

Abstract

Near-IR oxazine dyes are reported that contain sulfonate esters which are rapidly cleaved by esterase activity to unmask highly polar anionic sulfonates. Strategies for the synthesis of these dyes included the development of milder dye condensation conditions with improved functional compatibility, and the use of an alkyl halide that allows for the introduction of esterase-labile sulfonates without the need for sulfonation of the target molecule.
Cells and organisms are most transparent to near-IR light (650-900 nm), making this region of the electromagnetic spectrum optimal for optical imaging.1 Unfortunately, most organic near-IR dyes are not suitable for live intracellular applications. Dyes that are capable of diffusing across cell membranes typically give high background staining of cells because they accumulate in intracellular membranes and organelles.2 Moreover, most near-IR dyes are cationic, which favors accumulation in the negatively-polarized membranes of mitochondria.3 On the other hand, highly polar water-soluble sulfonated near-IR dyes (e.g., Alexa and Cy dyes) are unable to diffuse across cell membranes.
One potential strategy to overcome this problem is to design cell-permeable near-IR dyes that can be selectively unmasked to a more water-soluble form upon cellular entry. Toward this end, we recently reported a chemically-stable but esterase-labile sulfonate ester, AcOTFMB (Figure 1), that enables the delivery and unmasking of dansyl sulfonate within the cytoplasm of cells.4
Figure 1. Esterase-labile protecting groups for sulfonates.

Yellow-green fluorescent AcOTFMB dansyl sulfonate esters are stable to nucleophilic attack due to the electron-withdrawing properties of the trifluoromethyl group. The acetoxy trigger can be readily removed by esterase activity. Subsequent rapid 1,6-elimination affords the blue-fluorescent dansyl sulfonate anion.
Most sulfonate esters are potent electrophiles. Exceptions include neopentyl sulfonates (steric block of reactivity) and α-trifluoromethylated sulfonates (electronic deactivation).5 We have utilized these classes of sulfonate esters as platforms for the design of protecting groups that can be removed under specific conditions. In particular, we found that the incorporation of an acetoxy group into trifluoromethylbenzyl (TFMB) sulfonate esters maintained stability to nucleophiles but rendered these groups highly labile to esterase activity (Figure 1).4
While this approach was facile for the small dansyl fluorophore, it was unclear whether the AcOTFMB group could be incorporated into near-IR dyes, which are considerably larger and more challenging to synthesize. Here we report the successful synthesis of oxazine near-IR fluorophores bearing AcOTFMB esterase-labile sulfonate esters. We demonstrate that these dyes are chemically stable but are readily cleaved to the free sulfonate by esterase activity, while the corresponding TFMB esters are unaffected.
Examples of sulfonated near-IR fluorophores include cyanines,6 oxazines,7 and some rhodamine8 and BODIPY9 derivatives. Among these dye classes, near-IR oxazines have the advantages of a compact structure, excitation and bright fluorescence in the 650-700 nm region,10 and high photostability.11 To determine whether the AcOTFMB protection strategy was amenable to this class of near-IR fluorophores, we first synthesized the sulfonated oxazine dye 22 as shown in Scheme 1. Reaction of m-anisidine with acetone to form the dihydroquinoline 1 was facile in the presence of 2 mol % In(OTf)3.12 This product was then N-methylated and sulfonated to afford 3.7a Diazonium coupling yielded 4, and subsequent acid-catalyzed condensation of this arylazo compound with a m-aminophenol13 yielded the sulfonated dye 22, which exhibits maximal absorption at 673 nm and emission at 689 nm in phosphate-buffered saline (PBS).
Scheme 1.

Synthesis of a sulfonated oxazine dye
With this sulfonated near-IR fluorophore in hand, the next synthetic challenge was determining how and when to incorporate the TFMB and AcOTFMB sulfonate esters. Our initial attempts to directly introduce TFMB and AcOTFMB sulfonate esters into 22 by formation and reaction of the sulfonyl chloride were unsuccessful. We therefore installed the sulfonate esters into the dye precursors (Scheme 2). Thus, the sodium sulfonate salt 3 was converted into the allylic sulfonyl chloride 5 and subsequently treated with the corresponding alcohol and Et3N at 0°C to afford the sulfonate esters 6 and 7.
Scheme 2.

