

Abstract
Mechanisms of ligand binding to the PTH/PTHrP receptor (PTHR) were explored using PTH fragment analogs as radioligands in binding assays. In particular, the modified amino-terminal fragment analog, 125I-[Aib1,3,Nle8,Gln 10,homoarginine11,Ala12 Trp14,Tyr15]rPTH(1–15)NH2, 125I-[Aib1,3,M]PTH(1–15), was used as a radioligand that we hypothesized to bind solely to the juxtamembrane (J) portion of the PTHR containing the extracellular loops and transmembrane helices. We also employed 125I-PTH(1– 34) as a radioligand that binds to both the amino-terminal extracellular (N) and J domains of the PTHR. Binding was examined in membranes derived from cells expressing either wild-type or mutant PTHRs. We found that the binding of 125I-[Aib1,3,M]PTH(1–15) to the wild-type PTHR was strongly (∼90%) inhibited by guanosine 5′-O-(3-thio)triphosphate (GTPγS), whereas the binding of 125I-PTH(1–34) was only mildly (∼25%) inhibited by GTPγS. Of these two radioligands, only 125I-[Aib1,3,M]PTH(1–15) bound to PTHR-delNt, which lacks most of the receptor's N domain, and again this binding was strongly inhibited by GTPγS. Binding of 125I-[Aib1,3,M]PTH(1–15) to the constitutively active receptor, PTHR-H223R, was only mildly (∼20%) inhibited by GTPγS, as was the binding of 125I-PTH(1–34). In membranes prepared from cells lacking GαS via knockout mutation of Gnas, no binding of 125I-[Aib1,3,M]PTH(1–15) was observed, but binding of 125I-[Aib1,3,M]PTH(1–15) was recovered by virally transducing the cells to heterologously express GαS. 125I-PTH(1–34) bound to the membranes with or without GαS. The overall findings confirm the hypothesis that 125I-[Aib1,3,M]PTH(1–15) binds solely to the J domain of the PTHR. They further show that this binding is strongly dependent on coupling of the receptor to GαS-containing heterotrimeric G proteins, whereas the binding of 125I-PTH(1–34) can occur in the absence of such coupling. Thus, 125I-[Aib1,3,M]PTH(1–15) appears to function as a selective probe of GαS-coupled, active-state PTHR conformations.
PTH and PTHrP play critical roles in calcium homeostasis, tissue development, and bone remodeling. PTH and PTHrP use for these functions the PTH/PTHrP receptor (PTHR), a class 2 G protein-coupled receptor (GPCR). For both PTH and PTHrP, which in humans are polypeptides of 84 and 141 amino acids, respectively, the principal determinants of receptor-binding affinity map to the carboxy-terminal portion of the fully active (1–34) ligand fragment, whereas the principal determinants of receptor activation map to the amino-terminal portion of the ligand. Both PTH(1–34) and PTHrP(1–34) induce strong coupling of the PTHR to the adenylyl cyclase/cAMP pathway mediated by GαS but can also induce coupling to signaling pathways involving other G proteins, including Gαq/11 (1, 2), Gαi (3), and Gα12/13 (4).
The mechanisms by which PTH(1–34) and PTHrP-(1–34) bind to the PTHR and induce receptor activation have been investigated through the complementary approaches of receptor mutagenesis/chimerization (5–8) and photo-affinity cross-linking (9–11). The combined results of these studies have given rise to a bipartite model of ligand-receptor interaction. By this two-domain model, the carboxy-terminal portion of PTH(1–34) binds to the amino-terminal extracellular (N) domain of the receptor in an initial docking step, and then the amino-terminal portion of the ligand engages the extracellular loop/transmembrane domain, or juxtamembrane (J) region of the receptor to induce the conformational changes involved in receptor activation and G protein coupling (12–14). Accumulating data suggest that a similar two domain-binding mechanism may be used by other class 2 GPCRs, including the calcitonin receptor (15, 16), the secretin receptor (17,18), the vasoactive intestinal peptide receptor (19), and the corticotropin-releasing factor receptor (20–22), each of which binds a peptide ligand approximately equal in length to PTH(1–34).
Although the amino-terminal portion of PTH contains the principal determinants of receptor activation, amino-terminal fragments of PTH, such as PTH(1–14), elicit only extremely weak (EC50 = 200 μM) cAMP responses in PTHR-expressing cells, and exhibit no binding activity in competition assays performed with 125I-PTH(1–34) radioligand (23). The weak activities of such amino-terminal PTH fragments, as compared with the nanomolar potencies of PTH(1–34) ligands, can largely be explained by the absence of the docking interactions that normally occur between the carboxy-terminal portion of PTH(1–34) and the N domain of the receptor, as postulated by the two-domain model outlined above. In a recent series of structure-activity relationship studies performed on the PTH(1–14) fragment, we identified a number of modifications that together enhance binding affinity and cAMP-stimulating potency of the fragment on the PTHR by as much as five orders of magnitude. One such analog resulting from this work, [Aib1,3,Gln10,Har11,Ala12, Trp14]PTH(1–14)NH2, is as potent as PTH(1–34) for stimulating cAMP formation in PTHR-expressing cells (24–27). We have hypothesized that the modifications of these PTH(1–14) analogs improve binding interactions specifically to the J domain of the PTHR because the analogs exhibit the same potency on a PTHR derivative, PTHR-delNt, that lacks most of the receptor's N domain, as they do on the intact PTHR. In contrast, unmodified PTH(1–34) is approximately 100-fold less potent on PTHR-delNt than it is on the intact PTHR (26, 27). In recent extensions of this work, we derived inactive congeners of the modified PTH(1–14) analogs, substituted at positions 1–3, that competitively inhibit the agonist action of not only modified PTH(1–14) analogs, but also that of unmodified PTH(1–34) (28), consistent with the evolving model of the PTH-PTHR binding and activation mechanism.
In further extensions of our PTH(1–14) analog work, we sought to develop an amino-terminal PTH analog that could be used as a PTHR radioligand for exploring the interactions that occur specifically between the amino-terminal pharmacophoric region of PTH and the juxtamembrane region of the receptor. We have thus developed [Aib1,3,Nle8,Gln10,Har11,Ala12,Trp14,Tyr15]PTH-(1–15)NH2, herein termed [Aib1,3,M]PTH(1–15), which, when radioiodinated on tyrosine15, binds effectively as a radioligand to both the intact PTHR and to PTHR-delNt, as shown in recent whole-cell binding assays (27, 28). Here, we use this PTH(1–15) analog radioligand, along with 125I-PTH(1–34), and, in some experiments, the antagonist radioligand, 125I-PTHrP(5–36) (28), to further analyze the ligand-binding mechanisms used by the PTHR. The results show that, in contrast to the binding of 125I-PTH(1–34), the binding of 125I-[Aib1,3,M]PTH(1–15) to the PTHR is strongly dependent on the G protein-coupling/activation status of the receptor.
Results
Binding Time Courses in HKRK-B7 Cell Membranes
We first examined the time courses of receptor association for the agonist radioligands 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15), and that of the antagonist radioligand 125I-PTHrP(5–36) in membranes prepared from HKRK-B7 cells. These cells, derived from the porcine kidney cell line LLC-PK1, are stably transfected to express the human PTHR at a density of approximately 950,000 copies per cell (29). As shown in Fig. 1, 125I-PTH(1–34) reached a maximum level of binding by approximately 40 min (t1/2 = 12 min), 125I-[Aib1,3,M]PTH(1–15) reached a maximum level by approximately 90 min (t1/2 = 37 min) and 125I-PTHrP-(5–36) reached a maximum level within 5 min (t1/2 = 1.5 min). In these experiments, each radioligand was used at approximately the same concentration (∼0.03 nM) and with the same concentration of PTHR (membrane protein = 20 ng/μl). The differences in the equilibration times observed for these radioligands thus may reflect, at least in part, differences in the mechanisms by which each radioligand binds to the PTHR.
Fig. 1.
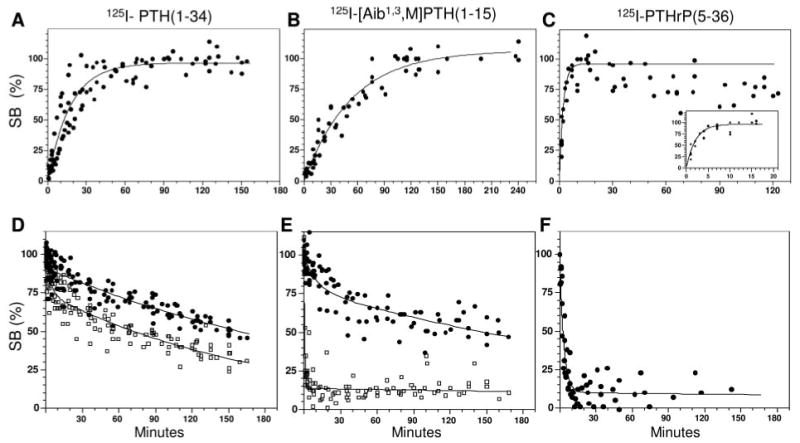
Radioligand Association and Dissociation Time Courses in HKRK-B7 Cell Membranes
Panels A–C show the time courses of association of 125I-PTH(1–34) (A), 125I-[Aib1,3,M]PTH(1–15) (B) and 125I-PTHrP(5–36) (C) with the PTHR in HKRK-B7 cell membranes. The inset of panel C displays the early time point data for 125I-PTHrP(5–36) on an expanded time scale. Panels D–F show the dissociation of complexes formed by the same radioligands and HKRK-B7 cell membranes after a 90 min preincubation phase. The agonists, 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15) were assessed in the absence (closed circles) and presence (open squares) of GTPγS (5 × 10−5 m, added at t = 0). Each graph shows aggregate data, expressed as a percent of the maximum, specifically bound radioactivity (SB), from four or more experiments. In Panels A–C, the total maximum binding (specific plus nonspecific) and nonspecific binding levels were 12,319 ± 1,518 cpm and 356 ± 52 cpm, respectively, for 125I-PTH(1–34) (total radioactivity was 23,631 ± 1,849 cpm, n = 5); 3,073 ± 505 cpm and 346 ± 85 cpm, respectively, for 125I-[Aib1,3,M]PTH(1–15) (total was 30,108 ± 1,674 cpm, n = 4) and 3,364 ± 233 cpm and 456 ± 60 cpm, respectively, for 125I-PTHrP (5–36) (total was 23,583 ± 1,899 cpm, n = 4). In Panels D–F, the total maximum binding at t = 0, and nonspecific binding were 13,347 ± 1,332 cpm and 1,203 ± 206 cpm, respectively, for 125I-PTH(1–34) (total radioactivity was 31,557 ± 2,178 cpm, n = 10); 3,012 ± 515 cpm and 647 ± 105 cpm, respectively, for 125I-[Aib1,3,M]PTH(1–15) (total was 38,522 ± 3,911 cpm, n = 7) and 4263 ± 811 cpm and 700 ± 110 cpm, respectively, for 125I-PTHrP(5–36) (total was 25,062 ± 1,610 cpm, n = 4). Values are means (±sem). All reactions contained a membrane protein concentration of 20 ng/μl.
