

Abstract
Thirty novel α- and β-D-2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleoside analogs were synthesized and evaluated for in vitro antiviral activity. Several α- and β-7-deazapurine nucleoside analogs exhibited modest anti-HCV activity and cytotoxicity. Four synthesized 7-deazapurine nucleoside phosphoramidate prodrugs (18–21) showed no anti-HCV activity, whereas the nucleoside triphosphates (22–24) demonstrated potent inhibitory effects against both wild-type and S282T mutant HCV polymerases. Cellular pharmacology studies in Huh-7 cells revealed that the 5′-triphosphates were not formed at significant levels from either the nucleoside or the phosphoramidate prodrugs, indicating that insufficient phosphorylation was responsible for the lack of anti-HCV activity. Evaluation of anti-HIV-1 activity revealed that an unusual α-form of 7-carbomethoxyvinyl substituted nucleoside (10) had good anti-HIV-1 activity (EC50 = 0.71 ± 0.25 μM; EC90 = 9.5 ± 3.3 μM) with no observed cytotoxicity up to 100 μM in four different cell lines.
Keywords: HCV, antiviral, 7-deazapurine, nucleoside, nucleotide, prodrug, Mitsunobu
Hepatitis C virus (HCV) is an important pathogen affecting nearly 170 million people worldwide.1 HCV infections become chronic in about 50% of cases,2 and about 20% of these chronic patients develop liver cirrhosis that can lead to hepatocellular carcinoma.3 Because current therapies such as interferon-alpha (IFN-α) and ribavirin carry limited efficacy and are associated with significant side-effects even when used with the newly approved HCV protease inhibitors Incivek and Victrelis, there is a need for more effective anti-HCV agents that can be used in IFN-α/ribavirin sparing regimens.4
Nucleosides that target HCV NS5B have demonstrated advantages of broader activity against various HCV genotypes and a higher barrier to development of resistant viruses when compared to known HCV protease inhibitors.5 A number of modified nucleoside analogs have been reported to have anti-HCV activity.6 Among the sugar-modifications, 2′-C-methyl featured nucleosides have shown the most promise, and several of these nucleosides and nucleoside monophosphate prodrugs are at various stages of clinical trials as anti-HCV agents.7 Although the base-modifications have not produced any clinically significant nucleosides, 7-deazapurine base-modified nucleosides have exhibited some usefulness with 2′-C-methyl-7-deazapurine nucleosides demonstrating the most potent anti-HCV activity in an HCV replicon.8 Based on the potent activity of some 2′-deoxy-2′-fluoro-2′-C-methyl nucleosides,9 we hypothesized that the combination of 2′-deoxy-2′-fluoro-2′-C-methyl ribosyl sugars and 7-deazapurine bases could produce potent anti-HCV nucleosides. Therefore, a series of novel 2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleoside analogs were designed and synthesized. To better understand the anti-HCV assay data, some phosphoramidate prodrugs and triphosphates of selected nucleosides were also prepared. These nucleosides and their phosphoramidate prodrugs were evaluated in a Huh-7 cell-based HCV replicon for anti-HCV activity and cytotoxicity. The compounds were also tested for anti-HIV-1 activity and cytotoxicity in three additional cell lines. In addition, some selected nucleosides were evaluated for anti-HBV activity. Herein we report the synthesis and biological evaluation of a series of 2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleoside analogs along with the phosphoramidate prodrugs and triphosphates of selected compounds.
