

Abstract
Invariant natural killer T cells (iNKT cells) play a prominent role during infection and other inflammatory processes, and these cells can be activated through their T cell receptors by microbial lipid antigens. However, increasing evidence shows that they are also activated in situations where no foreign lipid antigens are present, suggesting a role for lipid self-antigen. We now demonstrate that an abundant endogenous lipid, β-D-glucopyranosylceramide (β-GlcCer), is a potent iNKT cell self-antigen in mouse and human, and that its activity depends on N-acyl chain composition. Furthermore, β-GlcCer accumulates during infection and in response to Toll-like receptor agonists, contributing to iNKT cell activation. Thus, we propose that recognition of β-GlcCer by the invariant TCR translates innate danger signals into iNKT cell activation.
Invariant natural killer T (iNKT) cells are a subset of αβ T cells that recognize lipid antigens presented by the non-polymorphic CD1d molecule. In contrast to peptide-specific diverse αβ T cell receptor (TCR) major histocompatability complex-restricted (MHC)-restricted T cells, these cells bear an invariant TCR α-chain, utilizing TCR Vα14 and Jα18 paired with a limited Vβ chain repertoire in mouse, or Vα24 and Jα18 paired with Vβ11 in human. How iNKT cells, with an invariant TCR, restricted by a non-polymorphic antigen presenting molecule are activated in a wide variety of infectious and non-infectious pathological processes remains poorly understood1-2.
An enormous effort has been focused on the identification of lipids that are cognate antigens for the iNKT cell TCR. The discovery of the pharmacologic antigen α-galactosylceramide (α-GalCer)3, as well as glycolipid antigens from the bacteria Borrelia burgdorferi4 and Sphingomonas5-6, each with a primary α-linked monohexose, suggested the possibility that the major structures recognized by iNKT cells might be α-linked glycolipids. However, recognition of such lipids fails to explain the role of iNKT cells during the majority of infections or during inflammation, since primary α-glycosidic linkages have not been demonstrated to occur in most microbes or in mammalian glycolipids. Furthermore, iNKT cells have been shown to play a major role in situations where foreign lipid antigens would not be present at all, including autoinflammatory conditions, viral infection, or stimulation by Toll-like receptors (TLRs)7-10. These observations support a central role for lipid self-antigen in the activation of iNKT cells.
The rapid activation of iNKT cells by antigen presenting cells (APC) exposed to lipopolysaccharide (LPS) or other TLR agonists is dramatic7-10, and provides a robust and salient model for understanding the response of iNKT cells to innate signals. It is now clear that two signals are likely required for the physiological activation of iNKT cells, with the primary signal provided through the TCR by a CD1d-lipid complex, and a second signal provided by APC-derived cytokines, dominantly IL-127, 11. Accumulation of a stimulatory lipid self-antigen has been proposed to provide the TCR-mediated signal to iNKT cells following TLR-agonist stimulation9-10, but no specific lipid-antigen has yet been identified as responsible.
Phospholipids, including lyso-phosphatidylcholine (lyso-PC), have been proposed to be self-antigens for iNKT cells, but their stimulatory activity is weak, and has only been demonstrated for a subset of iNKT cells12-13. Isoglobotrihexosylceramide (iGB3) contains a terminal α-linked carbohydrate, and has been demonstrated to activate mouse iNKT cells via CD1d. Based largely on the observation that Hexb−/− (the Hexb gene product converts iGB4 to iGB3) mice have defective iNKT cell development, this lipid was proposed as a relevant self-antigen14. However, a subsequent report demonstrated that iGB3 synthase-deficient mice have a normal iNKT cell phenotype15, and it has been suggested that the iNKT cell defect in Hexb−/− mice might be due to altered lysosomal function rather than reduced iGB3 levels16. Furthermore, iGB3 is present at almost undetectable levels in murine lymphoid tissues17-18, and it has also been reported that humans do not express the relevant synthase, and are thus unable to synthesize iGB319.
In this study, we screened a panel of naturally occurring glycosphingolipids (GSLs) for antigenic activity on iNKT cells. β-GlcCer, previously reported as non-antigenic20, potently activates iNKT cells from both mouse and human through a cognate TCR interaction. Furthermore, β-GlcCer, the precursor of most GSLs outside of the central nervous system, accumulates in APC after stimulation with lipolysaccharide (LPS) and in vivo following bacterial infection. Blocking β-GlcCer synthesis in bone marrow-derived dendritic cells (BMDC) reduced both autoreactivity and iNKT cell activation in response to LPS or whole bacteria, whereas blocking the subsequent step in GSL synthesis, the conversion of β-GlcCer to β-lactosylceramide (β-LacCer), had no effect, or an opposite effect. Our data reveal β-GlcCer as a potent, physiologically relevant self-antigen for iNKT cells that is upregulated in response to microbial danger signals.
Results
Antigenic activity among a panel of GSLs
In order to identify self-lipids that stimulate iNKT cells, we planned first to identify the lipids present in CD1d on APC, and then screen those lipids for activity. We recently reported the dominant GSLs eluted from CD1d on APC, and the results are summarized in Supplementary Table 121. β-GlcCer would not have been detected by that analysis, as carbohydrate head groups were derivatized following GSL digestion with ceramide glycanase, an enzyme that does not cleave monohexose from a ceramide backbone. For this reason, as we transitioned to the activity-determination phase of our iNKT cell antigen discovery efforts, we also included β-GlcCer. Two assay systems were used to screen for lipid activity, a single TCR specificity iNKT cell hybridoma co-cultured with CD1d-trasfected RAW macrophages, and a primary iNKT cell line cultured with primary CD11c+ BMDC. We tested the GSLs listed in Supplementary Table 1 as well as phospholipids, including phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylserine (PS), lyso-phosphatidylcholine (lyso-PC), lyso-phosphatidylethanolamine (lyso-PE), lyso-phophatidylserine (lyso-PS), and lyso-phosphatidylinositol (lyso-PI). From the panel of lipids tested, none of the higher order GSLs or phospholipids were stimulatory. In contrast, the simplest GSL, β-GlcCer, demonstrated reproducible activity for iNKT cells (Fig. 1a,b and data not shown). Using a primary murine iNKT cell line, the activity of β-GlcCer was further demonstrated to elicit the production of both interferon-γ (IFN-γ) and IL-4 (Fig. 1c,d), a characteristic of TCR-mediated, but not cytokine-mediated iNKT cell activation8. As has been previously reported for other iNKT cell TCR-mediated signals, the ability of β-GlcCer to induce iNKT cell IFN-γ production could be enhanced by the addition of IL-12 (Fig. 1e), an indirect stimulus for iNKT cells that we have proposed to be a critical second signal during infection7, 11. The chemical composition of bovine milk β-GlcCer was confirmed by proton nuclear magnetic resonance spectroscopy (NMR), two-dimensional correlation spectroscopy (COSY), and total correlation spectroscopy (TOCSY) NMR, and no α-anomeric carbohydrate was detected by these analyses (data not shown). From these results, we concluded that an early biosynthetic GSL, β-GlcCer, activates iNKT cells in a CD1d-dependent manner.
Figure 1.
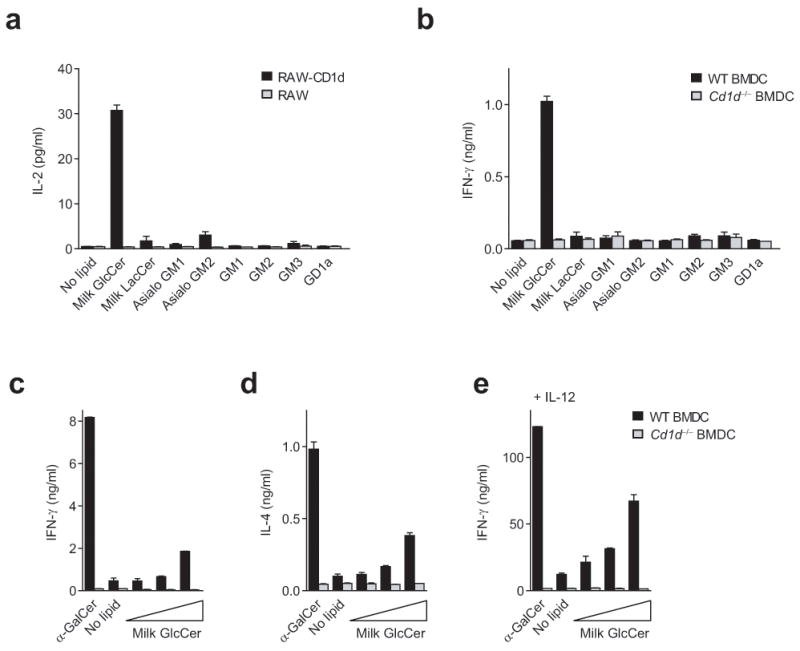
iNKT cell reactivity to a panel of GSLs. (a) RAW or CD1d-transfected RAW cells were co-cultured with the DN32 iNKT cell hybridoma in the presence of 10 μg/ml of the indicated lipid, and production of IL-2 was assessed by enzyme-linked immunosorbent assay (ELISA). (b) Murine CD11c+ BMDC were co-cultured with a primary iNKT cell line in the presence of 10 μg/ml of the indicated lipid. (c) IFN-γ and (d) IL-4 production in the absence of IL-12, and (e) IFN-γ production in the presence of 20 pg/ml IL-12, by a primary murine iNKT cell line co-cultured with CD11c+ BMDC in the presence of 10 ng/ml α-GalCer, or a milk β-GlcCer 5-fold dose titration with a top concentration of 20 μg/ml. IFN-γ and IL-4 production were measured by ELISA. Data are presented as mean and range of duplicate wells, and are representative of at least three independent experiments.