The time courses of dissociation of the complexes formed between each radioligand and the PTHR in HKRK-B7 cell membranes was then examined. A preincubation of radioligand and membranes of 90 min was used to allow for ligand·receptor complex formation, after which the dissociation phase was initiated by the addition of excess unlabeled peptide ligand. To gain information on the extent to which G protein interactions might modulate the stability of the complexes formed by the agonist radioligands, 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15), the dissociation times of these complexes were assessed in the absence and presence of the nonhydrolyzable guanine nucleotide analog, guanosine 5′-O-(3-thio)t-riphosphate (GTPγS) (5 ×10−5 m). The data were fit to a biexponential decay equation so as to accommodate potential rapid (t1/2 ≤ 5 min) and slow components in the resulting curves. As shown in Fig. 1D, in the absence of GTPγS, only approximately 9% (time course parameter values provided were derived from the curve fits of the aggregate data shown in the figures) of the complexes formed with 125I-PTH(1–34) dissociated rapidly, whereas the remaining fraction dissociated slowly (t1/2 = ∼180 min). The addition of GTPγS increased the fraction of 125I-PTH(1–34)·PTHR complexes that dissociated rapidly to 25%, whereas the remaining 75% of the complexes dissociated slowly (t1/2 = ∼120 min). In the absence of GTPγS, the dissociation profile obtained with 125I-[Aib1,3,M]PTH(1– 15) was similar to that obtained with 125I-PTH(1–34), in that 21% of the complexes dissociated rapidly and the remaining complexes dissociated slowly (t1/2 = ∼230 min; Fig. 1E). Upon addition of GTPγS, however, most (86%) of the 125I-[Aib1,3,M]PTH(1–15)·PTHR complexes dissociated rapidly (Fig. 1E). With the antagonist, 125I-PTHrP(5–36), assessed only in the absence of GTPγS, nearly all (90%) of the complexes dissociated rapidly (Fig. 1F).
Equilibrium Binding in HKRK-B7 Cell Membranes
The above dissociation data suggested that the two agonist radioligands, 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1– 15) form complexes with the PTHR that have distinct sensitivities to GTPγS. We further explored this possibility by comparing the effects of GTPγS, at varying concentrations, on the binding levels attained by the two radioligands in reactions performed in HKRK-B7 cell membranes under approximate equilibrium conditions (90-min incubations). As shown in Fig. 2 and Table 1, at the maximum concentration of 1 × 10−5 m, GTPγS only partially (22 ± 3%) inhibited the binding of 125I-PTH(1–34), whereas it nearly fully (88 ± 1%) inhibited the binding of 125I-[Aib1,3,M]PTH(1–15). These data are consistent with the above dissociation data because they confirm that the binding of 125I-[Aib1,3,M]PTH(1–15) to the PTHR is highly sensitive to GTPγS, whereas the binding of 125I-PTH(1–34) is not.
Fig. 2.
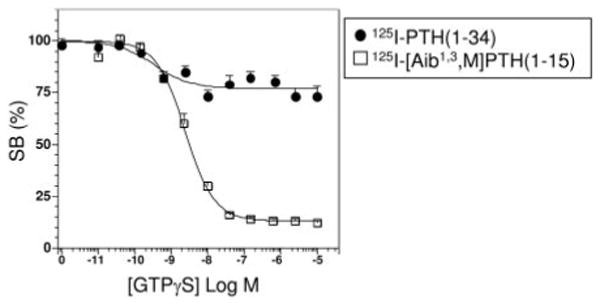
Effect of GTPγS on PTH Radioligand Binding to HKRK-B7 Cell Membranes
The effects of GTPγS on the binding of 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15) to the PTHR in HKRK-B7 cell membranes were assessed in near-equilibrium reactions (90-min incubations). Shown are data (mean ± sem) from six experiments, each performed in duplicate. Data are expressed as a percent of the total radioactivity specifically bound in the absence of GTPγS (SB). Values of total bound (specific plus nonspecific), nonspecifically bound (determined in wells containing 1 × 10−6 m unlabeled PTH(1–34) ligand), and total added radioactivity, were 6,487 ± 790 cpm, 2,387 ± 409 cpm, and 21,761 ± 1,394 cpm, respectively, for 125I-PTH(1–34) and 4,350 ± 521 cpm, 735 ± 147 cpm, and 34,123 ± 2,205 cpm, respectively, for 125I-[Aib1,3,M]PTH(1–15). Membrane protein concentrations were 10 ng/μl for 125I-PTH(1–34) and 100 ng/μl for 125I-[Aib1,3,M]PTH(1–15).
Table 1. Competition Binding in HKRK-B7 Cell Membranes.
| 125I-PTH(1–34) [IC50 (nm)] | 125I-[Aib1,3,M]PTH(1–15) [IC50 (nm)] | |
|---|---|---|
| PTH(1–34) | 0.81 ± 0 | 0.82 ± 0.17 |
| n | 9 | 9 |
| [Aib1,3,M]PTH(1–15) | 284 ± 61 | 0.92 ± 0.17 |
| n | 9 | 9 |
| GTPγS | 1.2 ± 0.4 (22%) | 2.9 ± 0.7 (88%) |
| n | 6 | 6 |
Competition binding assays were performed in membranes prepared from HKRK-B7 cells using either 125I-PTH(1–34) or 125I-[Aib1,3,M]PTH(1–15) as radioligand. Values in parentheses (%) indicate the maximum level of inhibition attained by GTPγS. Values are means (±sem) of data from the number of experiments (n), each performed in duplicate.
We further compared the binding of PTH(1–34) and [Aib1,3,M]PTH(1–15) ligands to the PTHR by performing competition studies using 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15) as tracer radioligands and varying concentrations of unlabeled PTH(1–34) and [Aib1,3, M]PTH(1–15) as competitors. As shown in Fig. 3A, unlabeled [Aib1,3,M]PTH(1–15) was 350-fold weaker than PTH(1–34) in inhibiting the binding of 125I-PTH(1– 34) to the PTHR in HKRK-B7 cell membranes (IC50s = 284 ± 61 nM and 0.81 ± 0.08 nM, respectively, P = 0.0008, Table 1). In contrast, [Aib1,3,M]PTH(1–15) was as potent as PTH(1–34) in inhibiting the binding of 125I-[Aib1,3,M]PTH(1–15) tracer radioligand to these membranes (IC50s = 0.92 ± 0.17 nm and 0.82 ± 0.17 nm, respectively, P = 0.3; Fig. 3B and Table 1). The weak capacity of [Aib1,3,M]PTH(1–15) to inhibit the binding of 125I-[Aib1,3,M]PTH(1–34) to the PTHR is consistent with the hypothesis that the PTH(1–15) analog binds solely to the J domain of the PTHR, and that PTH(1–34) binds to both the N and J domains of the receptor, as outlined above, and examined further below.
Fig. 3.
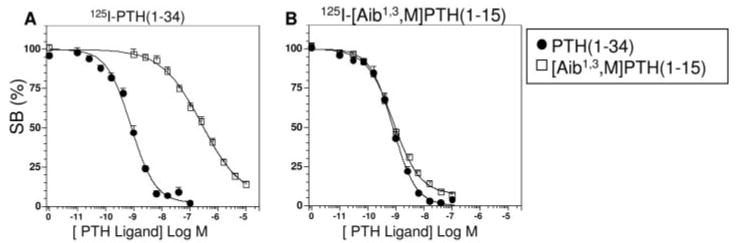
Competition Binding in HKRK-B7 Cell Membranes
Competition binding experiments were performed using membranes prepared from HKRK-B7 cells, either 125I-PTH(1–34) (A) or 125I-[Aib1,3,M]PTH(1–15) (B) as tracer radioligands, and unlabeled PTH(1–34) (filled circles) or [Aib1,3,M]PTH(1–15) (open squares) as competitor ligands. Each graph shows data (means ± sem) from nine experiments, each performed in duplicate. Data are expressed as a percent of the total radioactivity specifically bound (SB) in the absence of unlabeled ligand. In the experiments of panel A, the values of total binding (specific plus nonspecific), nonspecific binding, and total radioactivity added were 4,996 ± 628 cpm, 1,663 ± 271 cpm and 26,729 ± 1,995 cpm, respectively. In panel B, the corresponding values were 3,409 ± 416 cpm, 593 ± 108 cpm and 28,411 ± 2,625 cpm, respectively. Reactions contained a membrane protein concentration of 10 ng/μl (A) or 100 ng/μl (B).