The synthesis of 2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleosides was challenging due to either a lengthy synthesis or poor yields followed by a difficult separation based on using two different synthetic approaches. One approach is more linear in which the 2′-C-methyl and 2′-fluoro substituent are added to a 7-deazapurine ribonucleoside, and the other approach is more convergent where the completed moieties are brought together by a sugar-base condensation. Since the 2′-C-methyl-2′-fluoro sugar lactone 1 can be readily prepared using a recently published method,10 it appeared advantageous to choose the sugar-base condensation approach. We first attempted to utilize Vörbrügen sugar-base condensations under various conditions. Two sugars (1-O-acetyl-3,5-di-O-benzoyl-2-deoxy-2-fluoro-2-C-methylriboside and its 1-bromo analog) and two 7-deazapurine bases (6-chloro-7-deazapurine and 6-chloro-7-flouro-7-deazapurine) were used for the condensation, and the silylating agent was either N,O-bis(trimethylsilyl)acetamide (BSA) or hexamethyldisilazane (HMDS). The solvent was chosen from CH2Cl2, CHCl3, ClCH2CH2Cl, or CH3CN, in the presence or absence of a Lewis acid catalyst (trimethylsilyl trifluoromethanesulfonate (TMSOTf), SnCl4, or trimethylsilyl iodide). The reaction temperature ranged from room temperature to 70°C, and the reaction time varied from a few hours to several days. However, all these condensation conditions failed to generate detectable amounts of the desired product. Another reported method11 for condensation of 7-deazapurines was tested using 1-chloro-3,5-di-O-benzoyl-2-deoxy-2-fluoro-2-C-methylriboside with the catalysts of tris(2-(2-methoxyethoxy)ethyl)amine (TDA-1) and KOH, without success. The direct SN2 substitution using 1-bromo-3,5-di-O-benzoyl-2-deoxy-2-fluoro-2-C-methylriboside and 7-deazapurine under basic conditions (NaH) also did not work. Ultimately, Mitsunobu reaction conditions were explored, which resulted in successful couplings (Scheme 1). The required lactol 2 was prepared from lactone 1 by careful LiAl(t-BuO)3H reduction. Under relatively standard Mitsunobu conditions,12 the condensation of several different 7-substituted 6-chloro-7-deazapurines11b,13 with lactol 2 produced 6-chloro-7-deazapurine nucleosides 3. In the case that the 7-deazapurine contained an amino moiety, the amino group was protected with a pivaloyl group. The coupling products were obtained in about 40–60% yield, as an approximate 1:1 mixture of α and β isomers. Heating of 3 with NH4OH and 1,4-dioxane in a sealed steel vessel resulted in amination and deprotection, giving rise to the 7-deazapurine nucleosides 4a–g and 5a–g.14 When MeNH2 was used instead of NH4OH, 6-methylamino-7-deazapurine nucleosides 4h and 5h were obtained. Deprotection of 3 with NaOMe/MeOH produced 6-methoxy products 4i and 5i. The α and β isomers were isolated by flash chromatography, and the configuration was assigned based on NOE experiments. The 1H-NMR signals for 2′-C-methyl group are also very distinctive between the α and β isomers: all the α-isomers had a doublet at around 1.29 ppm with a coupling constant JH,F of 21.95 Hz, whereas the β-isomers doublets were around 0.94 ppm with JH,F of 22.33 Hz.
Scheme 1. Synthesis of α- and β-D-2′-deoxy-2-′-fluoro-2′-C-methyl-7-deazapurine nucleosides.

Reagents and conditions: (a) LiAl(t-BuO)3H, THF, 0 °C, 3 h, 99%; (b) 7-deazapurine (R3 = H or NHPiv), Ph3P, DEAD, THF, rt, 2 d, 40–60%; (c) i) NH4OH, 1.4-dioxane, 120 °C, 20 h, 60–80%; or ii) MeNH2, 1,4-dioxane, 120 °C, 20 h, 63–67%; or iii) MeOH, NaOMe, reflux, 3 h, 55–91%.
7-Iodo-7-deazapurine nucleosides (4e, 4g, 5e and 5g) were used as building blocks for the preparation of 7-carbon substituted nucleosides under the catalytic effects of various palladium reagents. For example, using Pd(PPh3)4 as a catalyst,15 4e and 5e were reacted with tributyl(vinyl)tin, giving 7-vinyl substituted nucleosides 6 and 7, respectively, in moderate to good yield (Scheme 2). Hydrogenation of the vinyl compound (6 or 7) resulted in the 7-ethyl nucleoside (8 or 9). Using Pd(OAc)2 and methyl acrylate, 4e and 5e were converted to 7-carbomethoxyvinyl-7-deazapurine nucleosides 10 and 11 in excellent yield (Scheme 3). Coupling of 7-iodo compounds (4e, 4g, 5e, or 5g) with triethylsilylacetelene in the presence of Pd(Ph3P)2Cl2 and CuI produced, upon desilylation with K2CO3 in methanol, moderate yield of 7-ethynyl-substituted nucleosides 12–15. Similarly, 7-phenylethynyl-substituted nucleosides 16 and 17 were also prepared in very good yield by replacing triethylsilylacetelene with phenylacetelene (Scheme 4).
