by testing the effects of iNOS because, historically, this was the first protein to be identified as a mediator of late PC and because a multitude of studies have demonstrated that iNOS is a necessary mediator of late PC induced by diverse stimuli, including ischemia, physical exercise, NO donors, various GPCR agonists, and endotoxin derivatives, indicating that this enzyme plays a central role in cardioprotection and may well be the final common pathway for any kind of late PC stimulus (3, 6). Although these studies have demonstrated that iNOS is necessary for late PC to occur, it remains to be determined whether a selective increase in iNOS activity via gene transfer is sufficient to replicate the salubrious effects of late PC. Initially, we employed a second-generation adenoviral vector (Ad5/ iNOS) deleted in the E1, E2A, and E3 regions (22). A total of 107 plaque-forming units of Ad-iNOS was injected intramyocardially in the soon-to-be ischemic region of mouse hearts. Three days later, the injected region exhibited an increase in iNOS protein content, iNOS activity, and nitroxide (NO2/NO3) levels, indicating effective transduction of myocytes with functionally competent iNOS. Immunohistochemistry confirmed that the transgene was expressed in cardiac myocytes. In another series of experiments, mice received either Ad5/iNOS or Ad5/LacZ and, 3 days later, were subjected to a 30-min coronary occlusion followed by reperfusion. Although the intramyocardial injection of iNOS resulted in transduction of only ~20% of the risk region, there was a marked reduction in infarct size in Ad5/iNOS-transduced mice compared with Ad5/LacZ-transduced mice (22). These data demonstrate that iNOS gene therapy results in effective limitation of infarct size 3 days later and suggest that paracrine effects of NO secreted by transduced cells amplify the effects of gene transfer. We subsequently extended our follow-up to several weeks after Ad5/iNOS gene transfer and found that, as late as 8 wk after injection of Ad5/iNOS, there was increased iNOS protein expression and activity in the myocardium, associated with persistent limitation of infarct size (21). Serial echocardiographic assessment of left ventricular (LV) function revealed no detrimental effects of iNOS gene therapy on LV function or dimensions up to 8 wk later. We have recently completed preliminary studies at 1 yr after recombinant adeno-associated virus (rAAV)-mediated LacZ or iNOS gene transfer, which suggest persistent protection at this time point. Together, these studies (21, 22) demonstrate that iNOS gene transfer affords effective cardioprotection, and that the magnitude is equivalent to that afforded by late PC but the duration is much longer (at least several weeks). These results provide proof of principle for gene therapy against ischemia-reperfusion injury that increases local myocardial NO levels without hemodynamic effects, thereby obviating the need for continuous intravenous infusion of NO donors.
Because in the setting of ischemic PC the beneficial effects of iNOS upregulation are mediated by COX-2 (29, 37), we sought to determine the role of COX-2 in the beneficial effects of iNOS gene therapy (22). We found that, 3 days after Ad5-mediated iNOS gene transfer, there was upregulation of COX-2 in the same region in which iNOS was upregulated. The increased expression of COX-2 was associated with increased myocardial content of COX-2 by-products (PGF2 and 6-keto-PGF1α), a pattern reminiscent of that observed after ischemic PC (28). When iNOS-transduced mice were pretreated with the COX-2 inhibitor NS-398 before the 30-min coronary occlusion, the beneficial effects of iNOS gene transfer were completely abrogated (22). Similarly, when Ad5-mediated iNOS gene transfer was performed in COX-2–/– mice, no reduction in infarct size was observed (13). Collectively, these studies (13, 22) demonstrate that 1) iNOS gene transfer results in increased myocardial COX-2 protein expression and increased PGE2 and PGI2 levels and 2) pharmacological inhibition or genetic ablation of COX-2 abrogates the infarct-sparing effects of iNOS gene transfer, demonstrating that increased biosynthesis of COX-2-derived prostanoids, such as PGE2 and PGI2, represents an essential mechanism of iNOS-dependent cardioprotection. Together with our previous studies of ischemic PC (29, 37), these observations (13, 22) reveal a close functional coupling between cardiac iNOS and cardiac COX-2; that is, upregulation of iNOS leads to secondary upregulation of COX-2, which in turn mediates the cytoprotective effects of iNOS.
How does iNOS gene transfer lead to upregulation of COX-2? We tested the role of NF-κB because 1) this transcription factor is known to be activated by NO (38), 2) the promoter of the COX-2 gene contains NF-κB-binding sequences (2), and 3) NF-κB is critical for the development of the late phase of ischemic PC (38). We used transgenic mice that express cardiac-specifically a mutant of IκBα (IκBαS32A,S36A) that cannot be serine-phosphorylated and thus inhibited; overexpression of this dominant-negative IκBα mutant prevents NF-κB activation (9). In IκBαS32A,S36A mice, gene therapy with Ad5/iNOS failed to increase myocardial COX-2 protein content and to limit infarct size, demonstrating an obligatory role of NF-κB in iNOS-dependent COX-2 upregulation and in iNOS-dependent protection (13).