Binding to an Amino-Terminally Truncated PTHR in COS-7 Cell Membranes
To analyze further the structural domains of the receptor involved in determining the binding affinity for PTH(1–34) and [Aib1,3,M]PTH(1–15), we used a PTHR construct, PTHR-delNt, that lacks most of the receptor's N domain (27). In prior studies performed in intact COS-7 cells expressing PTHR-delNt, we found that 125I-[Aib1,3,M]PTH(1–15) bound nearly as well as it did to the wild-type PTHR, and, as expected, 125I-PTH(1–34) failed to bind (reflecting the importance of the N domain of the PTHR in initiating high-affinity binding of this ligand) (28) (and data not shown). In our initial experiments performed with membranes prepared from COS-7 cells transfected with PTHR-delNt, however, we observed little or no binding of 125I-[Aib1,3,M]PTH(1–15) (as well as no binding of 125I-PTH(1–34) radioligand). In an effort to increase binding of 125I-[Aib1,3,M]PTH(1–15) to PTHR-delNt in membranes, we cotransfected the cells with a mutant GαS protein, GαS(α3/β5) (30) that has been shown to increase the maximum binding capacity of the PTHR, as assessed with a 125I-PTHrP(1–36) radioligand analog (12). This effect of the mutant GαS is presumably due to its capacity to couple to the receptor, and to thus stabilize a high-affinity receptor state, more efficiently (without increasing basal signaling) than does wild-type GαS (30). This strategy indeed increased binding of 125I-[Aib1,3,M]PTH(1–15) to PTHR-delNt in COS-7 cell membranes and resulted in maximum binding levels that were approximately 3-fold above those observed in membranes prepared from mock-transfected COS-7 cells without, as expected, resulting in detectable 125I-PTH(1–34) binding].
In competition assays performed with these PTHR-delNt/GαS(α3/β5)-expressing membranes, the binding of 125I-[Aib1,3,M]PTH(1–15) was potently inhibited by unlabeled [Aib1,3,M]PTH(1–15) and only weakly inhibited by unlabeled PTH(1–34) (IC50s = 0.62 ± 0.08 nm and 350 ± 40 nm, respectively; P = 0.007, Fig. 4A and Table 2). These data, when considered together with the competition data obtained with the wild-type PTHR (Fig. 3), confirm the hypothesis that [Aib1,3,M]PTH(1–15) binds predominantly, if not exclusively, to the PTHR J domain. They also verify that unmodified PTH(1–34) depends strongly on interactions to the N domain of the receptor to achieve its high binding affinity for the intact PTHR.
Fig. 4.
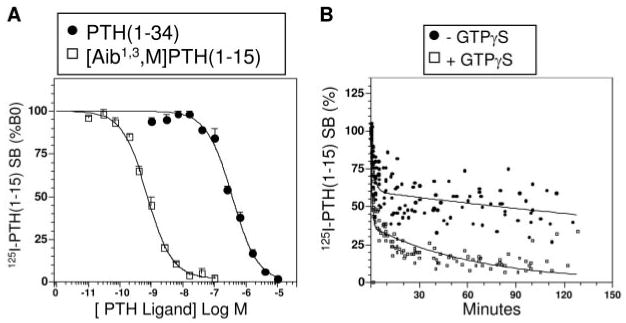
Ligand Binding to an Amino-Terminally Truncated PTHR in COS-7 Cell Membranes
Membranes were prepared from COS-7 cells transiently cotransfected with the amino-terminally truncated PTHR, PTHR-delNt, and, to increase binding capacity, with GαS-α3β5 (30), and used in competition binding (A) and time course dissociation assays (B). Panel A shows the capacity of unlabeled PTH(1–34) and [Aib1,3,M]PTH(1–15) to inhibit the binding of 125I-[Aib1,3,M]PTH(1–15) to the membranes. The data (means ± sem) are from three representative experiments each performed in duplicate. A summary of these and all related competition data obtained in COS-7 cells is reported in Table 2. Data are expressed as a percent of the total radioactivity specifically bound (SB) in the absence of unlabeled ligand (B0). Panel B shows the time courses of dissociation of 125I-[Aib1,3,M]PTH(1–15)·PTHR-delNt complexes in the absence (filled circles) and presence of GTPγS (5 × 10−5 m, open squares). Data are expressed as a percent of the maximal specific binding observed at t = 0. Shown are aggregate data from nine experiments. In the experiments of panel A, values of total bound (specific plus nonspecific), nonspecifically bound and total added radioactivity were 2,421 ± 465 cpm, 460 ± 101 cpm and 21,311 ± 1,006 cpm, respectively. In panel B, the values of maximal total bound radioactivity at t = 0, nonspecific binding, and total radioactivity added were 2,602 ± 486 cpm; 166 ± 24 cpm, and 26,382 ± 2,456 cpm, respectively. Reactions contained a membrane protein concentration of 100 ng/μl.
Table 2. Competition Binding in COS-7 Cell Membranes.
| PTHR-WT [IC50 (nm)] | PTHR-delNt [IC50 (nm)] | PTHR-H223R [IC50 (nm)] | |
|---|---|---|---|
| 125I-PTH(1–34) | |||
| PTH(1–34) | 3.5 ±1.0 | N.D. | 2.8 ± 1.1 |
| n | 10 | 4 | |
| [Aib1,3,M]PTH(1–15) | 300 ±140 | N.D. | N.D. |
| n | 4 | ||
| GTPγS | 15,000 ± 7,000 (33 ± 3) | N.D. | 3,700 ± 600 (17 ± 3) |
| n | 6 | 6 | |
| 125I-[Aib1,3,M]PTH(1–15) | |||
| PTH(1–34) | 0.091 ± 0.038 | 350 ± 40 | N.D. |
| n | 3 | 3 | |
| [Aib1,3,M]PTH(1–15) | 0.13 ± 0.02 | 0.62 ± 0.08 | 2.0 ± 0.6 |
| n | 9 | 6 | 4 |
| GTPγS | 1,300 ± 200 (85 ± 2) | 6,700 ± 2,600 (88 ± 1) | 11,000 ± 8,000 (21 ± 2) |
| n | 6 | 6 | 6 |
Competition binding assays were performed in membranes prepared from COS-7 cells transiently transfected with the indicated PTH receptor and either 125I-PTH(1–34) or 125I-[Aib1,3,M]PTH(1–15) as radioligand. PTHR-delNt was cotransfected with GαS(α3/β5) to increase maximum binding of 125I-[Aib1,3,M]PTH(1–15). Values in parentheses (%) indicate the maximum level of inhibition attained by GTPγS. Values are means (±sem) of data derived from the number of experiments (n), each performed in duplicate. N.D., Not done; PTHR-WT, wild-type PTHR.
We then assessed whether or not the absence of the receptor's N domain affected the stability and GTPγS sensitivity of the complexes formed between the receptor (PTHR-delNt) and 125I-[Aib1,3,M]PTH(1–15). As shown in Fig. 4B, in the absence of GTPγS, 40% of the 125I-[Aib1,3,M]PTH(1–15)·PTHR-delNt complexes dissociated rapidly (t1/2 = ∼1 min), and the remaining 60% dissociated slowly (t1/2 = ∼300 min). The addition of GTPγS increased the fraction of rapidly dissociating complexes to 63%, and caused a modest increase in the dissociation rate for the remaining complexes (t1/2 = ∼45 min). Removal of the receptor's N domain therefore does not prevent the formation of stable complexes with the 125I-[Aib1,3,M]PTH(1–15) radioligand, nor the sensitivity of these complexes to GTPγS.
Binding to a Constitutively Active Mutant PTHR
The results so far suggested that 125I-[Aib1,3,M]PTH(1– 15) binds selectively to a conformation of the PTHR J domain that is stabilized by the interaction of the receptor with a heterotrimeric G protein. Because heterotrimeric G proteins are thought to interact more efficiently with active-state receptor conformations than with inactive-state receptors (31), the results led us to further hypothesize that 125I-[Aib1,3,M]PTH(1–15) binds preferentially to an active-state PTHR conformation. To explore this, we used the constitutively active mutant PTHR, PTHR-H223R (32), which, like constitutively active GPCRs in general (31, 33), is thought to have a higher propensity to adopt an active-state conformation than does the wild-type receptor. We thus predicted that 125I-[Aib1,3,M]PTH(1–15) would bind to PTHR-H223R in a more stable, and less GTPγS-sensitive fashion than it does to the wild-type PTHR. Membranes were prepared from COS-7 cells transiently transfected with the wild-type PTHR or with PTHR-H223R and analyzed for binding 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15). In homologous competition binding assays, PTHR-H223R exhibited the same apparent binding affinity for PTH(1–34) as did the wild-type PTHR (2.8 ± 1.1 nm and 3.5 ±1.0 nm, respectively, P = 0.3; Table 2). Unexpectedly, PTHR-H223R exhibited an approximate 15-fold lower apparent affinity for [Aib1,3,M]PTH(1–15) than did the PTHR (2.0 ± 0.6 nm and 0.13 ± 0.02 nm, respectively, P = 0.03; Table 2). The reason for this lower apparent binding affinity for [Aib1,3,M]PTH(1–15) on PTHR-H223R is not clear at present. In any case, the maximum specific binding levels attained by both 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15) radioligands on PTHR-H223R were sufficient to permit analysis of the effects of GTPγS on this binding and the stability of the resultant ligand-receptor complexes.
As shown in Fig. 5, the binding of each radioligand to PTHR-H223R was indeed less sensitive to GTPγS than was its binding to the wild-type PTHR. Thus, GTPγS maximally (1 × 10−4 m) inhibited the binding of 125I-PTH(1–34) to the wild-type PTHR by 33 ± 3% and to PTHR-H223R by 17 ± 3% (P = 0.0007; Fig. 5A). Likewise, GTPγS maximally inhibited the binding of 125I-[Aib1,3,M]PTH(1–15) to the wild-type PTHR by 85±2% and to PTHR-H223R by 21 ± 2% (P = < 0.0001; Fig. 5B). As with the wild-type PTHR, the binding of 125I-[Aib1,3,M]PTH(1–15) to PTHR-delNt was nearly fully (88 ± 1%) inhibited by GTPγS (Fig. 5B).