Scheme 2. Synthesis of 7-deaza-7-vinyl and -7-ethyl-purine nucleosides.
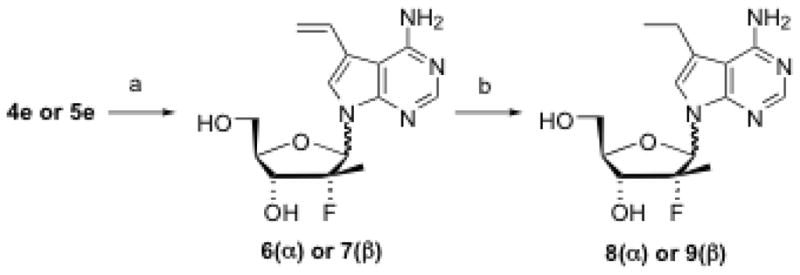
Reagents and conditions: (a) Tributyl(vinyl)tin, Pd(PPh3)4. DMF, 100 °C, 5 h, 53–75%; (b) Pd-C, H2, MeOH, rt, 20 h, 60–70%.
Scheme 3. Synthesis of 7-carbomethoxyvinyl-7-deazapurine nucleosides.
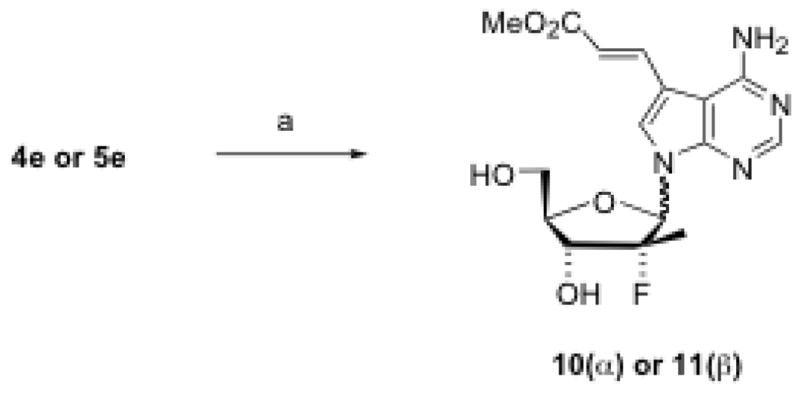
Reagents and conditions: (a) Pd(OAc)2, PPh3, 1,4-dioxane, Et3N, methyl acrylate, reflux, 16 h, 83–100%.
Scheme 4. Synthesis of 7-ethynyl-substituted nucleosides.

Reagents and conditions: (a) i) Pd(Ph3P)2Cl2, THF, Cul, Et3N, triethylsilylacetelene, 45 °C, 20 h, 78–86%; ii) K2CO3, MeCO, rt, 20 h, 30–71%; (b) Pd(Ph3P)2Cl2, THF, Cul, Et3N, phenylacetelene, 45 °C, 20 h, 89–95%.
Some nucleosides were selected for preparation of phenyl phosphoramidate prodrugs using a published procedure.16 Thus, two 6-amino-7-deazapurine nucleosides (5b and 5e) and two 2,6-diamino-7-deazapurine nucleosides (5f and 5g) were converted to their corresponding phosphoramidates 18–21 (Scheme 5). For an HCV polymerase enzyme kinetics study, three 5′-triphosphates (22–24) were also prepared (Scheme 6) and purified by HPLC [DIONEX NucleoPac PA-100 (9 mm × 250 mm) column; buffer A, 0.05 M TEAB; buffer B, 0.5 M TEAB; flow rate, 7.5 mL/min; gradient, increasing buffer B from 0% at 0 min to 50% at 10 min, and then 100% at 12 min and maintained to 17 min].
Scheme 5. Synthesis of nucleoside phosphoramidate prodrugs.

