

Abstract
Novobiocin analogs lacking labile glycosidic ether have been designed, synthesized and evaluated for Hsp90 inhibitory activity. Replacement of the synthetically complex noviose sugar with simple aromatic side chains produced analogs that maintain moderate cytotoxic activity against MCF7 and SkBR3 breast cancer cell-lines. Rationale for the preparation of des-noviose novobiocin analogs in addition to their synthesis and biological evaluation are presented herein.
Heat shock protein 90 kDa (Hsp90) is a remarkably versatile molecular chaperone that plays a key role in homeostasis.1,2 However, Hsp90 is unregulated in malignancies and facilitates the maturation, stabilization and/or activation of more than 50 oncogenic proteins, including several kinases and transcription factors.3–6 Thus, Hsp90 has attracted the attention of both industrial and academic laboratories as a promising target for the development of cancer chemotherapeutics.
Hsp90 exists as an obligate homodimer and possesses two nucleotide binding regions: 1) an N-terminal ATP-binding pocket that manifests ATPase activity, and produces the requisite energy for client protein maturation, and 2) a C-terminal nucleotide binding pocket that exhibits allosteric control over substrate and co-chaperones. 2,7–10 Traditional inhibitors of Hsp90 disrupt the N-terminal ATP binding pocket, resulting in client protein degradation and inhibition of cell proliferation. At present, no N-terminal Hsp90 inhibitor has been approved by the FDA. Detriments related to N-terminal inhibition have include; induction of the pro-survival heat shock response, as well as undesired toxicity profiles. 1,11,12 Thus, new opportunities for Hsp90 modulation, including C-terminal inhibition, have been sought after for Hsp90 disruption.13–21
Natural products identified as C-terminal Hsp90 inhibitors include the coumarin family of antibiotics such as novobiocin, clorobiocin and coumermycin A1 (Figure 1).22,23 Structure-activity relationship (SAR) studies on novobiocin, which was originally identified as a bacterial DNA gyrase inhibitor, transformed this broad spectrum antibiotic into a potent cytotoxic Hsp90 inhibitor.13 In addition, SAR studies revealed a class of C-terminal Hsp90 inhibitors that manifest cytotoxic activity at concentrations significantly lower than that which is needed for induction of the heat shock response, unlike the N-terminal inhibitors in clinical trials.24,25 Furthermore, evidence suggests that C-terminal inhibition may improve selective client protein degradation, producing a potential improvement over N-terminal inhibitors.24 Thus, C-terminal inhibitors of Hsp90 represent a promising option for Hsp90 modulation and may possess attributes that overcome limitations manifested with the N-terminal inhibitors.
Figure 1.

Representative Natural Product Hsp90 C-terminal inhibitors.
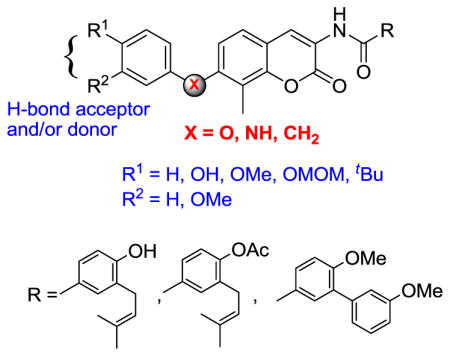
In pursuit of the development of more potent novobiocin analogs, we recently reported derivatives that contain sugar surrogates in lieu of the stereochemically complex noviose (Figure 2). These studies resulted in the attainment of compounds that exhibit nanomolar inhibitory activity against both breast and prostate cancer cell-lines.22,25–27 In continuation of previous optimization efforts; it was desired to replace the noviose sugar with substituted phenyl rings via various linkages (-O, -NH and –CH2). Rationale for the design of these analogs was three-fold: 1) Aryl ethers of novobiocin represent a novel class of uninvestigated analogs; 2) Elimination of the stereochemically complex sugar moiety (which requires 11-steps to prepare28), and 3) Removal of the hydrolytic sensitive glycosidic linkage, which may limit its utility for therapeutic applications.29,30 As a result of these studies, a non-labile and synthetically accessible class of novobiocin analogs was pursued.