Given that COX-2 is necessary for the beneficial effects of iNOS gene therapy, we tested whether selective upregulation of COX-2 (without iNOS gene therapy) would be sufficient to confer a protected phenotype. To this end, we have used rAAV vectors. rAAVs offer a number of important advantages compared with recombinant adenovirus (rAd) vectors. rAAVs are not associated with any known human disease, infect both mitotic and postmitotic cells, and are smaller than rAds, which may favor extravasations and tissue transduction. Most importantly, rAAVs lead to chromosomal, not episomal, integration of the transgene and do not induce synthesis of any viral proteins, so that there is virtually no immune/inflammatory reaction to rAAV-mediated gene transfer (34). As a result of these last two features, rAAV-mediated gene transfer results in long-lasting (at least 1 yr in several models), possibly permanent, transgene expression, which enables potentially indefinite production of the therapeutic protein (34). Thus the hypothesis driving our study was that the durable transgene expression afforded by rAAV vectors may enable chronic, possibly permanent, prophylactic cardioprotection. We utilized an rAAV helper-free system to prepare rAAV vectors for injection, thereby ensuring that there would be no contamination of the injectate with helper virus. Because of their long-term nature these studies are laborious and time intensive, and they are still ongoing. Nevertheless, we have obtained preliminary results (20) demonstrating that, 3 mo after rAAV-mediated COX-2 gene transfer, there is robust upregulation of COX-2 protein expression in the transduced myocardium in mice, which is associated with protection against myocardial infarction. We are currently conducting studies with other rAAV vectors encoding iNOS, HO-1, and ecSOD. The goal of all of these studies is to test whether rAAV-mediated gene transfer can confer permanent protection against infarction.
Our interest in ecSOD gene therapy stems from the fact that, in contrast to all other antioxidant proteins, which are intracellular, ecSOD is a secreted protein that acts on the extracellular matrix, thereby producing a paracrine effect on nontransduced cells. By intercepting extracellular O2•–, ecSOD prevents formation of cytotoxic ONOO– and also protects NO from inactivation, thereby increasing NO bioavailability. Thus ecSOD limits oxidative and nitrative stress and facilitates diffusion of NO to adjacent cells. In preliminary studies we have found that, 3 mo after rAAV-mediated ecSOD gene transfer, infarct size is reduced vis-à-vis mice transduced with rAAV/LacZ, and myocardial ecSOD levels are increased. We have recently completed preliminary studies with rAAV-mediated ecSOD gene transfer, which suggest persistent expression of ecSOD in cardiac myocytes at 8 mo and persistent protection against infarction at 12 mo.
Taken together, the studies summarized above (13, 20–22) indicate that cardiac transfer of the genes that mediate late PC (i.e., iNOS, COX-2, ecSOD) confers a cardioprotected phenotype that emulates late PC. rAAV-mediated transfer of these genes dramatically extends the duration of the protection (for at least 3 mo) compared with late PC (which lasts for only 3 days), with no inflammatory reaction. Thus it is possible to genetically reprogram the heart in a manner that confers prolonged (possibly even permanent) protection against ischemia-reperfusion injury (chronic prophylactic cardioprotection). At present, the major limitation to translating these basic studies to humans is the lack of effective techniques for transducing significant numbers of myocytes in vivo. Nevertheless, as new technologies emerge, it seems likely that methods for transducing cardiac myocytes will be developed that could be applied to patients with ischemic heart disease. Once such techniques become available, it will be important to test whether transfer of the genes that the heart utilizes to protect itself during late PC can confer long-lasting or permanent protection against infarction in patients. This, in my mind, is the ultimate goal of studying the phenomenon of cardiac PC.
In conclusion, the study of ischemic PC has had two major implications. First, it has revealed that the heart possesses a remarkable phenotypic plasticity (i.e., it responds to stress by changing its phenotype in a teleologically useful manner). Second, genetic exploitation of ischemic PC may offer unprecedented potential for protecting the ischemic myocardium. Twenty years after its discovery, it is clear that ischemic PC has been a major paradigm shift in the biology of myocardial ischemia, although the therapeutic expectations it raised remain to be fulfilled.
Acknowledgments
GRANTS This study was supported in part by National Heart, Lung, and Blood Institute R01 Grants HL-55757, HL-68088, HL-70897, HL-76794, and HL-78825.