Reagents and conditions: (a) Phenyl L-ethoxyalaninyl phosphorochloridate, 1-methylimidazole,THF, rt, 14 h, 11–47%.
Scheme 6. Synthesis of nucleoside triphosphates.

Reagents and conditions: M = NH(n-Bu)3; (a) i) POCl3, PO(OCH3)3, 2,4,6-collidine, 0 °C, 2 h; ii) (Bu3N)2P2O7H2, Bu3N, rt, 30 min; iii) TEAB, rt, 45 min.
All synthesized nucleosides and phosphoramidate prodrugs were evaluated for in vitro anti-HIV and anti-HCV activities, and toxicity. For determination of anti-HCV activity and toxicity (decreased rRNA), the compounds were tested in an HCV replicon system (96 h) using a standard published protocol.17 The anti-HIV activity and cytotoxicity were assayed as previous described.18 The three synthesized triphosphates (22–24) were tested against both wild-type and S282T mutant HCV NS5B polymerases.19 In addition, some selected compounds were also evaluated against HBV in hepatocytes.20
The results of antiviral activities and toxicities of these nucleoside analogs and prodrugs are summarized in Table 1. Among the 34 compounds tested in HCV replicon cells, 12 compounds showed modest anti-HCV activity with EC50 values less than 10 μM. However, their EC90 values were all greater than 10 μM, except one compound, 7-vinyl-7-deaza-adenine nucleoside (7, β-form), which demonstrated anti-HCV activity with an EC90 of 7.6 μM, comparable to the control compound, 2′-C-methyl-cytidine (NM-107). However, compound 7 also exhibited toxicity in the ribosomal RNA assay and in three independent cell lines, so it appears that the antiviral activity is secondary to cytotoxic effects. Interestingly, its α-isomer 6, also showed moderate anti-HCV activity and strong toxicity across all cell lines. Similarly, both α and β forms of 7-iodo substituted nucleosides (4e and 5e) also showed modest anti-HCV activity and toxicity. It seems that the activity and toxicity are associated mainly with the 7-position substituent, and not related to the α or β configuration. Moreover, these antiviral activities are almost certainly due to the secondary toxicity of the compounds, which could cause cell death or cytostatic effects, thus slowing down or stopping viral replication.
Table 1.
In vitro antiviral activity and cytotoxicity of 2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleosides (4a–17) and phosphoramidate prodrugs (18–21).a
| Compd # | Anti-HCV (μM) | rRNA (μM) | Anti-HIV-1 (μM) | Cytotoxicity, IC50 (μM) in: | ||||
|---|---|---|---|---|---|---|---|---|
| EC50 | EC90 | CC50b | EC50 | EC90 | PBM | CEM | VERO | |
| 4a | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 4b | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | 8.1 | > 100 |
| 4c | < 10 | > 10 | > 10 | > 100 | > 100 | > 100 | 96.0 | > 100 |
| 4d | < 10 | > 10 | > 10 | > 100 | > 100 | 89.1 | 76.0 | 71.1 |
| 4e | 7.0 | 24.9 | > 33 | > 100 | > 100 | > 100 | 40.9 | 65.9 |
| 4f | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 4g | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 4h | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 4i | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 5a | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 5b | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 5c | < 10 | > 10 | > 10 | 82.4 | > 100 | 18.5 | > 100 | > 100 |
| 5d | < 10 | > 10 | < 10 | > 100 | > 100 | 74.2 | 50.5 | 53.1 |