β-GlcCer in primary lymphoid tissues
To determine whether β-GlcCer could be detected in tissues that might be sites of iNKT cell activation, polar lipids extracted from mouse thymus, spleen, liver, and BMDC were analyzed by thin layer chromatography (TLC). We readily observed a monohexosyl ceramide in thymus, spleen, and BMDC (Fig. 2a). A detectable, but lower level of monohexosyl ceramide was present in the liver polar lipid extract. Using borate-impregnated TLC, we determined that, as expected, the monohexosyl ceramide band in thymus, spleen, and BMDC was almost exclusively β-GlcCer, and not β-GalCer (Supplementary Fig. 1). To estimate the β-GlcCer content in these tissues, we used TLC-based densitometric analysis of the tissue lipid extracts compared to a β-GlcCer standard titration. The β-GlcCer content of mouse thymus, spleen, and CD11c+ BMDC was estimated as 4.6 ± 1.1, 3.6 ± 0.8, and 3.9 ± 0.1 μg per mg of polar lipids. In order to determine whether the β-GlcCer detected in mammalian lymphoid tissues had stimulatory activity for iNKT cells, we used preparative-scale high performance liquid chromatography (HPLC) coupled with mass spectrometry (MS) to purify β-GlcCer from mouse spleen and thymus crude polar lipid extracts (Supplementary Fig. 2). The purified β-GlcCer-containing fractions were found to stimulate an iNKT cell hybridoma in co-culture with CD1d-transfected RAW cells (Fig. 2b). In the assay shown, the β-GlcCer concentrations were estimated as 18.0 ± 4.0 and 23.0 ± 5.5 μg/ml for thymus and spleen β-GlcCer fractions, respectively. For spleen polar lipid extracts, β-LacCer and phospholipid-containing fractions were purified by the same method used to purify β-GlcCer, and were unable to stimulate the iNKT cell hybridoma (Fig 2b). Thus, β-GlcCer is present in mammalian lymphoid tissues, and when purified from these tissues, activates iNKT cells in a CD1d-dependent manner.
Figure 2.
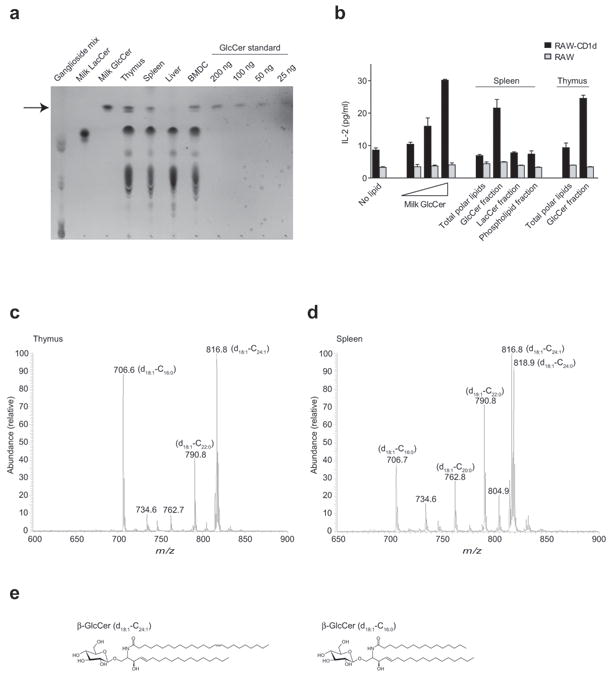
β-GlcCer is present in primary lymphoid tissues and activates iNKT cells. (a) TLC analysis of polar lipids extracted from murine thymus, spleen, whole liver, and BMDC along with GSL standards and milk β-GlcCer dose titration. The relative mobility of β-GlcCer is marked by an arrow. (b) The indicated lipid fractions from mouse thymus and spleen were enriched from total polar lipid extracts by normal-phase preparative HPLC as described in Supplementary Fig. 2. Enriched fractions were assayed for activity by co-culture with the iNKT hybridoma DN32 and RAW or CD1d-transfected RAW cells as APC, with a milk β-GlcCer 5-fold dose titration to a top concentration of 20 μg/ml shown for comparison. IL-2 ELISA data are presented as mean and range of duplicate wells, and are representative of two separate experiments. (c) Thymus and (d) spleen purified β-GlcCer were subjected to ESI-MS analysis in electrospary-positive mode. Major β-GlcCer ions are depicted with a lithium adduct, and fatty acid composition as determined by CID-MS is shown in parentheses. (e) Structures of two abundant β-GlcCer forms detected by ESI-MS.
Because β-GlcCer has been previously reported as non-antigenic for iNKT cells20 we questioned whether the activity of bovine milk and mammalian β-GlcCer that we observed might depend on specific lipid structures attached to the carbohydrate head group present in these purified materials. To address this possibility, the fatty acid compositions of β-GlcCer in bovine milk, mouse spleen, and mouse thymus were determined by electrospray ionization mass spectrometry (ESI-MS) in the positive-ion mode (Fig. 2c,d and Supplementary Fig. 3). Structural assignments were confirmed by collision-induced dissociation tandem mass spectrometry (CID-MS) as previously described22. In all three samples analyzed, the major ceramide backbone consisted exclusively of sphingenine (d18:1). In mouse thymus, the major N-acyl chains were C24:1, C22:0, and C16:0. In mouse spleen, the major N-acyl chains detected were C24:1, C24:0, C22:0, C20:0 and C16:0. In bovine milk, the major N-acyl chains detected were C24:0, C23:0, C22:0, C20:0, and C16:0. The structures of two abundant β-GlcCer forms observed in thymus and spleen are depicted (Fig. 2e). For all samples, other β-GlcCer molecular species were also detected in smaller quantities, confirming the expected diversity of N-acyl chains in mammalian GSLs. These results demonstrate that the β-GlcCer present in mammalian tissues contains multiple N-acyl chain structures, with C24:1 being the most abundant in lymphoid tissues.
β-GlcCer C24:1 is a potent mouse iNKT cell antigen
To functionally test the role of N-acyl chain composition on the activity of β-GlcCer, we studied a panel of synthetic β-GlcCer compounds, all containing sphingenine as the sphingosine base, with N-acyl chains varying from 8 to 24 carbons, either fully saturated or containing one unsaturation (Supplementary Fig. 4). β-GlcCer C24:1, C18:1, C18:0, and C16:0 were all detected in primary mouse tissue by ESI-MS, while the 12 and 8-carbon N-acyl chain forms of this lipid were not detected (Fig. 2c,d), and have not, to our knowledge, been reported to occur in mammals. β-GlcCer C24:1, C12:0, and β-GlcCer C18:1 activated an iNKT hybridoma in vitro, while β-GlcCer C18:0 and C16:0 showed no activity (Fig. 3a). Lyso-β-GlcCer (d18:1) and free ceramide backbones corresponding to each synthetic β-GlcCer were not active (data not shown). We next compared the potency of β-GlcCer C24:1 to the microbial antigen GSL-1 from Sphingomonas5-6, and two previously proposed self-antigens, iGB314 and lyso-PC12. The iGB3 used was d18:1-C26:0, and while not matched to β-GlcCer C24:1 in N-acyl chain structure, the C26:0 acyl chain would be expected to impart maximal activity based on data from α-GalCer analogs23-24. Assayed using a primary iNKT cell line in co-culture with CD11c+ BMDC, the antigenicity of β-GlcCer C24:1 was less than GSL-1 but greater than iGB3, while as previously reported, we could not detect activity for lyso-PC in mouse25 (Fig. 3b). Although we could not detect β-GalCer at potential sites of peripheral iNKT cell activation (Supplementary Fig. 1), and a β-GalCer deficient mouse has been reported not to have a demonstrable iNKT cell defect20, given the structural similarity of β-GalCer to β-GlcCer, we hypothesized that this lipid might also activate iNKT cells. Purified bovine brain β-GalCer, β-GalCer d18:1-C24:1, and as previously demonstrated, the non-physiological lipid β-GalCer d18:1-C12:0 26-27, activated mouse iNKT cells in a CD1d-dependent manner, albeit less potently than each corresponding β-GlcCer (Supplementary Fig. 5).
Figure 3.