Fig. 5.
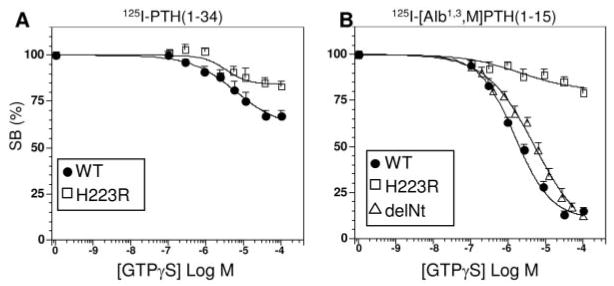
Capacity of GTPγS to Inhibit PTH Radioligand Binding to Wild-Type and Mutant PTHRs in COS-7 Cell Membranes
Shown are the effects of varying concentrations of GTPγS on the binding of 125I-PTH(1–34) (A) and 125I-[Aib1,3,M]PTH(1–15) (B) to membranes prepared from COS-7 cells transiently transfected with the wild-type PTHR (filled circles), the constitutively active mutant, PTHR-H223R (open squares) or PTHR-delNt (cotransfected with GαS -α3β5; open triangles, panel B only). Data (means ± sem) are from six experiments, each performed in duplicate, and are expressed as a percent of the total radioactivity specifically bound in the absence of GTPγS (SB). In panel A, the total amount of 125I-PTH(1–34) radioactivity bound (specific plus nonspecific) to the wild-type PTHR was 4,499 ± 828 cpm and that to PTHR-H223R was 4,361 ± 833 cpm; nonspecific binding was 574 ± 106 cpm and total radioactivity added was 23,651 ± 1,649 cpm. In panel B, the total amount of 125I-[Aib1,3,M]PTH(1–15) radioactivity bound to the wild-type PTHR was 3,994 ± 1,460 cpm, that to PTHR-H223R was 3,158 ± 1,063 cpm, and that to PTHR-delNt was 1,998 ± 264 cpm; nonspecific binding was 430 ± 51 cpm and total radioactivity added was 25,942 ± 2,185 cpm. Reactions contained a membrane protein concentration of 100 ng/μl.
In time course dissociation assays performed in the absence of GTPγS, the fraction of stable complexes (t1/2 > 5 min) formed by either radioligand with PTHR-H223R was approximately 30% higher than the corresponding fraction observed with the wild-type PTHR: 92% vs. 66% for 125I-PTH(1–34) (Fig. 6, A and B, filled symbols), and 84% vs. 51% for 125I-[Aib1,3,M]PTH(1–15) (Figs. 6, C and D, filled symbols). The addition of GTPγS caused little or no change in the fraction of stable complexes formed by either radioligand and PTHR-H223R (Fig. 6 B and D), whereas it decreased the fraction of stable complexes formed by 125I-PTH(1–34) and the wild-type PTHR by approximately 10%, and those formed by 125I-[Aib1,3,M]PTH(1–15) and the wild-type PTHR by approximately 30% (Fig. 6, A and C). The overall data obtained with PTHR-H223R are thus consistent with the hypothesis that 125I-[Aib1,3,M]PTH(1–15) binds preferentially to active-state conformations of the PTHR (which can be stabilized by the activating receptor mutation, His223→Arg, and/or coupling of the receptor to a heterotrimeric G protein).
Fig. 6.
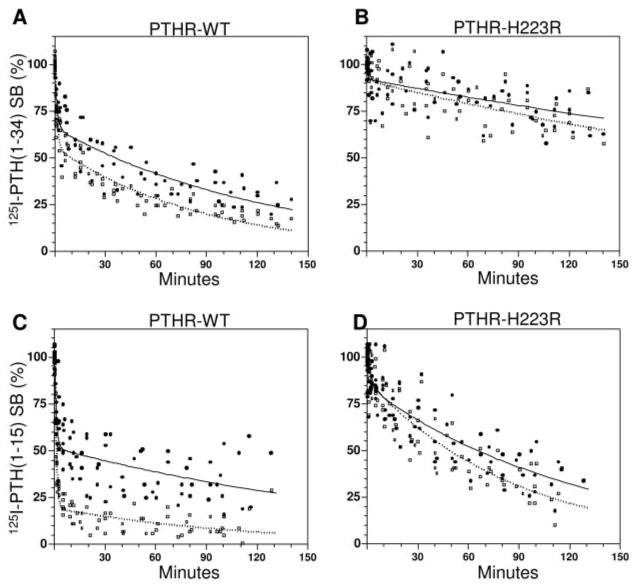
Dissociation of Radioligands from the Wild-Type PTHR and a Constitutively Active Mutant PTHR in COS Cell Membranes
Membranes were prepared from COS-7 cells transiently transfected with either the wild-type PTHR (PTHR-WT) (A and C) or with PTHR-H223R (B and D) and, after a 90-min ligand-receptor complex formation phase, the dissociation of complexes formed with either 125I-PTH(1–34) (A and B) or 125I-[Aib1,3,M]PTH(1–15) (C and D) was assessed either in the absence (filled circles) or presence of GTPγS (5 × 10−5 m final concentration, open squares, dashed lines). Shown are aggregate data, expressed as a percent of the maximal specific binding observed at t = 0 (SB), from six (A and B) or eight (C and D) experiments. Values of maximal total binding (specific plus nonspecific) at t = 0 for 125I-PTH(1−34) on the PTHR, and on PTHR-H223R, were 7,742 ± 1,867 cpm and 6,073 ± 787 cpm, respectively; the corresponding values for 125I-[Aib1,3,M]PTH(1−15) were 3,857 ± 688 cpm and 2,955 ± 471 cpm, respectively. Values of nonspecific binding and total radioactivity were 728 ±182 cpm and 37,024 ± 10,423 cpm, respectively, for 125I-PTH(1−34), and 223 ± 44 cpm and 27,805 ± 1,744 cpm, respectively, for 125I-[Aib1,3,M]PTH(1−15). Reactions contained a membrane protein concentration of 100 ng/μl.
Binding to Membranes Lacking GαS
The adenylyl cyclase/cAMP pathway is generally considered to be the principal signaling cascade to mediate the biological actions of the PTHR in vivo. Our current binding data obtained using cell membrane preparations are largely consistent with such a linkage for [Aib1,3,M]PTH(1–15), in that they demonstrate GTPγS sensitivity. They also suggest, however, that GαS-containing G protein heterotrimers are not essential for high-affinity binding of PTH(1–34) because GTPγS insensitivity was observed for this radioligand. We thus sought to specifically investigate the role that this Gα-subunit plays in modulating the receptor's affinity for PTH ligands. To do this, we used membranes prepared from a line of mouse embryonic fibroblast cells (34) that lack functional GαS due to homozygous disruption of the Gnas gene. These cells do not express detectable levels of the PTHR (34) and so were infected with an adenovirus vector encoding the PTHR to augment receptor expression. Parallel sets of cells were infected either with or without a second adenovirus vector encoding GαS, to thus obtain GαS+ and GαS− cells, each expressing the PTHR. The binding of 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15) to membranes prepared from these cells was then assessed over a range of membrane protein concentrations. Figure 7A shows that 125I-PTH(1–34) bound nearly as well to membranes derived from the GαS− cells as it did to those derived from the GαS+ cells. In contrast, Fig. 7B shows that 125I-[Aib1,3,M]PTH(1–15) bound only to membranes prepared from the GαS+ cells. These results demonstrate that GαS plays a major role in determining the PTHR's capacity to bind 125I-[Aib1,3,M]PTH(1–15) but is not required for binding 125I-PTH(1–34).
Fig. 7.
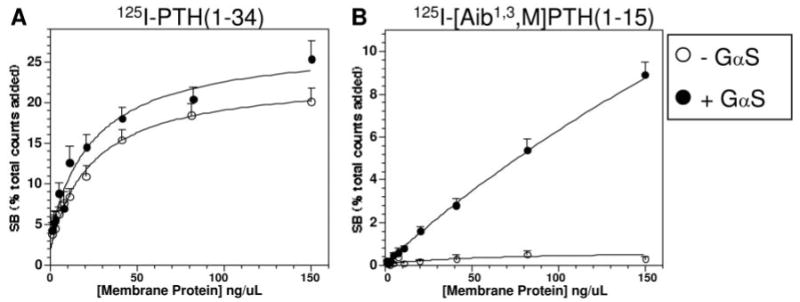
Radioligand Binding to the PTHR in Membranes from GαS− Cells
Membranes were prepared from mouse embryonic fibroblast cells that lack GαS due to homozygous disruption of Gnas and heterologously express via adenovirus-mediated transduction either the PTHR alone (open circles) or the PTHR and functional GαS (filled circles), and then tested at varying membrane protein concentrations for the capacity to bind 125I-PTH(1–34) (A) or 125I-[Aib1,3,M]PTH(1–15) (B). Reaction incubations were performed for 90 min. The amount of specifically bound radioactivity observed (SB) for each radioligand is expressed as a percent of the total amount of radioactivity added to the well. Shown are data (means ± sem) from seven experiments, each performed in duplicate. The mean values of total binding (specific plus nonspecific) at the highest concentration of protein tested for the membranes derived from the GαS− and GαS+ cells were 7,450 ± 1,166 cpm and 9,196 ± 1,003 cpm, respectively, for 125I-PTH(1–34), and 577 ±105 cpm and 5,177 ± 491 cpm, respectively, for 125I-[Aib1,3,M]PTH(1–15). The corresponding values of nonspecific binding (averaged from reactions performed at each protein concentration in the presence of 1 × 10−6 m unlabeled ligand), and total radioactivity added were 1,155 ± 245 cpm and 27,343 ± 5,541 cpm, respectively, for 125I-PTH(1–34), and 347 ± 46 cpm and 44,777 ± 5,887 cpm for 125I-[Aib1,3,M]PTH(1–15).