Figure 2.

Non-hydrolysable novobiocin analogues.
Syntheses of aryl ethers 13–21 were initiated from previously reported coumarin scaffold 1,22 via the oxidative coupling with commercially available boronic acids 2–4 in the presence of Cu(OAc)2 and pyridine to yield intermediates 5–7 in excellent yields.31,32 Removal of the carboxybenzyl (Cbz) protecting group and subsequent coupling with acid chloride 11 or 1225–27 provided compounds 13, 15, 17 or 19–21, respectively. Finally, hydrolysis of 13, 15 and 17 under mild conditions produced the free phenols 14, 16 and 18.
In parallel, a series of NH-linked aryl analogs 30–32 were synthesized for direct comparison to their aryl ether counterparts. Furthermore, these analogs incorporated a hydrogen bond donor in lieu of the ether acceptor and may therefore provide enhanced interactions with the C-terminal binding pocket.
The NH-linked aryl analogs 30–32, were synthesized from commercially available 3-nitro-2-methyl phenol 23, which was transformed to the corresponding amino phenol 24, upon reduction of the nitro functionality. Intermediate 26 was afforded in reasonable yield via a Buchwald N-arylation cross coupling33,34 of compound 24 with 4-iodo anisole 25 in the presence of Pd(dppf)Cl2 and KOtBu in dioxane. Phenol 26 was condensed with enamine 2725–27 in acetic acid under reflux conditions to provide N-arylated coumarin 28 in acceptable yield. Hydrogenolysis of the Cbz protecting group and subsequent coupling with acid chlorides 11 or 12, provided analogs 30 and 32, in good yields, respectively. Finally, methanolysis of acetate 30 yielded the corresponding phenol, 32.
To further investigate the influence of surrogates on the activity of aryl-containing replacements, linkers containing of a methylene in lieu of the ether or amine were also pursued. These analogs were prepared in an effort to provide direct comparison the aryl ethers/amines previously described. Methylene linked analogues 43–54 were prepared in 3–5 steps commencing with the coumarin 1, which was converted to triflate 33 in the presence of Tf2O and pyridine. Utilizing Pd-mediated borylation chemistry,35 intermediate 34 was obtained in one-step. Interestingly, the vinylogous amide was deprotected simultaneously under the enlisted conditions, which was required for benzamide formation. Subsequent coupling of amine 34 with acid chloride 11 or 12 provided analogs 35 and 36 respectively, in excellent yield.
Finally, palladium-catalyzed Sp2-Sp3 coupling36–38 of 35 with benzyl bromides 37–42 in the presence of K2CO3 yielded compounds 43, 45, 47, 49 and 51, respectively, in respectable yields. Methanolysis of aryl acetates 43, 45, and 47 yielded compounds 44, 46 and 48, respectively. In addition, analogs 50 and 52 were produced via acid mediated hydrolysis of the acetate and MOM groups present in 49 and 51 in single step. Intermediate 36 was also coupled with benzyl bromides 37 and 38 to yield compounds 53 and 54, respectively.
Upon preparation of these non-hydrolysable novobiocin analogs with various linkers (-O, -NH and -CH2), the compounds were evaluated for anti-proliferative activity against SkBR3 and MCF7 breast cancer cell-lines. In general, the ether analogs containing a prenylated aryl side chain (13–18) maintained moderate efficacy, whereas compounds exhibiting the biaryl side chain (19–21) displayed IC50 values >100 μM. This observation is noteworthy, as the presence of the prenylated functionality in noviosylated novobiocin analogs attenuates the antiproliferative activity, suggesting that simple replacement of the sugar moiety with an aryl appendage significantly alters the mode of binding. Furthermore, analysis of analogs 13–18 suggests a cell-line sensitivity to the appended methyl ether. The 3′-OMe analogs (15 and 16) were slightly more active than the corresponding 4′-OMe analogs (13 and 14) against SKBr3 cell lines; however, 13 and 14 were more active against MCF-7 cell lines. The aryl ether analogs also indicate that steric bulk at the 4′-position attenuates activity, as all aryl ethers containing a 4′t-BuOH moiety (17, 18, and 21) exhibited inferior anti-proliferative activities, suggesting spacial limitations at this region of the binding pocket.