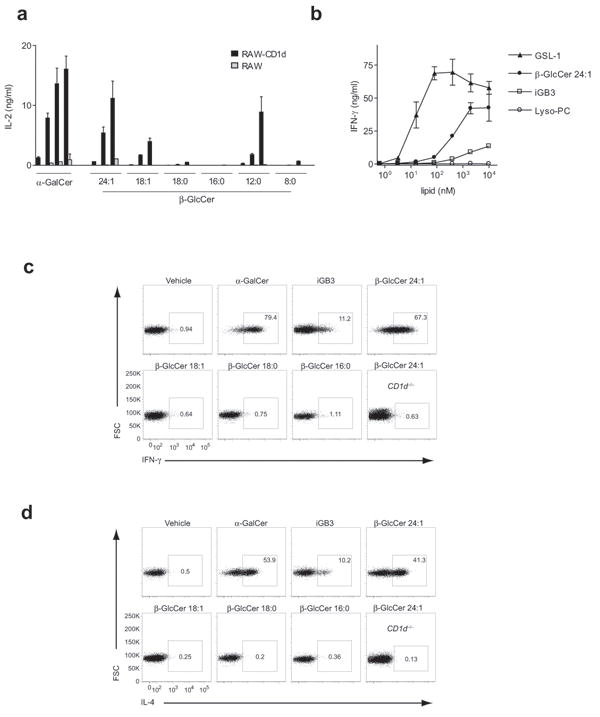
iNKT cell reactivity to a β-GlcCer panel with differing N-acyl chains. (a) A β-GlcCer panel in co-culture with RAW or CD1d-transfected RAW cells and the iNKT cell hybridoma DN32. IL-2 production was determined by ELISA to a 5-fold dose titration of the indicated lipid with a top concentration of 10 μg/ml, or 10 ng/ml for α-GalCer. (b) IFN-γ production by a primary murine iNKT cell line in co-culture with wild-type CD11c+ BMDC, comparing β-GlcCer C24:1, reported iNKT cell lipid self-antigens, and a microbial GSL antigen. IL-2 and IFN-γ ELISA data are presented as mean and range in duplicate wells, and are representative of three separate experiments. (c) IFN-γ and (d) IL-4 cytokine capture assay in liver mononuclear cells following intravenous injection of the indicated lipid. Mice were injected intravenously with 25 μg of the indicated lipid, or 1 μg for α-GalCer. The TCRβ+PBS-57 (α-GalCer analog)-loaded tetramer+ gate is shown except for the last plot in each panel, where the total TCRβ+ gate is shown for a β-GlcCer C24:1-injected CD1d-deficient mouse. The percentage of iNKT cells producing IFN-γ or IL-4 is indicated for each plot. Results are representative of at three independent experiments. The structures of the synthetic lipids used here are depicted in Supplementary Fig. 4, and as all contained a d18:1 shingenine base, they are abbreviated in this and other figures with only the N-acyl chain composition listed. For example, “β-GlcCer 24:1” represents β-D-glucopyranosylceramide d18:1-C24:1.
To examine self-antigen activity in vivo, we injected mice intravenously with a subset of the β-GlcCer synthetic panel, as well as iGB3. Two hours after injection with β-GlcCer C24:1, two-thirds of liver iNKT cells were IFN-γ positive, and greater than 40% were IL-4 positive, a substantially higher percentage than in mice injected with iGB3 (Fig. 3c,d). In liver mononuclear T cells from CD1d-deficient mice that lack NKT cells, there was no detectable response to β-GlcCer C24:1 injection. We also observed potent activation of CD11c+ dendritic cells and B cells in vivo 24 hrs following β-GlcCer C24:1 injection, most likely a result of trans-activation (Supplementary Fig. 6). We concluded from these studies that the activity of β-GlcCer is dependent on the N-acyl chain structure, and the abundant d18:1-C24:1 form of β-GlcCer is a potent antigen for iNKT cells in vivo.
In addition to iNKT cells with an invariant TCR-α, there are CD1d-restricted T cells within the diverse αβ TCR compartment, referred to as diverse NKT cells. Since there is currently no method available to identify primary diverse NKT cells, we screened a panel of ten individual diverse NKT cell hybridomas for activity by co-culture with primary murine BMDC and the synthetic β-GlcCer panel described above. Two of these ten diverse NKT cell hybridomas, VII68 and XV1928, displayed reactivity to a subset of β-GlcCer compounds (Supplementary Fig. 7 and data not shown). We concluded from these studies that β-GlcCer also activates diverse NKT cells, but that this reactivity may depend on different N-acyl chain structure than for iNKT cells.
β-GlcCer C24:1 mediates a cognate TCR interaction
To confirm that β-GlcCer was directly mediating iNKT cell stimulation via CD1d, we tested a synthetic β-GlcCer panel and milk β-GlcCer in an APC-free system using a primary iNKT cell line with purified, plate-bound CD1d loaded with each lipid. In this system, milk β-GlcCer, β-GlcCer C24:1, and β-GlcCer C12:0 activated a primary iNKT cell line (Fig. 4a), demonstrating direct, CD1d-dependent activation of iNKT cells by β-GlcCer. Next, we asked if CD1d tetramers loaded with β-GlcCer C24:1 could bind iNKT cells directly. Indeed, β-GlcCer C24:1-loaded tetramers stained a portion of iNKT cells from both C57Bl/6 and BALB/c mice, identified by sequential double staining with a PBS-57 (α-GalCer analog)-loaded CD1d tetramer (Fig. 4b). The staining appeared to be limited to the portion of iNKT cells that stained most brightly with the PBS-57-loaded tetramer, suggesting that β-GlcCer C24:1-loaded tetramer staining was brightest for the iNKT cell population with the highest affinity TCRs. The TCR Vβ-chain repertoire of mouse iNKT cells is limited, and those cells utilizing Vβ-chains Vβ2, Vβ7, Vβ8.1, Vβ8.2, and Vβ8.3 have been found to have higher affinity for α-GalCer-loaded CD1d tetramers compared to less frequently used Vβ-chains (Vβ6, Vβ9, Vβ10, Vβ14)29. Consistent with the affinity hierarchy described for α-GalCer-loaded tetramers, β-GlcCer C24:1-loaded tetramers identified the most frequently used iNKT cell TCR Vβ-chains (Fig 4c,d).
Figure 4.
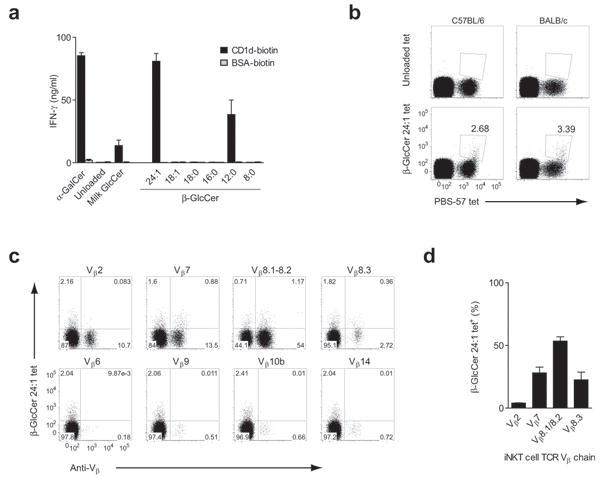
β-GlcCer presented by CD1d activates iNKT cells via cognate TCR interaction. (a) CD1d was loaded with equal molar concentrations of the indicated lipid. Loaded CD1d, or mock-loaded bovine serum albumin (BSA), was plate-bound and used to stimulate a primary murine iNKT cell line. IFN-γ ELISA data are presented as mean and range in duplicate wells. (b) Freshly-isolated splenocytes from C57Bl/6 and BALB/c mice were co-stained with PBS-57 CD1d tetramer and unloaded or β-GlcCer C24:1-loaded CD1d tetramer. TCRβ+CD19-cells are shown, and the percentage of iNKT cells that stained positive with β-GlcCer C24:1-loaded tetramer is shown. (c) C57Bl/6 splenocytes were stained with β-GlcCer C24:1 CD1d tetramer and the TCR Vβ antibody indicated above each plot. The CD3 molecular complex+CD19-PBS-57 tetramer+ gate is shown in each plot, with quadrants based on unloaded tetramer staining. (d) The percentage of iNKT cells from the β-GlcCer C24:1 tetramer+ population bearing each indicated TCR Vβ, with data presented as mean + s.e.m. from three separate experiments.
Although β-GlcCer tetramer staining was limited to a fraction of mouse iNKT cells (Fig 4b), the majority of iNKT cells produced cytokines after stimulation with β-GlcCer in vivo (Fig. 3). Furthermore, iNKT cells profoundly downregulate their surface TCR expression following activation with a strong antigen, but not following indirect activation30. 24 hours following intravenous injection of β-GlcCer C24:1, iNKT cells were almost undetectable, suggesting TCR-dependent activation of almost all mouse iNKT cells (Supplementary Fig. 8). The difference, then, between iNKT cell activation and tetramer binding is likely due to a higher affinity requirement for tetramer binding relative to activation. We concluded from these tetramer studies that β-GlcCer C24:1 mediates a cognate interaction between the iNKT cell TCR and CD1d in mouse.