Discussion
In this study, we employed structurally distinct PTH radioligand analogs and membranes prepared from cells expressing altered forms of the PTHR, or altered with respect to their G protein expression status, to further dissect the mechanisms of ligand binding to the PTHR. Our main findings were that: 1) 125I-PTH(1– 34) and the modified amino-terminal agonist analog, 125I-[Aib1,3,M]PTH(1–15), dissociated from the PTHR with complex kinetics exhibiting rapid and slow components, whereas the antagonist, 125I-PTHrP(5–36), dissociated with primarily rapid kinetics; 2) GTPγS strongly destabilized 125I-[Aib1,3,M]PTH(1–15)·PTHR complexes but only mildly destabilized 125I-PTH(1– 34)·PTHR complexes; 3) removal of the receptor's amino-terminal domain strongly diminished the binding affinity of PTH(1–34) but only mildly affected the binding affinity of [Aib1,3,M]PTH(1–15); 4) both 125I-[Aib1,3,M]PTH(1–15) and 125I-PTH(1–34) formed more stable and less GTPγS-sensitive complexes with the constitutively active receptor, PTHR-H223R, than they did with the wild-type PTHR; and 5) for the wild-type PTHR, GαS was required for binding 125I-[Aib1,3,M]-PTH(1–15) but not for binding 125I-PTH(1–34). The data from these studies add to those acquired in prior binding studies performed on the PTHR using the membrane assay approach (12,35,36), as well as to those acquired in whole cells using the photo-affinity cross-linking (9,10, 37) and receptor mutational approaches (7), which together have provided much of the groundwork for current models of the PTH/PTHR interaction mechanism (14). Many areas of uncertainty remain in understanding this mechanism, including the precise roles that the various domains of the ligand and receptor play in the formation, stability and conformational dynamics of the ligand-receptor complex, and the role that G protein coupling plays in the ligand-binding process, particularly in terms of its effects on interactions that occur to the N and J domains of the receptor. The present studies were designed to address some of these issues.
One unexpected observation made in our studies was the pronounced difference in GTPγS sensitivities of the binding of 125I-PTH(1–34) to the PTHR, vs. the binding of 125I-[Aib1,3,M]PTH(1–15). This difference suggests that the PTHR can form at least two types of receptor conformations that display high affinity for agonist ligands. One type, revealed by the binding analyses of PTH(1–34), apparently does not require coupling to a heterotrimeric G protein for the formation of stable complexes with PTH(1–34) ligands. Another type, revealed with 125I-[Aib1,3,M]PTH(1–15), is dependent on coupling to a G protein heterotrimer, specifically one containing GαS. We also found that the constitutively active PTHR, PTHR-H223R, binds both [Aib1,3,M]PTH(1–15) and PTH(1–34) in a GTPγS-insensitive fashion (Figs. 5 and 6); this finding suggests yet another type of high-affinity receptor conformation that occurs in the presence of the His223→Arg mutation. These findings overall support the view that GPCRs in general (31, 38), and the PTHR in particular (39–41) can adopt at least several different conformations and thereby exhibit an ensemble of ligand-binding and/or signaling functionalities. In this regard, the slower equilibration times seen in our association time course studies for the agonists, 125I-PTH(1–34) and 125I-[Aib1,3,M]PTH(1–15), vs. the faster equilibration time seen for the antagonist 125I-PTHrP(5–36) (Fig. 1, A–C), could potentially reflect rate-limiting conformational changes involved in the binding of the agonists that are absent in the binding of the antagonist (42).
Our data also provide information on the structural domains of the ligand and receptor that are likely to play a role in the formation and/or stabilization of distinct conformational states of the PTHR. The data showing high-affinity, GTPγS-sensitive binding of [Aib1,3,M]PTH(1–15) to PTHR-delNt (Figs. 4, A and B, and 5B) indicate that neither the (16–34) portion of the ligand, nor the N-domain of the receptor is required for the formation of the high-affinity receptor conformation that is bound by 125I-[Aib1,3,M]PTH(1–15), nor for the stabilization of this conformation by G protein. Both the (16–34) portion of the ligand and the N domain of the receptor are, however, required for the formation/stabilization of the high-affinity receptor conformation that is bound by unmodified PTH(1–34) in the apparent absence of coupling to a G protein heterotrimer. This is shown by the failure of 125I-[Aib1,3,M]PTH(1–15) to form a stable complex with the PTHR in the presence of GTPγS (Fig. 1E), and by the failure of 125I-PTH(1–34) radioligand to bind detectably to PTHR-delNt. The importance of the PTHR N domain in determining the overall affinity with which PTH(1–34) binds to the PTHR is further shown by our competition studies performed in COS-7 cell membrane using 125I-[Aib1,3,M]PTH(1–15) as a radioligand. In these studies, the potency with which PTH(1–34) inhibited the binding of 125I-[Aib1,3,M]PTH(1–15) to PTHR-delNt was 4000-fold weaker than the potency with which it inhibited the binding of this radioligand to the intact PTHR (350 ± 40 nM vs. 0.091 ± 0.038 nM; P = 0.007; Table 2). Related to these observations, in binding studies performed with the intact PTHR in HKRK-B7 cell membranes, we observed that unlabeled [Aib1,3,M]PTH(1–15), at a concentration of 1 × 10−5 m, nearly fully inhibited the binding of 125I-PTH(1–34) radioligand (Fig. 3A). Given that [Aib1,3,M]PTH(1–15) occupies only the J domain binding site of the receptor, this finding suggests that interactions between 125I-PTH(1–34) and the PTHR N domain are not, by themselves, strong enough to enable high-affinity binding, or detection of simultaneous binding of 125I-PTH(1–34) and [Aib1,3,M]PTH(1–15) to the receptor, via binding to the N and J domain sites, respectively (12), at least under the reaction conditions used here.
One of the more interesting inferences that may be taken from this work is that 125I-[Aib1,3,M]PTH(1–15) is a selective probe of an active-state conformation of the PTHR J domain that is stabilized by coupling of the receptor to a heterotrimeric G protein, specifically one containing GαS. The data supporting this conclusion include the nearly complete disruption of both 125I-[Aib1,3,M]PTH(1–15)·PTHR and 125I-[Aib1,3,M]PTH(1– 15)·PTHR-delNt complexes by GTPγS (Figs. 1E, 4B, and 5B); the GTPγS-insensitive binding of 125I-[Aib1,3,M]PTH(1–15) to the constitutively active receptor, PTHR-H223R (Figs. 5B and 6D); the failure of 125I-[Aib1,3,M]PTH(1–15) to bind to membranes prepared from cells lacking GαS (Fig. 7A), and the restoration of this binding upon reintroduction of functional GαS (Fig. 7B). In contrast to these findings with 125I-[Aib1,3,M]PTH(1–15), the data with 125I-PTH(1–34), showing that as much as 75% of the complexes formed with the wild-type PTHR are stable in the presence of GTPγS (Figs. 1D and 5A), suggest that this intact radioligand can bind to a distinct high-affinity receptor conformation. In line with this interpretation, the GTPγS treatments used in our experiments appeared to adequately saturate the G proteins in the membrane preparations because they resulted in nearly full inhibition of the binding of 125I-[Aib1,3,M]PTH(1–15). A GTPγS-insensitive high-affinity receptor conformation was recently described for the related corticotropin-releasing factor receptor, and termed in this study R0 (43). Stable agonist ligand·receptor complexes in the presence of GTPγS have also been observed for several other class 2 GPCRs, including the calcitonin gene-related peptide receptor (44), the calcitonin receptor (45) and the glucagon receptor (46), each of which couples to GαS and binds a peptide ligand similar in size to PTH(1–34). The molecular nature of this putative GTPγS-insensitive, high-affinity ligand-receptor state (R0) is unclear, but it is intriguing to consider the possibility that the capacity for its formation is a general property of the class 2 GPCRs, and that it might play some role in the biological activities of these receptors, for example, by enabling catalytic G protein activation (47–49), or modulating receptor down-regulation/desensitization responses (45, 49). Such capacity might also provide a means to produce varied biological responses to structurally distinct ligands that act on the same receptor. In the case of the PTHR, such an effect could conceivably produce altered responses to PTH vs. PTHrP, and this might underlie the differing vitamin D-stimulating and bone-resorbing responses seen for PTH(1–34) and PTHrP(1–36) analogs in recent in vivo studies performed in humans (50, 51). The mechanisms involved here are still unclear.
In earlier studies performed in intact (52) or solubilized (53) membranes prepared from canine renal cortex, nonhydrolyzable GTP analogs were found to inhibit the binding of PTH(1–34) by approximately 75%; an extent considerably greater than that seen in our current studies performed in intact membranes prepared from cells transfected with the human PTHR (∼25%). Although we have not assessed binding to the canine receptor, a likely explanation for the difference in GTP analog sensitivity seen in these studies is the difference in the species of receptor used (canine vs. human). The basis for this interpretation is that we have performed studies in membranes prepared from ROS 17/2.8 cells, which endogenously express the rat PTHR at a moderate level (∼80,000 per cell), as well as in membranes prepared from COS-7 cells transfected to express the rat receptor at a much higher level (∼2,000,000 per cell), and have found (data not shown) that, in both cases, GTPγS inhibits the binding of 125I-PTH(1–34) by approximately 75%, an extent similar to that seen in the earlier canine receptor studies. These findings raise the point that structural factors, such as the species of receptor or type of ligand analog used, might influence the extent to which the R0 state may form. The canine studies are of further interest in that they show that at least some R0 state can be detected for the PTHR in the more native milieu of renal cortical membranes.