Methylene linked analogs, 43, 44, 46 and 48–54, were also evaluated for anti-proliferative activity. With the exception of 54 and 48, methylene linked compounds exhibited a relatively flat SAR, manifesting equipotency against both MCF-7 and SkBR3 cell lines. Compound 48, exhibits moderate activity (~13 μM) against MCF-7 cells, but is inactive up to 100 μM against SkBR3 cells. Although, novobiocin-based analogs are typically slightly more active against MCF-7 cells, the ~10-fold sensitivity to 48 is intriguing. Compound 53 represents the most active aryl linked compound identified in this series, and manifested IC50 values of ~4 μM and ~7 μM against SkBR3 and MCF-7 cancer cells, respectively. Surprisingly, the NH-linked analogs exhibit no anti-proliferative activity against MCF-7 or SkBR3 cell lines up to 100 μM, suggesting that replacement of the ether linker with an NH moiety precludes binding. This result suggests that a hydrogen bond donor at this location and/or the conformational induced via the NH linker may be detrimental to binding and novobiocin analogs containing such features should not be pursed.
In order to confirm inhibition of Hsp90, two representative compounds, 14 and 43, were evaluated for their ability to induce Hsp90 client protein degradation. As expected for a C-terminal Hsp90 inhibitor, client protein degradation was observed and indicative of C-terminal inhibition, no Hsp90 induction was seen. This is in contrast to N-terminal Hsp90 inhibitors, which induce the heat shock response at the same concentration needed to induce client protein degradation.
In conclusion, a series of non-hydrolyzable novobiocin analogs that contain non-hydrolysable linkers (-O, -NH and –CH2) was designed, synthesized and evaluated for Hsp90 inhibitory activity against SkBR3 and MCF7 breast cancer cell-lines. Replacement of the synthetically complex noviose sugar with simple ether or methylene linked aromatic side chains resulted in moderate anti-proliferative activity that is attributable to Hsp90 C-terminal inhibition. These non-hydrolyzable novobiocin analogs provide a class of C-terminal Hsp90 inhibitors that can be obtained through an expedient process, as the synthetically complex noviose has been eliminated.
Figure 3.

Western blot analyses of Hsp90 client protein degradation assays against MCF-7 cells. Concentrations (in μM) of 14 (top) and 43 (bottom) are indicated above each lane. GDA (geldanamycin, 500 nM) and DMSO were respectively employed as positive and negative controls.
Scheme 1.

Synthesis of O-linked non-labile novobiocin analogs.
Scheme 2.

Synthesis of N-linked non-labile novobiocin analogs.
Scheme 3.

Synthesis of methylene linked novobiocin analogs.
Table 1.
Antiproliferative activity of non-labile novobiocin analogs.