β-GlcCer is a self-antigen for human iNKT cells
Given the high degree of evolutionary conservation seen for both CD1d and iNKT cells1, we would expect a physiologically relevant self-antigen to activate both mouse and human iNKT cells. We asked whether β-GlcCer was a self-antigen in human by testing the synthetic β-GlcCer panel described above for activity on human iNKT cells. Similarly to mouse iNKT cells, β-GlcCer C24:1, C12:0, and to a lesser degree C18:1, activated three independent human iNKT cell clones31 when presented by human peripheral blood mononuclear cell (PBMC)-derived APC, and this activation was efficiently inhibited by a monoclonal antibody against CD1d (Fig. 5a,b). iGB3, on the other hand, did not activate iNKT cells in this system. Using a primary human iNKT cell line, we compared the antigenic potency of Sphingomonas GSL-1, β-GlcCer C24:1, iGB3, and lyso-PC. In human, β-GlcCer C24:1 was less potent than GSL-1, but far more potent than iGB3 or lyso-PC. In fact, in contrast to mouse iNKT cells, we could not detect activity with iGB3 using primary human iNKT cells. Lyso-PC did not stimulate iNKT cells to produce detectable IFN-γ or IL-4, but as has been reported12, we did detect a small amount of granulocyte-macrophage colony-stimulating factor (Fig 5c and data not shown).
Figure 5.
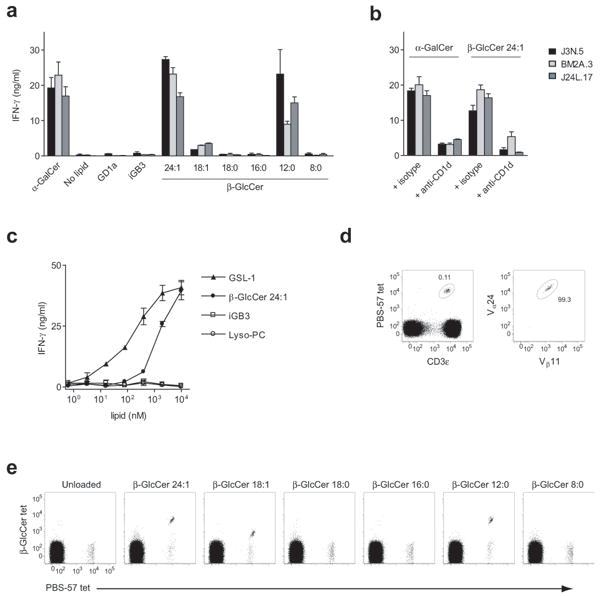
β-GlcCer is a cognate antigen for human iNKT cells. (a) Three human iNKT cell clones designated J3N.5, BM2A.3, and J24L.17, were co-cultured with human PBMC-derived monocytes in the presence of 10 μg/ml of the indicated lipid, or 10 ng/ml for α-GalCer, and (b) in the presence of an anti-CD1d or isotype control monoclonal antibody. (c) IFN-γ production by a primary human iNKT cell line in co-culture with PBMC-derived monocytes, comparing β-GlcCer C24:1 to reported iNKT cell lipid self-antigens and a microbial GSL antigen. IFN-γ production was assessed by ELISA, and is presented as mean and range in duplicate wells. (d) Left, anti-CD3ε and PBS-57 tetramer were used to identify iNKT cells. Right, CD3ε+/PBS-57 tetramer+ gated cells were co-stained with anti-Vβ24 and anti-Vβ11 to confirm invariant TCR chain usage. (e) PBMC were co-stained with PBS-57-loaded CD1d tetramer and CD1d tetramers loaded with the β-GlcCer N-acyl chain variant indicated above each plot. The CD3ε+ gate is shown. Note that CD1d tetramers loaded with β-GlcCer C24:1, C18:1, and C12:0 stain human iNKT cells. Data are representative of at least three separate experiments.
Having demonstrated the activity of β-GlcCer on human iNKT cell clones and a primary human iNKT cell line, we next cultured freshly-isolated PBMC overnight with β-GlcCer, without the addition of exogenous APC, and measured iNKT cell intracellular cytokine production. β-GlcCer C24:1 was able to stimulate cytokine production using this assay, and this effect was completely blocked by anti-CD1d monoclonal antibody (Supplementary Fig. 9a,b). We also used freshly-isolated human PBMC to assay iNKT cell proliferation in response to various β-GlcCer N-acyl chain variants. β-GlcCer C24:1, C12:0, and C18:1 in co-culture led to a marked expansion of iNKT cells over an 8 day period (Supplementary Fig. 9c).
To confirm the cognate interaction between β-GlcCer-loaded CD1d and the iNKT cell TCR in human, we undertook tetramer studies using lipid-loaded human CD1d. Staining with PBS-57-loaded tetramers allowed for unambiguous identification of iNKT cells, and the iNKT cells were confirmed to be Vα24+Vβ11+ (Fig. 5d). Double staining of freshly-isolated PBMC with β-GlcCer-loaded CD1d tetramers and PBS-57-loaded tetramers demonstrated that a substantial portion of human iNKT cells bind β-GlcCer C24:1, C12:0, and C18:1-loaded tetramers (Fig. 5e). In complementary experiments, we next examined if iNKT cell TCR tetramers32 could recognize CD1d loaded with β-GlcCer. We again found an N-acyl chain-dependent interaction, with β-GlcCer C24:1 and C12:0 mediating TCR tetramer binding (Supplementary Fig 10a). β-GlcCer-loaded CD1d tetramer staining was positive, but of variable intensity between subjects (Supplementary Fig. 10b), and for clarity, a subject with a high percentage of β-GlcCer CD1d tetramer-positive iNKT cells is shown in Fig. 5.
As was seen in the mouse, β-GlcCer tetramers recognized only a portion of the human iNKT cell population identified by α-GalCer-loaded tetramers. The percentage of cells recognized by β-GlcCer tetramers was not decreased by double-staining with α-GalCer tetramers under the staining conditions used, suggesting that tetramer competition was not a factor (data not shown). We hypothesized that, as in the mouse, β-GlcCer tetramers might identify human iNKT cells with high-affinity TCRs. To address this possibility, we used OCH, an α-GalCer analog that when loaded in human CD1d tetramers has been reported to bind a population of human iNKT cells with high-affinity TCRs32. Double staining with OCH- and β-GlcCer C24:1-loaded CD1d tetramers identified the same population, suggesting that β-GlcCer tetramers identified those iNKT cells with high-affinity TCRs (Supplementary Fig. 10c). It should be noted that robust human iNKT cell activation by β-GlcCer was seen with all human subjects and all iNKT cell clones tested, irrespective of the intensity of β-GlcCer CD1d tetramer staining, suggesting as in our mouse studies, that tetramer binding may have a more demanding threshold for recognition than NKT cell activation.
β-GlcCer contributes to iNKT cell self-reactivity
GSL self-lipid antigens have been proposed to contribute to iNKT cell activation in the absence of foreign lipid antigens33. Since β-GlcCer is relatively abundant in lymphoid tissues, and we found that it potently activates iNKT cells from both mouse and human, we postulated that this lipid might contribute to iNKT cell self-reactivity. We assessed the contribution of β-GlcCer to iNKT cell autoreactivity by perturbation of the pathways involved in the synthesis of this lipid. The pathways involved in the synthesis and degradation of β-GlcCer are shown in Supplementary Fig. 11a. Using a primary murine iNKT cell line, we found that, as previously shown10, 20, inhibition of GSL synthesis by either N-butyldeoxygalactonojirimycin (NB-DGJ) or D-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (D-PDMP) reduced iNKT cell autoreactivity (Fig 6a). To assess antigen processing and presentation in BMDC following treatment with GSL synthesis inhibitors, we used Gal–α-GalCer, an α-GalCer analog that requires lysosomal uptake and processing for antigenicity34 (Fig 6b). NB-DGJ and D-PDMP did not reduce the activation of iNKT cells by Gal–α-GalCer. Because both NB-DGJ and D-PDMP inhibit β-GlcCer synthesis, and consequently, the synthesis of all higher order ceramides based on β-GlcCer, we used siRNA silencing of either glucosylceramide synthase (Ugcg) or the downstream enzyme that converts β-GlcCer to β-LacCer, lactosylceramide synthase (B4galt6), in BMDC to isolate the β-GlcCer-dependent signal (Fig. 6c). siRNA silencing of Ugcg in BMDC reduced iNKT cell autoreactivity, while B4galt6 silencing increased autoreactivity (Fig 6d). siRNA silencing did not alter the ability of BMDC to present Gal–α-GalCer (Fig 6e), and did not alter CD1d surface levels as determined by flow cytometry (Fig 6f). Thus, modulation of β-GlcCer levels determines iNKT cell self-reactivity to BMDC.
Figure 6.