Overall, our findings are consistent with the notion that the interaction of the amino-terminal agonist pharmacophore of PTH with the juxtamembrane region of the PTHR plays a major role in inducing the conformational changes in the receptor that lead to G protein coupling, and that, reciprocally, coupling of the PTHR to a heterotrimeric G protein containing GαS stabilizes the interaction of the amino-terminal pharmacophore of the ligand with the J domain of the receptor. Our findings make clear that the N domain of the receptor is not required for the formation of a high-affinity, G protein-coupled, agonist-receptor complex, yet is important for the formation of high-affinity complexes with PTH(1–34), and may be particularly relevant in cells where the receptor exists largely in a G protein-uncoupled state, such as in cells transfected to overexpress the PTHR. Given that PTH(1–34) can bind stably to a GTPγS-insensitive state of the PTHR, the availability of a PTH ligand that binds preferentially to a G protein-coupled, active conformation of the PTHR, as appears to be the case for [Aib1,3,M]PTH(1–15), could facilitate efforts to develop a small-molecule agonist ligand for the PTHR. Such a reagent could serve as a powerful new probe of the PTH-receptor interaction mechanism and might eventually lead to new therapeutic approaches for PTH-related diseases, such as osteoporosis.
Materials and Methods
Peptides and Reagents
All peptides used have been described by us previously (26, 27,54) and were synthesized by the Protein and Peptide Core Facility at Massachusetts General Hospital (Boston, MA). The radioiodinated peptides, 125I-[Nle8,21,Tyr34]ratPTH(1–34)NH2, 125I-[Aib1,3,Nle8, Gln10,Har11,Ala12,Trp14,Tyr15]ratPTH(1–15)NH2 and 125I-[Ile5,Trp23,Tyr36]PTHrP(5–36)NH2, herein termed 125I-PTH(1–34), 125I-[Aib1,3,M]PTH(1–15) and 125I-PTHrP(5–36), respectively, were prepared using Na125I (specific activity: 2190 Ci/mmol; PerkinElmer/NEN Life Science Products, Boston, MA) and the oxidative chloramine-T procedure. Each radioligand was purified by reverse-phase HPLC to obtain an estimated specific activity of 2190 Cu/mmol.
Cell Culture
Cells were cultured in DMEM (Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 100 U/ml penicillin G, and 100 μg/ml streptomycin, and maintained at 37 C (or 33 C as noted below) in a humidified atmosphere containing 5% CO2. The cell line HKRK-B7 is a clonal derivative of the porcine kidney cell line, LLC-PK1; these cells stably express the recombinant human PTHR at a cell surface density of approximately 950,000 receptors per cell (29). HKRK-B7 cells were harvested for membrane preparations 2–3 d after the cell monolayers became confluent. GnasE2−/E2− cells, herein termed GαS− cells, are a line of mouse embryonic fibroblasts that lack GαS due to homozygous disruption of exon 2 of the Gnas gene (34). Because these GαS− cells do not express detectable levels of endogenous PTHRs (34), they were infected with an adenoviral transduction vector (Invitrogen Corp.; multiplicity of infection = 100) into which was inserted a cDNA encoding the wild-type human PTHR (virus construction details available upon request). Parallel sets of cells were coinfected with this PTHR vector and a second adenovirus vector (multiplicity of infection = 100 for each virus), into which was inserted a cDNA encoding functional rat GαS. The cells were cultured at 33 C in six-well plates (6-cm diameter wells) and harvested for membrane preparation 2 d after infection. COS-7 cells were transiently transfected in six-well plates using Fugene-6 (Roche Diagnostics, Indianapolis, IN) and CsCl-purified plasmid DNA (3 μl Fugene, 1 μg DNA, per well). Human PTHR-encoding plasmids (pCDNA1 vector; Invitrogen Corp.) contained either the wild-type PTHR (55), PTHR-delNt, in which the segment Tyr23 (the amino terminus of the mature PTHR) to Arg181 (∼10 amino acids amino-terminal of TM1) is replaced by an alanine (26), or the constitutively active mutant receptor, PTHR-H223R (32). To increase radioligand binding to PTHR-delNt, the COS-7 cells were cotransfected with a second plasmid (pcDNA1-Amp vector; Invitrogen Corp.) encoding the rat GαS mutant, GαS(α3/β5), in which five residues of the exposed α3/β5 loop are replaced by the corresponding residues of rat Gαi2 (N271K/K274D/R280K/T284D/I285T) (30). This mutant GαS has been shown to have increased capacity, relative to wild-type GαS, to stabilize receptor·G protein complexes (without increasing basal cAMP signaling), as shown for the β2-adrenergic receptor (30), and the PTHR (12). Cotransfections were performed using 1 μg of each DNA and 6 μl of Fugene-6 per well of a six-well plate.
Cell Membrane Preparation
Cell monolayers in T175 flasks (HKRK-B7 cells) or six-well plates (COS-7 and GαS− cells) were washed with hypoosmotic lysis buffer [10 mm Tris-HCl (pH 7.8), 4 mm EDTA] and collected using a Teflon policeman in the same buffer supplemented with a protease inhibitor cocktail (final concentrations: 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), 0.8 μm Aprotonin, 20 μM leupeptin, 40 μM Bestatin, 15 μm Pepstatin A, 14 μm E-64; Sigma-Aldrich Inc., St. Louis, MO). Three milliliters of this buffer were used for each T175 flask and 0.25 ml were used for each well of a six-well plate. The cells were then disrupted by passing 10 to 12 times through a steel ball-bearing/cylinder-based cell-shearing device (HGM Industries, Heidelberg, Germany) affixed at each end to a 10-ml syringe. The nuclei and cell fragments were removed by centrifugation at 800 × g for 10 min at 4 C, the membranes in the supernatant were collected by centrifugation at 14,000 × g for 30 min at 4 C, and the pellet was resuspended in membrane buffer [20 mM HEPES (pH 7.4), 0.1 m NaCl, 3 mM MgSO4, 20% glycerol, and the same protease inhibitor cocktail described above]. The membranes obtained from each T175 flask were resuspended in 1.0 ml of membrane buffer, and those from each well of a six-well plate were resuspended in 0.125 ml. The concentration of protein in the preparations was determined by the bicinchoninic acid protein detection system (Pierce, Rockford, IL) using BSA diluted in membrane buffer as a standard. Membrane aliquots (0.25 ml) were stored at −80 C and found to be stable at this temperature for at least 10 months.
Binding Assays
Binding reactions were performed at room temperature in assay buffer comprised of membrane buffer supplemented with BSA (3 mg/ml). Bound and free radioligand were separated by rapid vacuum filtration using 96-well vacuum filtration plates (Multiscreen-Durapore HV, low protein-binding, 0.65 μm membranes; Millipore Corp., Milford, MA) and a vacuum manifold. After rapid filtration of the sample and a single wash with 0.25 ml ice-cold assay buffer, the filters were air-dried, detached from the plate, and counted for γ radioactivity in a γ counter (Micromedic Model 600; Titertek Instruments, Huntsville, AL).
Binding Time Course Studies
Radioligand association and dissociation experiments were performed as bulk reactions in 15 ml round-bottom polystyrene snap-cap tubes (Falcon, Becton Dickinson, Franklin Lakes, NJ) in a total reaction volume of 3.0–5.0 ml. Reactions contained a total membrane protein concentration of 20–100 μg/ml, and a total radioactivity concentration of approximately 125,000 to 175,000 cpm/ml. Association reactions were initiated by the addition of membranes. At successive time-points thereafter, 0.2 ml aliquots (∼25,000–35,000 cpm) were withdrawn and immediately processed by vacuum filtration, as described above. Nonspecific binding was determined for each radioligand in parallel reaction tubes containing an excess of the corresponding unlabeled ligand (1 × 10−6 m). For dissociation reactions, membranes and radioligand were preincubated for 90 min to allow complex formation. The dissociation phase was then initiated by the addition of a saturating concentration of the unlabeled ligand (1 × 10−6 m final concentration) with or without GTPγS (Sigma-Aldrich Inc., St. Louis, MO) at a final concentration of 5 × 10−5 m. Immediately before this addition (t = 0), and at successive time points thereafter, 0.2 ml aliquots were withdrawn and rapidly processed by vacuum filtration, as described above. Nonspecific binding was determined for each radioligand in parallel reaction tubes containing an excess of the corresponding unlabeled ligand (1 × 10−6 m) in both the formation and dissociation phases of the reaction. In both the association and dissociation reactions, nonspecific binding was found to be radioligand-dependent and to not vary over the time course of the reactions, nor between membrane/receptor preparations; accordingly, a time-averaged value of nonspecific binding was calculated in each experiment for each radioligand from the aggregate of nonspecific binding values obtained at each time point and thus used to calculate specific binding for that radioligand at each time point. The resulting values of specifically bound radioactivity were expressed as a percent of the maximum radioactivity specifically bound for the respective radioligand (determined by the curve-fitting routine described below for the association reactions and from the observed binding at t = 0 for the dissociation experiments).
Competition Binding Studies
Peptide competition and GTPγS inhibition reactions were incubated directly in the wells of the 96-well vacuum filtration plates. Each well contained a total volume of 230 μl, a total membrane protein concentration of 10–100 μg/ml and a total amount of radioactivity of approximately 30,000 cpm. Reagents were added and mixed in the wells using an eightchannel repeating pipettor. Reactions were incubated for 90 min, at the end of which the plate was applied to the vacuum manifold, the samples were filtered, the filters were washed once with 0.25 ml ice cold assay buffer, air-dried, detached from the plate and counted for γ radioactivity. Nonspecific binding was determined for each radioligand in wells containing a saturating amount of unlabeled PTH ligand.