| 5e | 1.1 | > 33 | 10.4 | 94.0 | > 100 | 53.2 | 24.0 | 21.4 |
| 5f | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 5g | > 10 | > 10 | > 10 | 99.5 | >100 | 72.9 | 85.4 | 62.8 |
| 5h | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 5i | > 10 | > 10 | > 10 | 24.8 | 85.6 | > 100 | > 100 | > 100 |
| 6 | 5.6 | 18.4 | 8.3 | 35.5 | 65.2 | 21.0 | 16.0 | 14.6 |
| 7 | 1.8 | 7.6 | 4.5 | 14.7 | 44.8 | 4.8 | 3.5 | 10.1 |
| 8 | > 10 | > 10 | > 10 | 95.5 | > 100 | > 100 | > 100 | > 100 |
| 9 | < 10 | > 10 | > 10 | 19.0 | > 100 | 20.7 | > 100 | > 100 |
| 10 | > 10 | > 10 | > 10 | 0.71 | 9.5 | > 100 | > 100 | > 100 |
| 11 | > 10 | > 10 | > 10 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 12 | < 10 | > 10 | > 10 | 95.2 | > 100 | > 100 | > 100 | > 100 |
| 13 | < 10 | > 10 | > 10 | 59.1 | > 100 | 94.1 | > 100 | > 100 |
| 14 | > 10 | > 10 | > 10 | 7.1 | 59.1 | 54.0 | 26.5 | 61.7 |
| 15 | > 10 | > 10 | > 10 | 84.4 | > 100 | > 100 | > 100 | > 100 |
| 16 | > 10 | > 10 | > 10 | 7.7 | 38.0 | 40.8 | 15.7 | 51.3 |
| 17 | < 10 | > 10 | < 10 | 10.7 | 31.1 | 41.1 | 11.6 | 45.7 |
| 18 | > 10 | > 10 | > 10 | 39.4 | 98.8 | > 100 | > 100 | > 100 |
| 19 | > 10 | > 10 | > 10 | 58.1 | > 100 | 17.4 | 13.5 | 17.8 |
| 20 | > 10 | > 10 | > 10 | 88.0 | > 100 | > 100 | > 100 | > 100 |
| 21 | > 10 | > 10 | > 10 | 26.7 | > 100 | 67.4 | > 100 | 65.1 |
| NM-107 | 2.8 | 9.6 | > 10 | 47.5 | 100.0 | 47.4 | 54.4 | > 100 |
| AZT | > 10 | > 10 | > 10 | 0.0033 | 0.028 | > 100 | 14.3 | 56.0 |
All values are based on mean of replicate assays (N ≤ 2).
CC50: cytotoxic concentration that reduced the rRNA levels by 50% in 96 h.
Some 7-substituted nucleosides demonstrated anti-HIV-1 activity albeit again with cytotoxic effects in PBM cells, and in most cases all cell lines tested. These include 7-vinyl (6 and 7, α and β-isomers, respectively), 7-ethynyl (14, α-isomer), and 7-phenylethynyl (16 and 17, α and β-isomers, respectively) substituted nucleoside analogs. Only one compound, 7-carbomethoxyvinyl substituted nucleoside (10, α-isomer), exhibited strong anti-HIV-1 activity without any apparent cytotoxicity up to 100 μM. Again, it showed that the anti-HIV-1 activity and cytotoxicity are not related to the α- or β-configuration of these nucleoside analogs.
The enzymatic assay results of the nucleoside triphosphates (22–24) with HCV RNA-dependent-RNA-polymerases are presented in Table 2. These triphosphates were potent inhibitors of HCV NS5B polymerase. It is worth noting that these compounds had almost equal potency against wild-type and S282T mutant polymerases. This is in contrast to 2′-C-methylcytidine-TP (NM-107-TP), where a 17-fold increase in the S282T mutant compared to wild-type virus was observed.
Table 2.
Inhibitory activity of 2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleoside triphosphates against wild-type and S282T mutant HCV NS5B polymerases
| Nucleotide # | HCV NS5B pol IC50 (μM) | Fold resistance relative to wild-type HCV pol | |
|---|---|---|---|
| Wild-type | S282T | ||
| 22 | 15 | 15 | 1 |
| 23 | 4 | 6 | 1.5 |
| 24 | 3 | 6 | 2 |
| NM-107-TP | 3 | 51 | 17 |
Five selected nucleosides (4e, 10, 14, 16, and 17) were evaluated for their in vitro antiviral activity against wild-type HBV. Compounds 4e and 17 exhibited weak anti-HBV activity and toxicity to HepG2 cells (data not shown). Compounds 10, 14 and 16 showed no anti-HBV activity.