Footnotes
This lecture was presented at the 35th International Congress of Physiological Sciences in San Diego, CA, on April 2, 2005.
REFERENCES
- 1.Abete P, Ferrara N, Cioppa A, Ferrara P, Bianco S, Calabrese C, Cacciatore F, Longobardi G, Rengo F. Preconditioning does not prevent postischemic dysfunction in aging heart. J Am Coll Cardiol. 1996;27:1777–1786. doi: 10.1016/0735-1097(96)00070-8. [DOI] [PubMed] [Google Scholar]
- 2.Appleby SB, Ristimaki A, Neilson K, Narko K, Hla T. Structure of the human cyclo-oxygenase-2 gene. Biochem J. 1994;302:723–727. doi: 10.1042/bj3020723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- 4.Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 5.Bolli R, Becker L, Gross G, Mentzer R, Jr, Balshaw D, Lathrop DA. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95:125–134. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 6.Bolli R, Dawn B, Tang XL, Qiu Y, Ping P, Xuan YT, Jones WK, Takano H, Guo Y, Zhang J. The nitric oxide hypothesis of late preconditioning. Basic Res Cardiol. 1998;93:325–338. doi: 10.1007/s003950050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolli R, Manchikalapudi S, Tang XL, Takano H, Qiu Y, Guo Y, Zhang Q, Jadoon AK. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ Res. 1997;81:1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- 8.Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y, Dawn B. Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res. 2002;55:506–519. doi: 10.1016/s0008-6363(02)00414-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawn B, Xuan YT, Marian M, Flaherty MP, Murphree SS, Smith TL, Bolli R, Jones WK. Cardiac-specific abrogation of NF-κB activation in mice by transdominant expression of a mutant IκBα. J Mol Cell Cardiol. 2001;33:161–173. doi: 10.1006/jmcc.2000.1291. [DOI] [PubMed] [Google Scholar]
- 10.del Valle HF, Lascano EC, Negroni JA. Ischemic preconditioning protection against stunning in conscious diabetic sheep: role of glucose, insulin, sarcolemmal and mitochondrial KATP channels. Cardiovasc Res. 2002;55:642–659. doi: 10.1016/s0008-6363(02)00468-6. [DOI] [PubMed] [Google Scholar]
- 11.Fenton RA, Dickson EW, Meyer TE, Dobson JG., Jr Aging reduces the cardioprotective effect of ischemic preconditioning in the rat heart. J Mol Cell Cardiol. 2000;32:1371–1375. doi: 10.1006/jmcc.2000.1189. [DOI] [PubMed] [Google Scholar]
- 12.Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci USA. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Y, Luo C, Dawn B, Tan W, Wu WJ, Hunt G, Zhu X, Li Q. The cardioprotection afforded by iNOS gene therapy is mediated by COX-2 via an NF-κB-dependent pathway (Abstract) Circulation. 2004;110:III–29. [Google Scholar]
- 14.Guo Y, Wu WJ, Zhu XP, Li Q, Tang XL, Bolli R. Exercise-induced late preconditioning is triggered by generation of nitric oxide (Abstract) J Mol Cell Cardiol. 2001;33:A41. [Google Scholar]
- 15.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 16.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 17.Kremastinos DT, Bofilis E, Karavolias GK, Papalois A, Kaklamanis L, Iliodromitis EK. Preconditioning limits myocardial infarct size in hypercholesterolemic rabbits. Atherosclerosis. 2000;150:81–89. doi: 10.1016/s0021-9150(99)00389-5. [DOI] [PubMed] [Google Scholar]
- 18.Kristiansen SB, Lofgren B, Stottrup NB, Khatir D, Nielsen-Kudsk JE, Nielsen TT, Botker HE, Flyvbjerg A. Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia. 2004;47:1716–1721. doi: 10.1007/s00125-004-1514-4. [DOI] [PubMed] [Google Scholar]
- 19.Kuzuya T, Hoshida S, Yamashita N, Fuji H, Oe H, Hori M, Kamada T, Tada M. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res. 1993;72:1293–1299. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Guo Y, Luo C, Tan W, Wu WJ, Xu B, Siddiqui T, Rokosh GD, Bolli R. Long-term protection against myocardial infarction with cyclooxygenase-2 (COX-2) gene therapy via a recombinant adeno-associated viral (rAAV) vector (Abstract) Circulation. 2004;110:III–107. [Google Scholar]