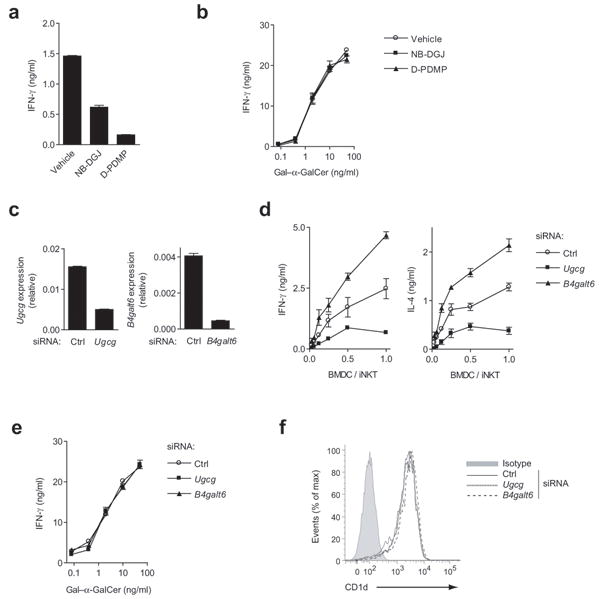
β-GlcCer contributes to iNKT cell self-reactivity. (a) CD11c+ BMDC were co-cultured with an iNKT cell line at a 1:5 ratio in the presence of inhibitors of β-GlcCer synthesis, NB-DGJ or D-PDMP, and iNKT IFN-γ production was determined by ELISA. (b) IFN-γ production by iNKT cells co-cultured with CD11c+ BMDC and Gal–α-GalCer, an antigenic lipid that requires lysosomal processing, was measured in the presence of β-GlcCer synthesis inhibitors. (c) siRNA silencing of Ugcg and B4galt6, assessed by quantitative PCR (qPCR) relative to Gapdh, was performed in CD11c+ BMDC, and after 48 hrs, BMDC were harvested and (d) co-cultured with a primary iNKT cell line at the indicated ratio. Autoreactivity was assessed by cytokine ELISA. (e) The presentation of Gal–α-GalCer by siRNA-silenced CD11c+ BMDC was assessed by measuring IFN-γ production by an iNKT cell line after co-culture. (f) CD1d surface expression on CD11c+ BMDC was measured by flow cytometry 48 hrs after the introduction of siRNA. ELISA data are presented as mean and range in duplicate wells, and data are representative of three separate experiments.
β-GlcCer mediates iNKT cell activation during infection
As we and others have previously shown, the iNKT response to LPS-exposed BMDC requires both a signal through CD1d and APC-derived IL-12 7-9-10. We hypothesized that β-GlcCer might be a prominent component of an LPS-induced, CD1d-mediated signal, and next investigated the pathways involved in the synthesis and degradation of β-GlcCer following TLR-agonist exposure. Using a recently published gene expression dataset35, we looked at the regulation of Ugcg and B4galt6 in murine BMDC in response to five TLR agonists. Four out of five of the stimuli led to the upregulation of Ugcg with a peak expression of 2-6 hrs following TLR agonist exposure. All stimuli led to a decrease in the expression of B4galt6 mRNA over the first 8 hrs of TLR agonist exposure (Supplementary Fig. 11b). Only minimal changes were seen in the expression of genes responsible for degradation of β-GlcCer and β-LacCer, Gba and Glb1 (data not shown). The LPS concentration used for generation of the dataset above was 100 ng/ml, which is substantially higher than we have found is required to stimulate iNKT cells in co-culture with BMDC. Using 1 ng/ml LPS, we determined the expression of the genes involved in β-GlcCer metabolism by qPCR, and again observed rapid upregulation of Ugcg and concomitant downregulation of B4galt6 (Fig. 7a). TLC analysis of polar lipid extracts from CD11c+ BMDC following LPS-treatment revealed an increase in the amount of β-GlcCer (Fig. 7b), quantified by densitometric analysis (Fig. 7c, with additional controls show in Supplementary Fig. 12a,b). Naturally-occurring β-GlcCer runs as a doublet by TLC in the utilized solvent system, and the potent antigen β-GlcCer C24:1 migrates in the accumulating upper band (Supplementary Fig. 12c).
Figure 7.
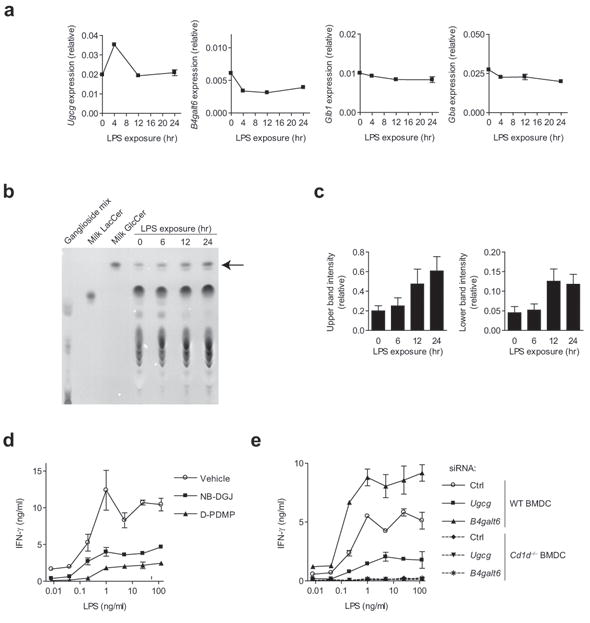
A role for β-GlcCer in the iNKT cell response to LPS-exposed BMDC. (a) CD11c+ BMDC were exposed to 1 ng/ml LPS for the indicated period of time, and the expression of genes involved in β-GlcCer metabolism was analyzed by qPCR. Expression data relative to Gapdh representative of three separate experiments is presented, error being the s.e.m. among triplicate samples. (b) TLC analysis of polar lipid extracts from LPS-treated CD11c+ BMDC, with the relative mobility of β-GlcCer marked by an arrow. (c) Densitometic analysis for the upper and lower β-GlcCer TLC bands, with error reported as s.d. of band intensity. (d) IFN-γ production by an iNKT cell line co-cultured with CD11c+ BMDC in the presence of LPS and the indicated inhibitor of β-GlcCer synthesis. (e) siRNA silencing was performed in CD11c+ BMDC, and after 48 hrs, BMDC were harvested and co-cultured with a primary iNKT cell line in the presence of LPS. ELISA data are presented as mean and range in duplicate wells. For all data in this figure, three independent experiments were performed.
Having demonstrated that β-GlcCer accumulates in BMDC after TLR-agonist stimulation, we next asked whether this lipid contributes to iNKT cell activation. We confirmed that, as has been previously shown for TLR agonists9-10, inhibition of GSL synthesis reduced the response of iNKT cells to LPS-exposed BMDC (Fig 7d). To isolate the β-GlcCer contribution to this GSL-dependent activation, we utilized siRNA silencing. We found that Ugcg silencing limited the iNKT cell response to LPS-treated BMDC, while B4galt6 silencing enhanced this response (Fig. 7e). By TLC analysis, a decrease in β-GlcCer levels was observed following NB-DGJ, D-PDMP, and Ugcg silencing, while an increase in β-GlcCer was seen following B4galt6 silencing (Supplementary Fig. 13). We could not detect changes in LPS-mediated IL-12 production or BMDC maturation, as assessed by flow cytometry following NB-DGJ, D-PDMP, siRNA targeting of Ugcg or B4galt6 as compared to control. D-PDMP did, however, lead to a slight decrease in CD1d surface levels on CD11c+ BMDC, potentially contributing to the decreased iNKT cell activation seen following treatment of BMDC with this inhibitor (Supplementary Fig. 14). Alteration in some lipid synthesis pathways has been shown to alter endosomal or lysosomal function, subsequently reducing iNKT cell activation16. For this reason, we used confocal fluorescence microscopy to assess the endosomal and lysosomal systems in CD11c+ BMDC following β-GlcCer synthesis inhibitor or siRNA treatment. No morphological abnormalities were observed in the endosomal or lysosomal systems (Supplementary Fig. 15). We concluded from these studies that the presentation of β-GlcCer by CD1d provides a substantial component of the TCR-mediated activation signal provided to iNKT cells by BMDC following TLR-agonist exposure.
Since bacteria might contain iNKT cell lipid antigens in addition to TLR-agonists, we investigated the relative functional contribution of the self-antigen β-GlcCer as an activating self-lipid antigen in the activation of iNKT cells by whole bacteria using a gene silencing approach. Ugcg silencing reduced the activation of iNKT cells in response to BMDC co-cultured with Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pneumonia, and Listeria monocytogenes (Fig. 8a). As was seen following LPS exposure in BMDC, B4galt6 silencing did not reduce iNKT cell activation. These results suggest that β-GlcCer contributes to iNKT cell activation during bacterial infection.
Figure 8.