Since the selected nucleoside 5′-triphosphates demonstrated potent inhibitory activity against HCV polymerases, it is surprising that the nucleoside phosphoramidate prodrugs 18–21 did not show any anti-HCV activity. In many cases, the phosphorylation step of nucleoside analogs to the monophosphate is the rate-limiting step. Therefore, a monophosphate prodrug approach would bypass initial phosphorylation and potentially increase the amount of nucleoside 5′-triphosphate produced intracellularly. In order to understand the mechanism of these nucleoside analogs, a cellular pharmacology study was conducted. The nucleoside 5f and its phosphoramidate prodrug 20 were separately incubated at 50 μM with Huh-7 cells at 37°C for 4 h, and then the cells were washed with phosphate-buffered saline. The intracellular metabolites were extracted with 60% methanol in water and identified by LC-MS/MS. In these experiments, only a trace amount of nucleoside triphosphate was found following incubation of the nucleoside 5f or its phosphoramidate prodrug 20. In the case of compound 5f, neither the 5′-monophosphate nor the diphosphate was detected. In contrast, with the prodrug 20 a large amount of the monophosphate and the parent nucleoside 5f were detected. In addition, no corresponding 7-deazaguanosine nucleoside or nucleoside phosphate was found after incubation of the two compounds, suggesting that no deamination had occurred. These results provide an explanation for the lack of antiviral activity of these nucleoside analogs and phosphoramidate prodrugs. This work confirmed that not only the first phosphorylation step from the nucleoside analog to monophosphate, but also the second phosphorylation step from the monophosphate to the diphosphate was problematic in Huh-7 cells at least for nucleoside 5f, and probably for all other nucleoside analogs synthesized. It is not clear if the third step of the phosphorylation, from the diphosphate to the triphosphate was also a problem. Nonetheless, a monophosphate prodrug approach is not sufficient, and a diphosphate prodrug, or most likely a triphosphate prodrug would be needed to exert the anti-HCV activity of these nucleoside analogs.
It is unusual that some α-nucleoside analogs exhibited antiviral activity and/or toxicity, as these nucleoside analogs are not typically efficiently phosphorylated to the triphosphate.21 Although it is possible that these compounds are acting as non-nucleoside inhibitors, it is more likely that they are unstable and degrade to the 7-deazapurine base. Moreover, the fact that both α and β nucleosides with the same 7-substitutent had similar antiviral activity and toxicity profiles suggests that the biological activity may be associated with the 7-substituent moieties of the nucleosides. The only compound with good anti-HIV-1 activity and no cytotoxicity, α-7-carbomethoxyvinyl nucleoside 10, is worth further investigation to determine its mechanism of action.
In conclusion, thirty novel α and β-D-2′-deoxy-2′-fluoro-2′-C-methyl-7-deazapurine nucleoside analogs were synthesized and evaluated for in vitro anti-HCV activity in a replicon assay. The key step in the synthesis, the sugar-base condensation, was achieved via a Mitsunobu reaction. Several α- and β-7-deazapurine nucleoside analogs exhibited modest anti-HCV activity however this activity was most likely due to their intrinsic toxicity. The four synthesized 7-deazapurine nucleoside phosphoramidate prodrugs 18–21 showed no anti-HCV activity, whereas the nucleoside triphosphates 22–24 demonstrated good inhibitory activity against both wild-type and S282T mutant HCV polymerases (IC50: 3–15 μM). These 7-deazapurine nucleosides and prodrugs were also evaluated for in vitro anti-HIV-1 activity and cytotoxicity. Only one compound, an α-form of 7-carbomethoxyvinyl substituted nucleoside (10), showed good anti-HIV-1 activity (EC50 and EC90 of 0.71 ± 0.25 μM and 9.5 ± 3.3 μM, respectively) with no cytotoxicity up to 100 μM. Some selected compounds were evaluated against HBV, and none showed significant in vitro anti-HBV activity. A cellular pharmacology study of the nucleosides and prodrugs in Huh-7 cells utilizing LC-MS/MS revealed that the triphosphates were not formed at therapeutically significant levels, indicating problems in the first and second phosphorylation steps for these nucleoside analogs by the cellular phosphorylation enzymes.