Data Calculations
Data were processed using least-squares, nonlinear regression analysis. Association time course data were analyzed using the mono-exponential equation: y = ymax·(1-e(−kobs·x)), where y is the radioactivity specifically bound, ymax is the maximum radioactivity specifically bound, x is time and kobs is the observed association rate constant. Dissociation time course data were analyzed using the biexponential decay equation: y = (span-1·e(−koff1·x))+(span-2·e(−koff2·x)), where span-1 and span-2 are the fractions of complexes with rapid (t1/2 ≤ 5 min) and slow (t1/2 > 5 min) dissociation rates, respectively; koff1 and koff2 are the corresponding dissociation rate constants (min−1); y is the specifically bound radioactivity and x is time. Values of t1/2 were calculated from the equation: t1/2 = 0.6932/k. Competition binding data were analyzed using the equation: y = ymin+ (ymax − ymin)/1 +(IC50/x)n, where y, ymin and ymax are the observed, calculated minimum and calculated maximum specific binding, respectively; x is inhibitor concentration (nm), IC50 is the concentration (nm) of inhibitor to achieve half of that inhibitor's maximal effect and n is the slope factor (range: −0.8 to −1.4). Paired data sets were statistically compared using the Student's t test (two-tailed) assuming unequal variances for the two sets.
Acknowledgments
We thank Catherine Berlot (Weis Center for Research, Danville, PA) for providing the GαS(α3/β5) expression plasmid.
This work was supported by Grant DK-11794 from the National Institutes of Health.
Abbreviations
- Aib
α-Amino-isobutyric acid
- G protein
heterotrimeric (αβγ) guanine nucleotide-binding protein
- GαS
stimulatory G protein α subunit that mediates activation of adenylyl cyclase
- GPCR
G protein-coupled receptor
- GTPγS
guanosine 5′-O-(3-thio)triphosphate
- Har
homoarginine
- J
juxtamembrane domain
- N
amino-terminal extracellular domain
- Nle
norleucine
- PTHR
PTH/PTHrP receptor. PTHR-delNt, a PTHR construct that has most (mature amino terminus to Arg181) of the amino-terminal domain replaced by an alanine
- PTHR-H223R
a constitutively active PTHR that contains the activating point mutation, His223→Arg
- TM
one of the seven helical transmembrane domains of a GPCR
Footnotes
Disclosure: T.D., A.L, M.J.M., M.B., and H.J. have nothing to declare. T.J.G. and J.T.P. are recipients of a research grant from Chugai Pharmaceutical Co. of Japan; the grant is for the study of PTH ligands.
References
- 1.Iida-Klein A, Guo J, Takemura M, Drake MT, Potts JT, Jr, Abou-Samra A, Bringhurst FR, Segre GV. Mutations in the second cytoplasmic loop of the rat parathyroid hormone (PTH)/PTH-related protein receptor result in selective loss of PTH-stimulated phospholipase C activity. J Biol Chem. 1997;272:6882–6889. doi: 10.1074/jbc.272.11.6882. [DOI] [PubMed] [Google Scholar]
- 2.Huang Z, Chen Y, Pratt S, Chen TH, Bambino T, Nissenson R, Shoback D. The N-terminal region of the third intracellular loop of the parathyroid hormone (PTH)/PTH-related peptide receptor is critical for coupling to cAMP and inositol phosphate/Ca2++ signal transduction pathways. J Biol Chem. 1996;271:33382–33389. doi: 10.1074/jbc.271.52.33382. [DOI] [PubMed] [Google Scholar]
- 3.Mahon M, Donowitz M, Yun C, Segre G. Na(+)/H(+) exchanger regulatory factor 2 directs parathyroid hormone 1 receptor signalling. Nature. 2002;417:858–861. doi: 10.1038/nature00816. [DOI] [PubMed] [Google Scholar]
- 4.Singh AT, Gilchrist A, Voyno-Yasenetskaya T, Radeff-Huang JM, Stern PH. Gα12/Gα13 subunits of heterotrimeric G proteins mediate parathyroid hormone activation of phospholipase D in UMR-106 osteoblastic cells. Endocrinology. 2005;146:2171–2175. doi: 10.1210/en.2004-1283. [DOI] [PubMed] [Google Scholar]
- 5.Juppner H, Schipani E, Bringhurst FR, McClure I, Keutmann HT, Potts JT, Jr, Kronenberg HM, Abou-Samra AB, Segre GV, Gardella T. The extracellular, amino-terminal region of the PTH/PTHrP receptor determines the binding affinity for carboxyl-terminal fragments of PTH(1–34) Endocrinology. 1994;134:879–884. doi: 10.1210/endo.134.2.8299582. [DOI] [PubMed] [Google Scholar]
- 6.Gardella TJ, Juppner H, Wilson AK, Keutmann HT, Abou-Samra AB, Segre GV, Bringhurst FR, Potts JTJ, Nussbaum SR, Kronenberg HM. Determinants of [Arg2]PTH-(1–34) binding and signaling in the transmembrane region of the parathyroid hormone receptor. Endocrinology. 1994;135:1186–1194. doi: 10.1210/endo.135.3.8070362. [DOI] [PubMed] [Google Scholar]
- 7.Bergwitz C, Gardella TJ, Flannery MR, Potts JTJ, Kronenberg HM, Goldring SR, Juppner H. Full activation of chimeric receptors by hybrids between parathyroid hormone and calcitonin: evidence for a common pattern of ligand-receptor interaction. J Biol Chem. 1996;271:26469–26472. doi: 10.1074/jbc.271.43.26469. [DOI] [PubMed] [Google Scholar]
- 8.Vilardaga J, Lin I, Nissenson R. Analysis of parathyroid hormone (PTH)/secretin receptor chimeras differentiates the role of functional domains in the PTH/PTH-related peptide (PTHrP) receptor on hormone binding and receptor activation. Mol Endocrinol. 2001;15:1186–1199. doi: 10.1210/mend.15.7.0665. [DOI] [PubMed] [Google Scholar]
- 9.Bisello A, Adams AE, Mierke D, Pellegrini M, Rosenblatt M, Suva L, Chorev M. Parathyroid hormone-receptor interactions identified directly by photocross-linking and molecular modeling studies. J Biol Chem. 1998;273:22498–22505. doi: 10.1074/jbc.273.35.22498. [DOI] [PubMed] [Google Scholar]
- 10.Behar V, Bisello A, Bitan G, Rosenblatt M, Chorev M. Photoaffinity cross-linking identifies differences in the interactions of an agonist and an antagonist with the parathyroid hormone/parathyroid hormone-related protein receptor. J Biol Chem. 2000;275:9–17. doi: 10.1074/jbc.275.1.9. [DOI] [PubMed] [Google Scholar]
- 11.Gensure R, Carter P, Petroni B, Juppner H, Gardella T. Identification of determinants of inverse agonism in a constitutively active parathyroid hormone/parathyroid hormone related peptide receptor by photoaffinity cross linking and mutational analysis. J Biol Chem. 2001;276:42692–42699. doi: 10.1074/jbc.M106215200. [DOI] [PubMed] [Google Scholar]
- 12.Hoare S, Gardella T, Usdin T. Evaluating the signal transduction mechanism of the parathyroid hormone 1 receptor: effect of receptor-G-protein interaction on the ligand binding mechanism and receptor conformation. J Biol Chem. 2001;276:7741–7753. doi: 10.1074/jbc.M009395200. [DOI] [PubMed] [Google Scholar]
- 13.Gardella TJ, Jüppner H. Molecular properties of the PTH/PTHrP receptor. Trends Endocrinol Metab. 2001;12:210–217. doi: 10.1016/s1043-2760(01)00409-x. [DOI] [PubMed] [Google Scholar]
- 14.Gensure RC, Gardella TJ, Juppner H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem Biophys Res Commun. 2005;328:666–678. doi: 10.1016/j.bbrc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 15.Dong M, Pinon DI, Cox RF, Miller LJ. Importance of the amino terminus in secretin family G protein-coupled receptors. Intrinsic photoaffinity labeling establishes initial docking constraints for the calcitonin receptor. J Biol Chem. 2004;279:1167–1175. doi: 10.1074/jbc.M305719200. [DOI] [PubMed] [Google Scholar]
- 16.Pham V, Wade JD, Purdue BW, Sexton PM. Spatial proximity between a photolabile residue in position 19 of salmon calcitonin and the amino terminus of the human calcitonin receptor. J Biol Chem. 2004;279:6720–6729. doi: 10.1074/jbc.M307214200. [DOI] [PubMed] [Google Scholar]
- 17.Dong M, Zang M, Pinon D, Li Z, Lybrand T, Miller L. Interaction among four residues distributed through the secretin pharmacophore and a focused region of the secretin receptor amino terminus. Mol Endocrinol. 2002;16:2490–2501. doi: 10.1210/me.2002-0111. [DOI] [PubMed] [Google Scholar]
- 18.Dong M, Li Z, Pinon DI, Lybrand TP, Miller LJ. Spatial approximation between the amino terminus of a peptide agonist and the top of the sixth transmembrane segment of the secretin receptor. J Biol Chem. 2004;279:2894–2903. doi: 10.1074/jbc.M310407200. [DOI] [PubMed] [Google Scholar]
- 19.Tan YV, Couvineau A, Van Rampelbergh J, Laburthe M. Photoaffinity labeling demonstrates physical contact between vasoactive intestinal peptide and the N-terminal ectodomain of the human VPAC1 receptor. J Biol Chem. 2003;278:36531–6536. doi: 10.1074/jbc.M304770200. [DOI] [PubMed] [Google Scholar]
- 20.Assil KI, Abou-Samra A. Sauvagine cross links to the second extracellular loop of the corticotropin releasing factor type 1 receptor. J Biol Chem. 2002;277:32558–32561. doi: 10.1074/jbc.M204964200. [DOI] [PubMed] [Google Scholar]
- 21.Hoare SR, Sullivan SK, Ling N, Crowe PD, Grigoriadis DE. Mechanism of corticotropin-releasing factor type I receptor regulation by nonpeptide antagonists. Mol Pharmacol. 2003;63:751–765. doi: 10.1124/mol.63.3.751. [DOI] [PubMed] [Google Scholar]