- 21.Li Q, Guo Y, Tan W, Stein AB, Dawn B, Wu WJ, Zhu X, Lu X, Xu X, Siddiqui T, Tiwari S, Bolli R. Gene therapy with iNOS provides long-term protection against myocardial infarction without adverse functional consequences. Am J Physiol Heart Circ Physiol. 2006;290:H584–H589. doi: 10.1152/ajpheart.00855.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Guo Y, Xuan YT, Lowenstein CJ, Stevenson SC, Prabhu SD, Wu WJ, Zhu Y, Bolli R. Gene therapy with inducible nitric oxide synthase protects against myocardial infarction via a cyclooxygenase-2-dependent mechanism. Circ Res. 2003;92:741–748. doi: 10.1161/01.RES.0000065441.72685.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- 24.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 25.Przyklenk K, Li G, Whittaker P. No loss in the in vivo efficacy of ischemic preconditioning in middle-aged and old rabbits. J Am Coll Cardiol. 2001;38:1741–1747. doi: 10.1016/s0735-1097(01)01603-5. [DOI] [PubMed] [Google Scholar]
- 26.Shinmura K, Bolli R. The risk for myocardial infarction with cyclooxygenase-2 inhibitors. Ann Intern Med. 2005;143:615–618. doi: 10.7326/0003-4819-143-8-200510180-00022. [DOI] [PubMed] [Google Scholar]
- 27.Shinmura K, Nagai M, Tamaki K, Bolli R. Gender and aging do not impair opioid-induced late preconditioning in rats. Basic Res Cardiol. 2004;99:46–55. doi: 10.1007/s00395-003-0436-5. [DOI] [PubMed] [Google Scholar]
- 28.Shinmura K, Tang XL, Wang Y, Xuan YT, Liu SQ, Takano H, Bhatnagar A, Bolli R. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proc Natl Acad Sci USA. 2000;97:10197–10202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shinmura K, Xuan YT, Tang XL, Kodani E, Han H, Zhu Y, Bolli R. Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning. Circ Res. 2002;90:602–608. doi: 10.1161/01.res.0000012202.52809.40. [DOI] [PubMed] [Google Scholar]
- 30.Tang XL, Stein AB, Shirk G, Bolli R. Hypercholesterolemia blunts NO donor-induced late preconditioning against myocardial infarction in conscious rabbits. Basic Res Cardiol. 2004;99:395–403. doi: 10.1007/s00395-004-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang XL, Takano H, Xuan YT, Sato H, Kodani E, Dawn B, Zhu Y, Shirk G, Wu WJ, Bolli R. Hypercholesterolemia abrogates late preconditioning via a tetrahydrobiopterin-dependent mechanism in conscious rabbits. Circulation. 2005;112:2149–2156. doi: 10.1161/CIRCULATIONAHA.105.566190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsang A, Hausenloy DJ, Mocanu MM, Carr RD, Yellon DM. Preconditioning the diabetic heart: the importance of Akt phosphorylation. Diabetes. 2005;54:2360–2364. doi: 10.2337/diabetes.54.8.2360. [DOI] [PubMed] [Google Scholar]
- 33.Ueda Y, Kitakaze M, Komamura K, Minamino T, Asanuma H, Sato H, Kuzuya T, Takeda H, Hori M. Pravastatin restored the infarct size-limiting effect of ischemic preconditioning blunted by hypercholesterolemia in the rabbit model of myocardial infarction. J Am Coll Cardiol. 1999;34:2120–2125. doi: 10.1016/s0735-1097(99)00440-4. [DOI] [PubMed] [Google Scholar]
- 34.Verma IM, Weitzman MD. Gene therapy: twenty-first century medicine. Annu Rev Biochem. 2005;74:711–738. doi: 10.1146/annurev.biochem.74.050304.091637. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Guo Y, Zhang SX, Wu WJ, Wang J, Bao W, Bolli R. Ischemic preconditioning upregulates inducible nitric oxide synthase in cardiac myocyte. J Mol Cell Cardiol. 2002;34:5–15. doi: 10.1006/jmcc.2001.1482. [DOI] [PubMed] [Google Scholar]
- 36.Wu WJ, Xuan YT, Tan W, Zhu X, Zhu Y, Guo Y. The loss of ischemic preconditioning in the senescent heart is associated with impaired upregulation of inducible nitric oxide synthase and cyclooxygenase-2 (Abstract) Circulation. 2003;108:IV–187. [Google Scholar]
- 37.Xuan YT, Guo Y, Zhu Y, Han H, Langenbach R, Dawn B, Bolli R. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J Mol Cell Cardiol. 2003;35:525–537. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xuan YT, Tang XL, Banerjee S, Takano H, Li RC, Han H, Qiu Y, Li JJ, Bolli R. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–1109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- 39.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]