Acknowledgments
We thank Emilie Fromentin, Aleksandr Obikhod, Sarah Solomon, and Jason Grier for excellent technical assistance. This work was supported in part by 2P30-AI-050409 (RFS), and the Department of Veterans Affairs (RFS), and by the Cancer Research Society (MG). Dr. Schinazi is a founder and major shareholder of RFS Pharma, LLC and his laboratory received no funding from RFS Pharma or vice versa. Dr. Götte received research grants from Tibotec, Merck, Gilead Sciences, AstraZeneca, Pfizer, and GlaxoSmithKline. All other authors have no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Wasley A, Alter MJ. Semin Liver Dis. 2000;20:1. doi: 10.1055/s-2000-9506. [DOI] [PubMed] [Google Scholar]
- 2.Cuthbert JA. Clin Microbiol Rev. 1994;7:505. doi: 10.1128/cmr.7.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Bisceglie AM. Hepatology. 2000;31:1014. doi: 10.1053/he.2000.5762. [DOI] [PubMed] [Google Scholar]
- 4.Di Bisceglie AM, Mchutchison J, Rice CM. Hepatology. 2002;35:224. doi: 10.1053/jhep.2002.30531. [DOI] [PubMed] [Google Scholar]
- 5.McCown MF, Rajyaguru S, Le Pogam S, Ali S, Jiang WR, Kang H, Symons J, Cammack N, Najera I. Antimicro Agents Chemother. 2008;52:1604. doi: 10.1128/AAC.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll SS, Olsen DB. Infect Disord Drug Targets. 2006;6:17. doi: 10.2174/187152606776056698. [DOI] [PubMed] [Google Scholar]
- 7.(a) Rodriguez-Torres M, Lalezari J, Gane EJ, DeJesus E, Nelson DR, Everson GT, Jacobson IM, RRK, McHutchison JG. 59th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD 2008); San Francisco. October 31-November 4, 2008. [Google Scholar]; (b) Lalezarl J, Asmuth D, Casiro A, Vargas A, Dubuc P, Liu W, Pletropaolo K, Zhou XJ, Sullivan-Bolyal J, Mayers D Group, tI-C-I. 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA, USA. 2009. [Google Scholar]; (c) Rodriguez-Torres M, Lawitz E, Flach S, Denning JM, Albanis E, Symonds W, Berrey MM. 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA, USA. 2009. [Google Scholar]
- 8.Eldrup AB, Prhavc M, Brooks J, Bhat B, Prakash TP, Song Q, Bera S, Bhat N, Dande P, Cook PD, Bennett CF, Carroll SS, Ball RG, Bosserman M, Burlein C, Colwell LF, Fay JF, Flores OA, Getty K, LaFemina RL, Leone J, MacCoss M, McMasters DR, Tomassini JE, Von Langen D, Wolanski B, Olsen DB. J Med Chem. 2004;47:5284. doi: 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]
- 9.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. J Med Chem. 2005;48:5504. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 10.Wang P, Chun BK, Rachakonda S, Du J, Khan N, Shi J, Stec W, Cleary D, Ross BS, Sofia MJ. J Org Chem. 2009;74:6819. doi: 10.1021/jo901345j. [DOI] [PubMed] [Google Scholar]
- 11.(a) Ugarkar BG, Castellino AJ, DaRe JS, Ramirez-Weinhouse M, Kopcho JJ, Rosengren S, Erion MD. J Med Chem. 2003;46:4750. doi: 10.1021/jm030230z. [DOI] [PubMed] [Google Scholar]; (b) Seela F, Peng X. J Org Chem. 2006;71:81. doi: 10.1021/jo0516640. [DOI] [PubMed] [Google Scholar]
- 12.But TYS, Toy PH. Chem Asian J. 2007;2:1340. doi: 10.1002/asia.200700182. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Seth PP, Ranken R, Swayze EE, Migawa MT. Nucleosides Nucleotides Nucleic Acids. 2004;23:161. doi: 10.1081/ncn-120027825. [DOI] [PubMed] [Google Scholar]