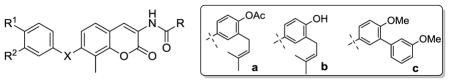 | ||||||
|---|---|---|---|---|---|---|
| Entry | X | R | R1 | R2 | SkBR3 | MCF7 |
| 13 | O | a | OMe | H | 19.51±1.8 | 18.20±2.1 |
| 14 | O | b | OMe | H | 42.72±2.1 | 6.51±0.7 |
| 15 | O | a | H | OMe | 13.68±1.1 | 15.69±2.3 |
| 16 | O | b | H | OMe | 33.14±0.9 | 62.8±4.4 |
| 17 | O | a | tBu | H | 72.39±1.9 | 74.99±4.9 |
| 18 | O | b | tBu | H | 68.27±3.8 | 20.55±0.8 |
| 19 | O | c | OMe | H | >100 | >100 |
| 20 | O | c | H | OMe | >100 | >100 |
| 21 | O | c | tBu | H | >100 | >100 |
| 30 | NH | a | OMe | H | >100 | >100 |
| 31 | NH | b | OMe | H | >100 | >100 |
| 32 | NH | c | OMe | H | >100 | >100 |
| 43 | CH2 | a | OMe | H | 23.49±0.7 | 11.5±0.9 |
| 44 | CH2 | b | OMe | H | 11.08 | 13.25±1.2 |
| 46 | CH2 | b | H | OMe | 14.83±1.3 | 19.65±2.0 |
| 48 | CH2 | b | OMe | OMe | >100 | 13.94±3.3 |
| 49 | CH2 | a | OMOM | OMe | 14.24±0.4 | 14.88±0.9 |
| 50 | CH2 | b | OH | OMe | 10.69±1.1 | 14.62±1.4 |
| 51 | CH2 | a | OMOM | H | 13.50±0.7 | 22.67±1.7 |
| 52 | CH2 | b | OH | H | 21.46±0.5 | 32.11±3.2 |
| 53 | CH2 | c | H | H | 4.09±0.2 | 7.11±1.6 |
| 54 | CH2 | c | OMe | H | >100 | >100 |
Acknowledgments
The authors gratefully acknowledge the support of this project by NIH CA120458, the Madison and Lila Self Graduate Fellowship (A.S.D.), the American Foundation of Pharmaceutical Education Pre-doctoral Fellowship (A.S.D.) and the Institute for Advancing Medical Innovation Pre-doctoral Fellowship (A.S.D) for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duerfeldt Adam S, Blagg BSJ. Bioorg Med Chem Lett. 2010;20:4983. doi: 10.1016/j.bmcl.2010.06.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peterson Laura B, Blagg BSJ. Future Med Chem. 2009;1:267. doi: 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powers MV, Workman P. Endocrine-Related Cancer. 2006;13:S125. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- 4.Maloney A, Workman P. Expert Opinion on Biol Therapy. 2002;2:3. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 5.Subbarao Sreedhar A, Kalmar E, Csermely P, Shen Y-F. FEBS Letters. 2004;562:11. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 6.Brandt GEL, Blagg BSJ. Curr Top Med Chem. 2009;9:1447. doi: 10.2174/156802609789895683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matts RL, Brandt GEL, Lu Y, Dixit A, Mollapour M, Wang S, Donnelly AC, Neckers L, Verkhivker G, Blagg BSJ. Bioorg Med Chem. 2011;19:684. doi: 10.1016/j.bmc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matts Robert L, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson Steve D, Verkhivker Gennady M, Blagg BSJ. ACS Chem Biol. 2011;6:800. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearl LH, Prodromou C. Annual Rev Biochem. 2006;75:271. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 10.Janin YL. Drug Discov Today. 2010;15:342. doi: 10.1016/j.drudis.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Holzbeierlein JM, Windsperger A, Vielhauer G. Curr Oncol Rep. 2010;12:95. doi: 10.1007/s11912-010-0086-3. [DOI] [PubMed] [Google Scholar]
- 12.Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB. Curr Top Med Chem. 2009;9:1479. doi: 10.2174/156802609789895728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burlison JA, Neckers L, Smith Andrew B, Maxwell A, Blagg Brian SJ. J Am Chem Soc. 2006;128:15529. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 14.Donnelly A, Blagg BSJ. Curr Med Chem. 2008;15:2702. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang YT, Blagg BSJ. J Org Chem. 2007;72:3609. doi: 10.1021/jo062083t. [DOI] [PubMed] [Google Scholar]