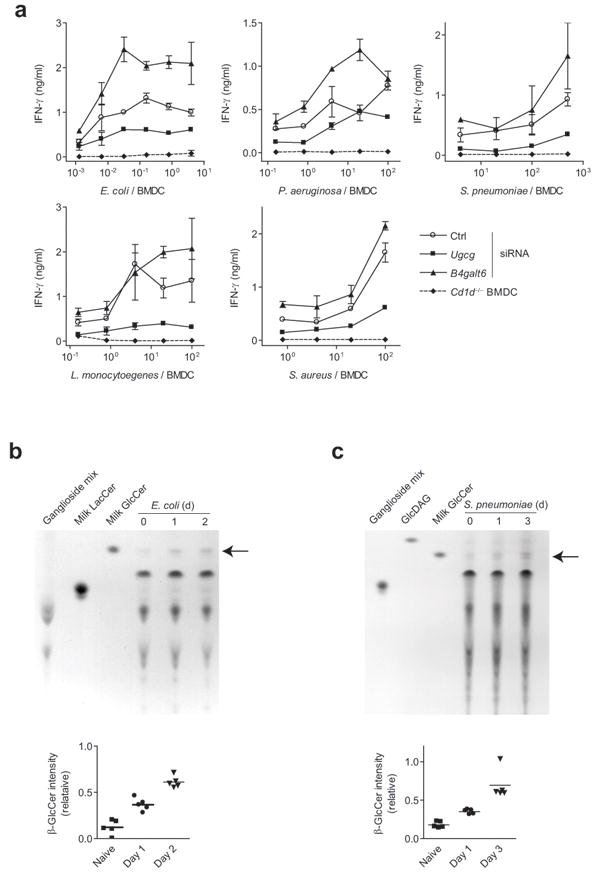
β-GlcCer contributes to microbial activation of iNKT cells. (a) A primary iNKT cell line was co-cultured with Ugcg, B4galt6, or control siRNA-silenced CD11c+ BMDC in the presence of heat-killed bacteria, and activation was assessed by IFN-γ production. ELISA data are presented as mean and range in duplicate wells, and are representative of three separate experiments. (b) Mice were infected with E. coli intravenously, and spleens harvested on days 1 and 2. Polar lipid extracts were extracted and analyzed by TLC. (c) TLC analysis of whole lung lipid extracts from days 1 and 3 following intranasal S. pneumoniae infection. A TLC solvent system that allowed separation of bacterial GlcDAG from β-GlcCer was utilized. The relative mobility of β-GlcCer is marked by an arrow. Densitometric quantification of β-GlcCer bands is shown, and each point represents data obtained from an individual mouse.
To extend the physiological significance of β-GlcCer accumulation in BMDC following LPS exposure, we next examined β-GlcCer levels in vivo during infection. As a model of gram-negative bacterial sepsis, we transferred E. coli intravenously and assessed β-GlcCer levels in the spleen. By 24 hrs following E. coli injection, β-GlcCer levels increased as a portion of total polar lipids, and remained elevated at 48 hrs (Fig. 8b). As a model of gram-positive bacterial infection, we investigated S. pneumoniae pulmonary infection, a model in which iNKT cells have been shown to play a prominent role11, 36. Because S. pneumoniae produces α-glucosyldiacylglycerol (GlcDAG), a lipid that co-migrates with β-GlcCer by TLC in some solvent systems, we modified our solvent system to separate these two lipids, and included GlcDAG as a lipid standard. As seen in the spleen following E. coli infection, we observed an increase in β-GlcCer levels in total lung polar lipid extracts of S. pneumoniae-infected mice (Fig. 8c). This increase was prominent by day 3, corresponding to iNKT cell activation in this model11. We concluded from these studies that β-GlcCer accumulates in involved organs following bacterial infection.
Discussion
Although iNKT cells are considered to be innate lymphocytes, they utilize machinery of the adaptive immune system to express TCRs of limited diversity. The nature of the specificity of these invariant TCRs, and how this specificity might regulate activation of an innate lymphocyte population, is fundamental to understanding the increasingly appreciated role of innate lymphocytes in immunity. Because iNKT cells are activated in contexts where foreign lipid antigens would not always be present to provide cognate TCR-mediated signals, self-lipid antigens have been proposed to participate in their activation. Identification of the involved self-lipid antigens has remained a central question in the field33. We now find that β-GlcCer, the simplest GSL, acts as a physiologically relevant self-lipid antigen for iNKT cells.
We demonstrate activity for both naturally-occurring and synthetic β-GlcCer in vitro using mouse iNKT cell hybridomas, mouse and human primary iNKT cell lines, and freshly-isolated human iNKT cells. By injecting β-GlcCer intravenously, we show activity in vivo on unperturbed mouse iNKT cells. β-GlcCer also activates in an APC-free system, using plate-bound, lipid-loaded CD1d. As an additional important proof of principle, β-GlcCer-loaded CD1d tetramers specifically stain a subset of iNKT cells from both mouse and human directly ex vivo, providing strong evidence that the iNKT cell TCR can directly bind β-GlcCer C24:1-loaded CD1d complexes. Based on these observations, we conclude that β-GlcCer activates both mouse and human iNKT cells via a direct cognate interaction between the iNKT cell TCR and β-GlcCer-loaded CD1d.
In mammals, GSLs exhibit diverse structures, varying in both fatty acyl chain and carbohydrate head group. Examination of each of these structural features for β-GlcCer provides important insight into the critical determinants of iNKT cell self-antigenicity. We found clear evidence that the activity of β-GlcCer varies with N-acyl chain structure. Such differences also have been observed for α-GalCer variants, and have been attributed to the kinetics, stability, and sub-cellular location of antigen loading23-24, 37. Importantly, β-GlcCer C24:1, the most potent β-GlcCer variant tested for iNKT cells, is the specific form found in mammalian lymphoid tissues in highest abundance.
Various approaches have been taken in the field to clarify the contribution of the lipid head group to iNKT cell activation. Two informative studies have looked at the role of the iNKT TCR CDR3β loop in iNKT cell autoreactivity. Matulis et al. demonstrated that naturally-occurring, high-affinity human iNKT cell TCR interactions with CD1d were dependent on the TCR CDR3β loop, and that β-GlcCer-loaded tetramers, similar to OCH-loaded tetramers, could discriminate between high and low affinity iNKT cell TCRs32. Mallevaey et al. studied a particularly autoreactive Vα14-Vβ6 iNKT TCR that was generated by random mutagenesis of mouse CDR3β, and showed that multiple cellular lipids could influence the binding of this TCR to CD1d, either positively or negatively. While several higher order GSLs reduced binding relative to unloaded tetramer, β-GlcCer and β-GalCer did not have this effect29. These studies, while not specifically implicating monohexosyl ceramides as an iNKT self-antigens, are consistent with the notion that β-GlcCer might support iNKT cell TCR binding to CD1d for autoreactive iNKT cells.
Two groups recently compared CD1d–β-GSL–TCR co-crystal structures with the previously-reported CD1d–α-GalCer–TCR structure38, and surprisingly, the primary β-linked sugar was found to be ‘molded’ by the TCR into a configuration similar to that seen with α-GalCer, thus providing a structural explanation for the antigenic activity of β-linked lipids39-40. In addition to iGB3, other antigenic primary β-linked GSLs have been previously described, including β-mannosylceramide, and β-GalCer C12:0 26-27, 41. Taken together with our data, these reports suggest that CD1d-bound, β-linked monohexosyl ceramides can fulfill the structural requirements of a self-antigen for an iNKT cell TCR. The ‘energetic penalty’ incurred with altering the conformation of a monohexosyl ceramide bound to CD1d to adopt a topology similar to that of α-GalCer is likely to be lower than that required for more complex GSLs, and our data suggest that the acyl chain composition of the lipid may also play a role in this process.
In addition to fulfilling structural requirements for antigenicity, a physiologically relevant self-antigen must be present at sites of iNKT cell activation. As with iGB3, β-GalCer and β-mannosylceramide would not be found in any significant amount at most sites of peripheral iNKT cell activation. We show that β-GlcCer C24:1, on the other hand, is easily detectable in lymphoid tissues, accumulates during infection, and therefore is likely to play a physiological role. This is of particular importance, since elution studies have demonstrated that CD1d presents lipids representative of the total cellular lipid profile or compartment surveyed21, 42-43.
Several lines of evidence have suggested the possibility that GSLs contribute to iNKT cell self-reactivity, although GSLs may not be the only lipid class contributing to iNKT cell autoreactivity25. Interestingly, one group has reported an iNKT cell-dependent immunomodulatory or inhibitory role for β-GlcCer in vivo 44. Using GSL synthesis inhibitors, previous studies have indirectly implicated GSLs as being important iNKT cell antigens in BMDC following TLR-agonist stimulation9-10, 20. Our identification of a specific antigenic β-GSL, β-GlcCer, allowed us to target the pathways involved in the synthesis of that lipid. Silencing Ugcg, the gene encoding β-GlcCer synthase, reduced both self-reactivity and the response of iNKT cells to BMDC in the presence of LPS or whole bacteria. Targeting B4galt6, the major enzyme involved in the conversion of β-GlcCer to β-LacCer, increased self-reactivity as well as the response of iNKT cells to LPS and some bacteria, likely a result of the observed accumulation of β-GlcCer. Our siRNA silencing results, controlled for antigen presentation, IL-12 production, APC activation status, and endosomal morphology, strongly suggest that β-GlcCer plays a significant role in the CD1d-dependent signal in APC during many infections. Moreover, β-GlcCer has been reported to accumulate in spleen, serum, and liver of LPS-exposed rodents45, suggesting that endosomal uptake of systemically circulating β-GlcCer by APC could provide another source of antigen. Thus, the induction of GSL biosynthesis, both in antigen presenting cells and systemically, provides a mechanism for danger-sensing by iNKT cells, mediated by β-GlcCer.