- 14.Selected synthetic procedures and spectroscopic data: 4-amino-7-iodo-1-(2-deoxy-2-fluoro-2-C-methyl-α-D-ribofuranosyl)-1H-pyrrolo[2,3-d]pyrimidine (4e) and 4-amino-7-iodo-1-(2-deoxy-2-fluoro-2-C-methyl-β-D-ribofuranosyl)-1H-pyrrolo[2,3-d]pyrimidine (5e). To a solution of 4-chloro-7-iodo-1H-pyrrolo[2,3-d]pyrimidine (339 mg, 1.21 mmol), 2-deoxy-2-fluoro-2-C-methyl-α/β-D-ribofuranose (500 mg, 1.34 mmol) and triphenylphosphine (698 mg, 2.66 mmol) in anhydrous THF (5 mL) was added diisopropyl azodicarboxylate (DIAD) (0.53 mL, 2.66 mmol), and the reaction mixture was stirred at rt for 2 d. The solvent was evaporated, and the residue was purified by flash chromatography on silica gel eluting with hexane-EtOAc (9:1 to 2:1) to give 390 mg (51%) of the nucleoside 3e as a white solid, which contained a 1:1 ratio of α and β anomers. The compound 3e (390 mg, 0.61 mmol) was placed in a steel vessel, and 1,4-dioxane (10 mL) was added followed by NH4OH (28%, 20 mL). The steel vessel was sealed and heated at 120°C for 14 h. After cooling to rt, the solvent was evaporated and the residue was purified by flash chromatography on silica gel eluting with CH2Cl2-MeOH (95:5 to 9:1) to give 108 mg (43%) of 4e and 82 mg (33%) of 5e both as a white solid. 4e: Rf 0.30 (CH2Cl2-MeOH 9:1); 1H-NMR (DMSO-d6) δ 8.12 (s, 1H, H-2), 7.38 (d, J = 3.47 Hz, 1H, H-6), 6.70 (brs, 2H, NH2), 6.38 (d, J = 20.79 Hz, 1H, H-1′), 5.71 (d, J = 6.93 Hz, 1H, OH-3′), 4.88 (t, J = 5.78 Hz, 1H, OH-5′), 4.12–4.10 (m, 1H, H-3′), 4.07–4.05 (m, 1H, H-4′), 3.70, 3.50 (2m, 2H, H-5′), 1.29 (d, J = 21.95 Hz, 3H, CH3). LC-MS calcd for C12H15FIN4O3 (M+1): 409.0, found: 409.0. 5e: Rf 0.42 (CH2Cl2-MeOH 9:1); 1H-NMR (DMSO-d6) δ 8.13 (s, 1H, H-2), 7.77 (s, 1H, H-6), 6.75 (brs, 2H, NH2), 6.32 (d, J = 18.10 Hz, 1H, H-1′), 5.65 (d, J = 6.93 Hz, 1H, OH-3′), 5.31 (t, J = 4.62 Hz, 1H, OH-5′), 4.14–4.12 (m, 1H, H-3′), 3.87, 3.67 (2m, 3H, H-4′, H-5′), 0.94 (d, J = 22.72 Hz, 3H, CH3). LC-MS calcd for C12H15FIN4O3 (M+1): 409.0, found: 409.0.
- 15.Okamoto A, Taiji T, Tanaka K, Saito I. Tetrahedron Lett. 2000;41:10035. [Google Scholar]
- 16.McGuigan C, Cahard D, Salgado A, De Clercq E, Balzarini J. Antimicro Agents Chemother. 1996;7:31. [Google Scholar]
- 17.Stuyver LJ, Whitaker T, McBrayer TR, Hernandez-Santiago BI, Lostia S, Tharnish PM, Ramesh M, Chu CK, Jordan R, Shi J, Rachakonda S, Watanabe KA, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2003;47:244. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie MY, Hart GC, Smith GA, Hahn EF. Antimicrob Agents Chemother. 1990;34:1061. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stuyver LJ, Lostia S, Adams M, Mathew JS, Pai BS, Grier J, Tharnish PM, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deval J, Powdrill MH, D’Abramo CM, Cellai L, Götte M. Antimicrob Agents Chemother. 2007;51:2920. doi: 10.1128/AAC.00186-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korba BE, Gerin JL. Antiviral Res. 1992;19:55. doi: 10.1016/0166-3542(92)90056-b. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Choudhury D, Chattopadhyaya J, Eriksson S. Biochem. 1999;38:16993. doi: 10.1021/bi9908843. [DOI] [PubMed] [Google Scholar]