- 16.Le Bras G, Radanyi C, Peyrat JF, Brion JD, Alami M, Marsaud V, Stella B, Renoir JM. J Med Chem. 2007;50:6189. doi: 10.1021/jm0707774. [DOI] [PubMed] [Google Scholar]
- 17.Matthews SB, Vielhauer GA, Manthe CA, Chaguturu VK, Szabla K, Matts RL, Donnelly AC, Blagg BSJ, Holzbeierlein JM. Prostate. 2010;70:27. doi: 10.1002/pros.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radanyi C, Le Bras G, Bouclier C, Messaoudi S, Peyrat JF, Brion JD, Alami M, Renoir JM. Biochem Biophy Res Comm. 2009;379:514. doi: 10.1016/j.bbrc.2008.12.102. [DOI] [PubMed] [Google Scholar]
- 19.Shelton SN, Shawgo ME, Matthews SB, Lu Y, Donnelly AC, Szabla K, Tanol M, Vielhauer GA, Rajewski RA, Matts RL, Blagg BSJ, Robertson JD. Mol Pharm. 2009;76:1314. doi: 10.1124/mol.109.058545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J Am Chem Soc. 2005;127:12778. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 21.Messaoudi S, Peyrat J-F, Brion J-D, Alam BMn. Expert Opinion on Therapeutic Patents. 2011 doi: 10.1517/13543776.2011.594041. [DOI] [PubMed] [Google Scholar]
- 22.Kusuma BR, Peterson LB, Zhao H, Vielhauer G, Holzbeierlein J, Blagg BSJ. J Med Chem. 2011;54:6234. doi: 10.1021/jm200553w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burlison JA, Blagg BSJ. Org Lett. 2006;8:4855. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 24.Ansar S, Burlison JA, Hadden MK, Yu XM, Desino KE, Bean J, Neckers L, Audus KL, Michaelis ML, Blagg BSJ. Bioorg Med Chem Lett. 2007;17:1984. doi: 10.1016/j.bmcl.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Donnelly AC, Zhao HP, Kusuma BR, Blagg BSJ. Med Chem Comm. 2010;1:165. [Google Scholar]
- 26.Zhao H, Kusuma BR, Blagg BSJ. ACS Med Chem Lett. 2010;1:311. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J Med Chem. 2011;54:3839. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Xiao M, Shen G, Blagg BSJ. J Org Chem. 2004;69:7375. doi: 10.1021/jo048953t. [DOI] [PubMed] [Google Scholar]
- 29.Haerle J, Guenther S, Lauinger B, Weber M, Kammerer B, Zechel DL, Luzhetskyy A, Bechthold A. Chem & Biol. 2011;18:520. doi: 10.1016/j.chembiol.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Olson SH, Slossberg LH. Tet Lett. 2003;44:61. [Google Scholar]
- 31.Kilitoglu B, Arndt H-D. Synlett. 2009:720. [Google Scholar]
- 32.Tan ES, Naylor JC, Groban ES, Bunzow JR, Jacobson MP, Grandy DK, Scanlan TS. ACS Chem Biol. 2009;4:209. doi: 10.1021/cb800304d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Decker M, Si YG, Knapp BI, Bidlack JM, Neumeyer JL. J Med Chem. 2010;53:402. doi: 10.1021/jm9013482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lipshutz BH, Chung DW, Rich B. Advanced Synthesis & Catalysis. 2009;351:1717. doi: 10.1002/adsc.200900323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H, Hendricks RT, Arora N, Nitzan D, Yee C, Lucas MC, Yang Y, Fung A, Rajyaguru S, Harris SF, Leveque VJP, Hang JQ, Le Pogam S, Reuter D, Tavares GA. Bioorg Med Chem Lett. 2010;20:4614. doi: 10.1016/j.bmcl.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Cano C, Barbeau OR, Bailey C, Cockcroft XL, Curtin NJ, Duggan H, Frigerio M, Golding BT, Hardcastle IR, Hummersone MG, Knights C, Menear KA, Newell DR, Richardson CJ, Smith GCM, Spittle B, Griffin RJ. J Med Chem. 2010;53:8498. doi: 10.1021/jm100608j. [DOI] [PubMed] [Google Scholar]
- 37.Ding CZ, Zhang YK, Li X, Liu Y, Zhang S, Zhou Y, Plattner JJ, Baker SJ, Liu L, Duan M, Jarvest RL, Ji J, Kazmierski WM, Tallant MD, Wright LL, Smith GK, Crosby RM, Wang AA, Ni ZJ, Zou W, Wright J. Bioorg Med Chem Lett. 2010;20:7317. doi: 10.1016/j.bmcl.2010.10.071. [DOI] [PubMed] [Google Scholar]
- 38.Kang S-W, Gothard CM, Maitra S, Atiatul W, Nowick JS. J Am Chem Soc. 2007;129:1486. doi: 10.1021/ja0677970. [DOI] [PubMed] [Google Scholar]