- 22.Hoare SR, Sullivan SK, Schwarz DA, Ling N, Vale WW, Crowe PD, Grigoriadis DE. Ligand affinity for amino-terminal and juxtamembrane domains of the corticotrophin releasing factor type I receptor: regulation by G-protein and nonpeptide antagonists. Biochemistry. 2004;43:3996–4011. doi: 10.1021/bi036110a. [DOI] [PubMed] [Google Scholar]
- 23.Luck M, Carter P, Gardella T. The (1–14) fragment of parathyroid hormone (PTH) activates intact and amino-terminally truncated PTH-1 receptors. Mol Endocrinol. 1999;13:670–680. doi: 10.1210/mend.13.5.0277. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu M, Carter P, Gardella T. Autoactivation of type 1 parathyroid hormone receptors containing a tethered ligand. J Biol Chem. 2000;275:19456–19460. doi: 10.1074/jbc.M001596200. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu M, Potts JJ, Gardella T. Minimization of parathyroid hormone: novel amino-terminal parathyroid hormone fragments with enhanced potency in activating the type-1 parathyroid hormone receptor. J Biol Chem. 2000;275:21836–21843. doi: 10.1074/jbc.M909861199. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu N, Guo J, Gardella T. Parathyroid hormone (1–14) and (1–11) analogs conformationally constrained by α aminoisobutyric acid mediate full agonist responses via the juxtamembrane region of the PTH 1 receptor. J Biol Chem. 2001;276:49003–49012. doi: 10.1074/jbc.M106827200. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu N, Dean T, Khatri A, Gardella T. Amino-terminal parathyroid Hormone fragment analogs containing α,-α-dialkyl amino acids at positions 1 and 3. J Bone Miner Res. 2004;19:2078–2086. doi: 10.1359/JBMR.040914. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu N, Dean T, Tsang JC, Khatri A, Potts JT, Jr, Gardella TJ. Novel parathyroid hormone (PTH) antagonists that bind to the juxtamembrane portion of the PTH/PTH-related protein receptor. J Biol Chem. 2005;280:1797–1807. doi: 10.1074/jbc.M408270200. [DOI] [PubMed] [Google Scholar]
- 29.Takasu H, Guo J, Bringhurst F. Dual signaling and ligand selectivity of the human PTH/PTHrP receptor. J Bone Miner Res. 1999;14:11–20. doi: 10.1359/jbmr.1999.14.1.11. [DOI] [PubMed] [Google Scholar]
- 30.Grishina G, Berlot CH. A surface-exposed region of G(sα) in which substitutions decrease receptor-mediated activation and increase receptor affinity. Mol Pharmacol. 2000;57:1081–1092. [PubMed] [Google Scholar]
- 31.Kenakin T. Principles: receptor theory in pharmacology. Trends Pharmacol Sci. 2004;25:186–192. doi: 10.1016/j.tips.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 32.Schipani E, Kruse K, Juppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- 33.Samama P, Cotecchia S, Costa T, Lefkowitz R. A Mutation-induced activated state of the β2-adrenergic receptor: extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- 34.Bastepe M, Gunes Y, Perez-Villamil B, Hunzelman J, Weinstein LS, Juppner H. Receptor-mediated adenylyl cyclase activation through XLαs, the extra-large variant of the stimulatory G protein α-subunit. Mol Endocrinol. 2002;16:1912–1919. doi: 10.1210/me.2002-0054. [DOI] [PubMed] [Google Scholar]
- 35.Hoare S, Usdin T. Quantitative cell membrane based radioligand binding assays for parathyroid hormone receptors. J Pharmacol Toxicol Methods. 1999;41:83–90. doi: 10.1016/s1056-8719(99)00024-6. [DOI] [PubMed] [Google Scholar]
- 36.Shimada M, Chen X, Cvrk T, Hilfiker H, Parfenova M, Segre GV. Purification and characterization of a receptor for human parathyroid hormone and parathyroid hormone related peptide. J Biol Chem. 2002;277:31774–31780. doi: 10.1074/jbc.M204166200. [DOI] [PubMed] [Google Scholar]
- 37.Gensure R, Gardella T, Juppner H. Multiple sites of contact between the carboxyl terminal binding domain of PTHrP (1–36) analogs and the amino terminal extracellular domain of the PTH/PTHrP receptor identified by photoaffinity cross linking. J Biol Chem. 2001;276:28650–28658. doi: 10.1074/jbc.M100717200. [DOI] [PubMed] [Google Scholar]
- 38.Maudsley S, Martin B, Luttrell LM. The origins of diversity and specificity in g protein-coupled receptor signaling. J Pharmacol Exp Ther. 2005;314:485–494. doi: 10.1124/jpet.105.083121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bisello A, Chorev M, Rosenblatt M, Monticelli L, Mierke DF, Ferrari SL. Selective ligand-induced stabilization of active and desensitized parathyroid hormone type 1 receptor conformations. J Biol Chem. 2002;277:38524–38530. doi: 10.1074/jbc.M202544200. [DOI] [PubMed] [Google Scholar]
- 40.Sneddon WB, Syme CA, Bisello A, Magyar CE, Rochdi MD, Parent JL, Weinman EJ, Abou-Samra AB, Friedman PA. Activation-independent parathyroid hormone receptor internalization is regulated by NHERF1 (EBP50) J Biol Chem. 2003;278:43787–43796. doi: 10.1074/jbc.M306019200. [DOI] [PubMed] [Google Scholar]
- 41.Vilardaga J, Frank M, Krasel C, Dees C, Nissenson R, Lohse M. Differential conformational requirements for activation of G proteins and regulatory proteins, arrestin and GRK in the G protein coupled receptor for parathyroid hormone (PTH)/PTH related protein. J Biol Chem. 2001;276:33435–33443. doi: 10.1074/jbc.M011495200. [DOI] [PubMed] [Google Scholar]
- 42.Teague SJ. Implications of protein flexibility for drug discovery. Nat Rev Drug Discov. 2003;2:527–541. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- 43.Hoare SR, Sullivan SK, Pahuja A, Ling N, Crowe PD, Grigoriadis DE. Conformational states of the corticotrophin releasing factor 1 (CRF1) receptor: detection, and pharmacological evaluation by peptide ligands. Peptides. 2003;24:1881–1897. doi: 10.1016/j.peptides.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Chatterjee TK, Fisher RA. Multiple affinity forms of the calcitonin gene-related peptide receptor in rat cerebellum. Mol Pharmacol. 1991;39:798–804. [PubMed] [Google Scholar]
- 45.Hilton JM, Dowton M, Houssami S, Sexton PM. Identification of key components in the irreversibility of salmon calcitonin binding to calcitonin receptors. J Endocrinol. 2000;166:213–226. doi: 10.1677/joe.0.1660213. [DOI] [PubMed] [Google Scholar]
- 46.Post SR, Miyazaki H, Tager HS. Identification of a Mg(2+)- and guanyl nucleotide-dependent glucagon receptor cycle by use of permeabilized canine hepatocytes. J Biol Chem. 1992;267:25776–25785. [PubMed] [Google Scholar]
- 47.Roberts DJ, Waelbroeck M. G protein activation by G protein coupled receptors: ternary complex formation or catalyzed reaction? Biochem Pharmacol. 2004;68:799–806. doi: 10.1016/j.bcp.2004.05.044. [DOI] [PubMed] [Google Scholar]
- 48.Rodbell M. The complex regulation of receptor-coupled G-proteins. Adv Enzyme Regul. 1997;37:427–435. doi: 10.1016/s0065-2571(96)00020-9. [DOI] [PubMed] [Google Scholar]
- 49.Michelangeli VP, Findlay DM, Moseley JM, Martin TJ. Mechanisms of calcitonin induction of prolonged activation of adenylate cyclase in human cancer cells. J Cyclic Nucleotide Protein Phosphor Res. 1983;9:129–141. [PubMed] [Google Scholar]
- 50.Horwitz MJ, Tedesco MB, Gundberg C, Garcia-Ocana A, Stewart AF. Short-term, high-dose parathyroid hormone-related protein as a skeletal anabolic agent for the treatment of postmenopausal osteoporosis. J Clin Endocrinol Metab. 2003;88:569–575. doi: 10.1210/jc.2002-021122. [DOI] [PubMed] [Google Scholar]
- 51.Horwitz MJ, Tedesco MB, Sereika SM, Garcia-Ocana A, Bisello A, Hollis BW, Gundberg C, Stewart AF. Safety and tolerability of subcutaneous PTHrP(1–36) in healthy human volunteers: a dose escalation study. Osteoporos Int. 2006;17:225–230. doi: 10.1007/s00198-005-1976-3. [DOI] [PubMed] [Google Scholar]
- 52.Teitelbaum AP, Nissenson RA, Arnaud CD. Coupling of the canine renal parathyroid hormone receptor to adenylate cyclase: modulation by guanyl nucleotides and N-ethylmaleimide. Endocrinology. 1982;111:1524–1533. doi: 10.1210/endo-111-5-1524. [DOI] [PubMed] [Google Scholar]
- 53.Nissenson RA, Mann E, Winer J, Teitelbaum AP, Arnaud CD. Solubilization of a guanine nucleotide-sensitive parathyroid hormone-receptor complex from canine renal cortex. Endocrinology. 1986;118:932–939. doi: 10.1210/endo-118-3-932. [DOI] [PubMed] [Google Scholar]
- 54.Carter P, Juppner H, Gardella T. Studies of the N-terminal region of a parathyroid hormone-related peptide(1–36) analog: receptor subtype-selective agonists, antagonists and photochemical crosslinking agents. Endocrinology. 1999;140:4972–4981. doi: 10.1210/endo.140.11.7102. [DOI] [PubMed] [Google Scholar]
- 55.Schipani E, Karga H, Karaplis AC, Potts JT, Jr, Kronenberg HM, Segre GV, Abou-Samra AB, Juppner H. Identical complementary deoxyribonucleic acids encode a human renal and bone parathyroid hormone (PTH)/PTH-related peptide receptor. Endocrinology. 1993;132:2157–2165. doi: 10.1210/endo.132.5.8386612. [DOI] [PubMed] [Google Scholar]