Two signals play an important role in iNKT cell activation during microbial infection, the first being a lipid antigen presented by CD1d to the iNKT cell TCR, and the second an inflammatory cytokine, such as IL-12. We now propose that β-GlcCer, an accumulating self-antigen following APC activation, provides a major TCR signal for iNKT cells. The ability of an iNKT cell to be activated by the integration of APC-dependent innate signals explains how these cells, with an invariant TCR, can be activated in multiple pathologic contexts in the absence of foreign lipid antigens. The recognition of the self-antigen β-GlcCer by an invariant TCR provides a clear example of the translation of an innate danger signal using the machinery of the adaptive immune system, a mechanism that may apply to other innate T lymphocytes.
Methods
Mice and human subjects
C57Bl/6 and BALB/c mice were from Jackson Laboratories. Cd1d−/− mice on a C57Bl/6 background were kindly provided by M. Exley (Beth Israel Deaconess Medical Center, Boston, MA). Animal studies were approved by the Dana-Farber Cancer Institute Animal Care and Use Committee. Human peripheral blood was obtained from healthy donors in accordance with Brigham & Women’s Hospital IRB approval.
Mouse in vitro NKT cell assay
All in vitro co-culture assays were performed in 96-well flat-bottom plates for 14-18 hrs. The iNKT cell hybridoma DN32, and CD1d-transfected RAW macrophage cells have been previously described21, 46, and were used at 5 × 104 each per well. The generation of CD11c+ BMDC and primary iNKT cell lines have also been previously described11, 47. iNKT cells were used at 2.5 - 5 × 104 per well, in a 5:1 ratio with BMDC unless otherwise noted. IL-12 was from Peprotech. LPS from Salmonella abortus equi was from Sigma-Aldrich. Heat-killed bacteria preparation and strains used have been previously described11. N-butyldeoxygalactonojirimycin (NB-DGJ, Calbiochem) was used at 50 μM. D-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (D-PDMP, Matreya) was used at 10 μM.
Human in vitro iNKT cell assay
Human iNKT cell clones have been previously described31. Human primary iNKT cell lines were generated by expanding freshly isolated PBMC in 50 U/ml IL-2 (Novartis) and 5 ng/ml IL-15 (Peprotech) in culture with 10 ng/ml α-GalCer for 14 days. iNKT cells were purified by PBS-57-loaded CD1d tetramer selection with magnetic beads (Miltenyi Biotec), and were > 99% CD3+PBS-57-tet+. Generation of human PBMC-derived monocytes has been previously described31. 5 × 104 iNKT cells were cultured with 5 × 104 PBMC-derived monocytes per well.
Intravenous injection of lipids
Lipids for injection were dried under nitrogen and sonicated in PBS pH 7.4, 0.05% Tween-20, and were injected in 200 μl via the tail vein. For analysis of cytokine elaboration, mice were euthanized 1 hr after injection for α-GalCer, and 2 hrs after injection for all other lipids. Livers were harvested after perfusion with ice-cold PBS, mechanically dissociated, and passed through a 70 μm filter before isolation of mononuclear cells by density centrifugation over Histopaque (Sigma-Aldrich).
In vivo infection
E. coli (ATCC stain 25922) was injected intravenously at 6 × 103 CFU / mouse, as determined by limiting dilution on LB agar plates. S. pneumoniae (stain URF918) was provided by K. Kawakami, and growth and intranasal infection has been previously described11. For S.pneumoniae infection, 1.5 × 103 CFU per mouse was used, as determined by limiting dilution on Todd-Hewitt agar plates containing 5% sheep red blood cells.
Antibodies and flow cytometry
Data was acquired using a FACSCanto II (BD Biosciences) and analyzed using FlowJo (Treestar). Doublets were excluded using FSC-A and FSC-H linearity. Anti-mouse antibodies, BD Bioscienes: TCRβ (H57-597), CD3 molecular complex (17A2), CD1d (1B1), Vβ2(B20.6), Vβ6(RR4-7), Vβ7(TR310), Vβ8.1/8.2(MR5-2), Vβ8.3(1B3.3), Vβ9(MR10-2), Vβ10b(B21.5), Vβ14(14-2), IFN-γ (XMG1.2), and CD3ε (145-2C11). Anti-human antibodies: CD3ε (UCHT1, BD Biosciences), Vα24 and Vβ11 (C15 and C21, Immunotech). For ELISA, anti-mouse IFN-γ and IL-4 (BD Biosciences), and anti-human IFN-γ (Pierce) antibody sets were used. Monoclonal anti-CD1d (42.1) has been previously described48, and mouse IgG1 isotype control was from BD Bioscienes. Human and mouse PBS-57-loaded CD1d tetramers were from the NIH tetramer facility. Mouse IFN-γ and IL-4 cytokine capture assays were performed according to the manufacturer’s instructions (Miltenyi Biotec). β-GlcCer tetramer binding was performed at room temperature for 30 min in PBS, 0.5%FBS, 2mM EDTA followed by the addition of PBS-57-loaded tetramer and lineage markers for 10 min, and washing prior to sample acquisition.
CD1d loading and tetramer assembly
Mouse and human biotinylated CD1d were from the NIH tetramer facility. For loading, lipids were dried under nitrogen, sonicated in 0.05% Tween-20 for mouse CD1d, or 0.025% Triton X-100 for human CD1d, and incubated with CD1d overnight at 37 °C. Mock-loaded, biotinylated BSA (Sigma-Aldrich) was used as a control for plate-bound CD1d assays. For plate-bound CD1d assay, a 50:1 molar loading ratio, and for tetramerization studies, a 200:1 molar ratio of lipid:CD1d was used. For tetramerization, streptavidin-phycoerythrin (Invitrogen) was added in a 1:4.5 molar ratio to lipid-loaded CD1d. For plate-bound CD1d assays, 0.25 μg of loaded CD1d was added to each well of a 96 well streptavidin-coated plate (Thermo Scientific), bound at 25 °C for 30 min, and washed extensively before adding iNKT cells.
Lipids
α-GalCer, OCH, Gal–α-GalCer, and GlcDAG were produced as previously described24, 49. β-D-GlcCer d18:1-C24:1(15Z), C18:1 (9Z), C18:0, C16:0, C12:0, and C8:0 as well as β-D-GalCer d18:1-C24:1(15Z), C12:0, and phospholipids were from Avanti Polar Lipids. iGB3 (d18:1-C26:0) was from Enzo Life Sciences. Sphingomonas GSL-1 was provided by the NIH tetramer facility. All other lipids were from Matreya, LLC.
TLC and mass spectrometry
Lipid extraction and analysis by TLC has been previously described50. Lipid fractions were run on silica TLC plates (EMD chemicals) and visualized with α-naphthol. For the majority of TLCs, the mobile phase was 60:30:6; CHCl3:CH3OH:H2O (v/v/v). For discrimination of GlcDAG and β-GlcCer, the mobile phase was 65:25:3.7; CHCl3:CH3OH: H2O (v/v/v). Densitometry of TLC plates was performed using Adobe Photoshop. The relative intensity value for each query band was calculated compared to a standard band, and normalized to the total intensity of each lane. Densitometric quantification of β-GlcCer in spleen, thymus, and BMDC polar lipid extracts was performed by extrapolation from a best fit line to a β-GlcCer standard curve. MS analysis including low-energy CAD MSn was performed on a linear ion-trap (LIT) mass spectrometer (Thermo Finnigan) with Xcalibur operating system, as previously described11.
qPCR analysis
RNA was extracted using the RNeasy system (Qiagen), and cDNA was synthesized with the Quantitect system (Qiagen). qPCR was performed using the Brilliant SYBR Green qPRC Master Mix (Agilent technologies) and the Stratagene MX3000P system. Primer sequences are listed in Supplementary information.
siRNA silencing
Pooled siRNA against Ugcg, B4galt6, and control siRNA were from Thermo Scientific. 1 × 106 CD11c+ sorted BMDC were nucleofected with 1 μM siRNA using the Nucleofector II device, program Y-001 (Lonza). After nucleofection, BMDC were rested for 24-48 hrs prior to use, in low-adherence Sumilon cell-tight culture dishes (Sumitomo Bakelite).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) R01AI063428. P.J.B was supported by NIH T32AI007306. E.Y.K. was supported by NIH T32HL007633. M.B. was supported by NIH K08AI077795. S.D.G. and J.P.S. were supported by the Higher Education Funding Council for England. F.H. was supported by NIH grants (P41-RR00954, P30 DK56341, and P60-DK20579). G.S.B. acknowledges support in the form of a Personal Research Chair from J. Bardrick, Royal Society Wolfson Research Merit Award, Medical Research Council, and The Wellcome Trust (084923/B/08/Z) for funding. NMR analysis was supported in part by NIH-funded Research Resource for Integrated Glycotechnology (NIH 5-P41-RR05351) to P. Azadi at the Complex Carbohydrate Research Center. We thank H. Kim for assistance with flow cytometry. We appreciate the ongoing support from the NIH tetramer facility.
Footnotes
Author contributions: P.J.B. and R.V.V.T. conceived of, performed, and interpreted data from experiments. P.J.B. primarily wrote the manuscript. M.B., A.T., F.H., J.P.S., S.D.G. and E.Y.K. assisted with experimental design and data interpretation, performed experiments, and edited the manuscript. G.S.B. assisted with design of experiments and synthesized critical materials. M.B.B assisted with design of experiments and data interpretation, supervised research, and significantly contributed to the manuscript.
References
- 1.Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. 2004;114:1379–1388. doi: 10.1172/JCI23594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawano T, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 4.Kinjo Y, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol. 2006;7:978–986. doi: 10.1038/ni1380. [DOI] [PubMed] [Google Scholar]
- 5.Kinjo Y, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- 6.Mattner J, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- 7.Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol. 2003;4:1230–1237. doi: 10.1038/ni1002. [DOI] [PubMed] [Google Scholar]
- 8.Nagarajan NA, Kronenberg M. Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol. 2007;178:2706–2713. doi: 10.4049/jimmunol.178.5.2706. [DOI] [PubMed] [Google Scholar]
- 9.Paget C, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity. 2007;27:597–609. doi: 10.1016/j.immuni.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Salio M, et al. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc Natl Acad Sci U S A. 2007;104:20490–20495. doi: 10.1073/pnas.0710145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brigl M, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med. 2011 doi: 10.1084/jem.20102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox LM, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol. 2009;7:e1000228. doi: 10.1371/journal.pbio.1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gumperz JE, et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity. 2000;12:211–221. doi: 10.1016/s1074-7613(00)80174-0. [DOI] [PubMed] [Google Scholar]
- 14.Zhou D, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science. 2004;306:1786–1789. doi: 10.1126/science.1103440. [DOI] [PubMed] [Google Scholar]
- 15.Porubsky S, et al. Normal development and function of invariant natural killer T cells in mice with isoglobotrihexosylceramide (iGb3) deficiency. Proc Natl Acad Sci U S A. 2007;104:5977–5982. doi: 10.1073/pnas.0611139104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gadola SD, et al. Impaired selection of invariant natural killer T cells in diverse mouse models of glycosphingolipid lysosomal storage diseases. J Exp Med. 2006;203:2293–2303. doi: 10.1084/jem.20060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, et al. Immunologic glycosphingolipidomics and NKT cell development in mouse thymus. J Proteome Res. 2009;8:2740–2751. doi: 10.1021/pr801040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Speak AO, et al. Implications for invariant natural killer T cell ligands due to the restricted presence of isoglobotrihexosylceramide in mammals. Proc Natl Acad Sci U S A. 2007;104:5971–5976. doi: 10.1073/pnas.0607285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christiansen D, et al. Humans lack iGb3 due to the absence of functional iGb3-synthase: implications for NKT cell development and transplantation. PLoS Biol. 2008;6:e172. doi: 10.1371/journal.pbio.0060172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stanic AK, et al. Defective presentation of the CD1d1-restricted natural Va14Ja18 NKT lymphocyte antigen caused by beta-D-glucosylceramide synthase deficiency. Proc Natl Acad Sci U S A. 2003;100:1849–1854. doi: 10.1073/pnas.0430327100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muindi K, et al. Activation state and intracellular trafficking contribute to the repertoire of endogenous glycosphingolipids presented by CD1d [corrected] Proc Natl Acad Sci U S A. 2010;107:3052–3057. doi: 10.1073/pnas.0915056107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu FF, Turk J. Structural determination of glycosphingolipids as lithiated adducts by electrospray ionization mass spectrometry using low-energy collisional-activated dissociation on a triple stage quadrupole instrument. J Am Soc Mass Spectrom. 2001;12:61–79. doi: 10.1016/S1044-0305(00)00194-X. [DOI] [PubMed] [Google Scholar]
- 23.Wun KS, et al. A molecular basis for the exquisite CD1d-restricted antigen specificity and functional responses of natural killer T cells. Immunity. 2011;34:327–339. doi: 10.1016/j.immuni.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu KO, et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci U S A. 2005;102:3383–3388. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pei B, et al. Diverse endogenous antigens for mouse NKT cells: self-antigens that are not glycosphingolipids. J Immunol. 2011;186:1348–1360. doi: 10.4049/jimmunol.1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ortaldo JR, et al. Dissociation of NKT stimulation, cytokine induction, and NK activation in vivo by the use of distinct TCR-binding ceramides. J Immunol. 2004;172:943–953. doi: 10.4049/jimmunol.172.2.943. [DOI] [PubMed] [Google Scholar]
- 27.Parekh VV, et al. Quantitative and qualitative differences in the in vivo response of NKT cells to distinct alpha- and beta-anomeric glycolipids. J Immunol. 2004;173:3693–3706. doi: 10.4049/jimmunol.173.6.3693. [DOI] [PubMed] [Google Scholar]
- 28.Cardell S, et al. CD1-restricted CD4+ T cells in major histocompatibility complex class II-deficient mice. J Exp Med. 1995;182:993–1004. doi: 10.1084/jem.182.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mallevaey T, et al. T cell receptor CDR2 beta and CDR3 beta loops collaborate functionally to shape the iNKT cell repertoire. Immunity. 2009;31:60–71. doi: 10.1016/j.immuni.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson MT, et al. The response of natural killer T cells to glycolipid antigens is characterized by surface receptor down-modulation and expansion. Proc Natl Acad Sci U S A. 2003;100:10913–10918. doi: 10.1073/pnas.1833166100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brigl M, et al. Conserved and heterogeneous lipid antigen specificities of CD1d-restricted NKT cell receptors. J Immunol. 2006;176:3625–3634. doi: 10.4049/jimmunol.176.6.3625. [DOI] [PubMed] [Google Scholar]
- 32.Matulis G, et al. Innate-like control of human iNKT cell autoreactivity via the hypervariable CDR3beta loop. PLoS Biol. 2010;8:e1000402. doi: 10.1371/journal.pbio.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gapin L. iNKT cell autoreactivity: what is ‘self’ and how is it recognized? Nat Rev Immunol. 2010;10:272–277. doi: 10.1038/nri2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prigozy TI, et al. Glycolipid antigen processing for presentation by CD1d molecules. Science. 2001;291:664–667. doi: 10.1126/science.291.5504.664. [DOI] [PubMed] [Google Scholar]
- 35.Amit I, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawakami K, et al. Critical role of Valpha14+ natural killer T cells in the innate phase of host protection against Streptococcus pneumoniae infection. Eur J Immunol. 2003;33:3322–3330. doi: 10.1002/eji.200324254. [DOI] [PubMed] [Google Scholar]
- 37.Im JS, et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity. 2009;30:888–898. doi: 10.1016/j.immuni.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borg NA, et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature. 2007;448:44–49. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- 39.Pellicci DG, et al. Recognition of beta-linked self glycolipids mediated by natural killer T cell antigen receptors. Nat Immunol. 2011 doi: 10.1038/ni.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu ED, Girardi E, Wang J, Zajonc DM. Cutting Edge: Structural Basis for the Recognition of {beta}-Linked Glycolipid Antigens by Invariant NKT Cells. J Immunol. 2011 doi: 10.4049/jimmunol.1101636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Konek JJ, et al. Mouse and human iNKT cell agonist beta-mannosylceramide reveals a distinct mechanism of tumor immunity. J Clin Invest. 2011;121:683–694. doi: 10.1172/JCI42314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox D, et al. Determination of cellular lipids bound to human CD1d molecules. PLoS One. 2009;4:e5325. doi: 10.1371/journal.pone.0005325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan W, Kang SJ, Evans JE, Cresswell P. Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol. 2009;182:4784–4791. doi: 10.4049/jimmunol.0803981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ilan Y. Alpha versus beta: are we on the way to resolve the mystery as to which is the endogenous ligand for natural killer T cells? Clin Exp Immunol. 2009;158:300–307. doi: 10.1111/j.1365-2249.2009.04030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Memon RA, et al. Regulation of sphingolipid and glycosphingolipid metabolism in extrahepatic tissues by endotoxin. J Lipid Res. 2001;42:452–459. [PubMed] [Google Scholar]
- 46.Lantz O, Bendelac A. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med. 1994;180:1097–1106. doi: 10.1084/jem.180.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiba A, et al. Rapid and reliable generation of invariant natural killer T-cell lines in vitro. Immunology. 2009;128:324–333. doi: 10.1111/j.1365-2567.2009.03130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Exley M, et al. CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology. 2000;100:37–47. doi: 10.1046/j.1365-2567.2000.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veerapen N, et al. Synthesis and biological activity of alpha-galactosyl ceramide KRN7000 and galactosyl (alpha1-->2) galactosyl ceramide. Bioorg Med Chem Lett. 2009;19:4288–4291. doi: 10.1016/j.bmcl.2009.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tatituri RV, et al. Inactivation of Corynebacterium glutamicum NCgl0452 and the role of MgtA in the biosynthesis of a novel mannosylated glycolipid involved in lipomannan biosynthesis. J Biol Chem. 2007;282:4561–4572. doi: 10.1074/jbc.M608695200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.