Incorporation of TFMB and AcOTFMB sulfonate esters into an oxazine dye
The sulfonate esters 6 and 7 were stable to diazonium coupling conditions in acidic aqueous methanol to afford 8 and 9. Subsequent acid-catalyzed condensation of 8 with N-ethyl-7-hydroxy-tetrahydroquinoline yielded the desired TFMB-protected sulfonated oxazine 23. However, the standard oxazine dye-formation conditions of HCl in hot aqueous ethanol led to substantial deprotection of AcOTFMB, presumably because carboxylic esters are labile to these conditions. Gratifyingly, dye formation in hot acetic acid readily afforded the desired oxazine dye 24 with the AcOTFMB sulfonate ester intact (Scheme 2).
To facilitate the synthesis of more dyes containing a protected sulfonate, we constructed TFMB and AcOTFMB-protected iodopropyl sulfonates 11 and 13 (Scheme 3). Treatment of 3-chlorosulfonyl chloride with the corresponding alcohols and Et3N at 0°C yielded the respective chloropropyl sulfonates 10 and 12. The chlorides were then selectively displaced with iodides. These compounds are especially notable because 1) their synthesis clearly demonstrates the remarkable stability of TFMB and AcOTFMB sulfonates to nucleophilic attack (chloro is displaced selectively even after overnight reflux with excess iodide), and 2) the iodide 13 can allow the introduction of an AcOTFMB-protected sulfonate into molecules with a suitable nucleophile, avoiding the need for sulfonation.
Scheme 3.

Synthesis of iodopropyl sulfonates.
To make use of 11 and 13 for the construction of near-IR dyes, we synthesized sulfonated analogs of the oxazine dye MR12114 (Scheme 4). The tetrahydroquinoline 15 was dissolved in DMF and heated with 11 or 13 in the presence of excess K2CO3 to yield 18 and 19. Diazonium coupling followed by acid-catalyzed condensation with N-ethyl-7-hydroxy-tetrahydroquinoline yielded the desired oxazine dyes 26 and 27. The corresponding free sulfonate dye was synthesized analogously by treatment of 15 with 1,3-propanesultone to afford 16 which was subsequently elaborated to yield the sulfonated dye 25 (Scheme 4). The excitation and emission wavelengths for this dye are similar to MR121, at 659 and 671 nm respectively.
Scheme 4.

Synthesis of sulfonated derivatives of MR121.
We next attempted to synthesize symmetrical oxazine dyes with two protected sulfonates, which would generate highly polar anionic molecules upon cleavage. Current synthetic approaches to such dyes require a phenolic intermediate such as 29 (Scheme 5).7,10a,13,14a While the synthesis of the TFMB-protected sulfonate ester 29 was facile, we were unable to isolate the corresponding AcOTFMB-protected phenolic intermediate (Scheme 5). Because the corresponding TFMB-protected phenol 29 is stable, we surmise that the phenol promotes intramolecular deacetylation and self-immolation of the AcOTFMB group (Scheme 5). Indeed, the corresponding AcOTFMB-protected anisole 19 is stable and readily isolated. Thus, derivatives of AcOTFMB bearing intramolecular nucleophiles may be prone to self-immolation.
Scheme 5.

Intramolecular nucleophiles can cause self-immolation of AcOTFMB sulfonates.
In order to access symmetrical bis-sulfonated dyes without the need for the isolation of a phenolic intermediate such as 29, we constructed the silyl ether 30. We hypothesized that the AcOTFMB group would be stable in the presence of the silyl ether, and that the acidic conditions of dye synthesis would unmask the silyl ether and allow dye formation (Scheme 6). We were gratified to find that the silyl ether 32 was indeed stable, and that the silyl ethers 31 and 32 readily formed the desired symmetric bis-sulfonated oxazine dyes 33 and 34 when treated with 20 and 21 under acidic conditions (Scheme 6).
Scheme 6.
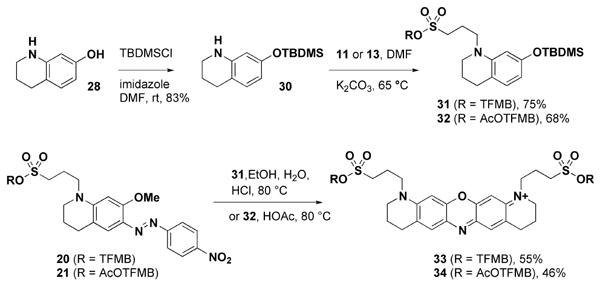
Bypassing phenols: formation of bis-sulfonated oxazine dyes from silyl ether and anisole starting materials.
This method of oxazine dye synthesis has three key advantages over the classical methods that utilize condensation of one or more phenolic compounds. 7,10a,13,14a First, during the N-alkylation step, protection of the phenol avoids any undesired O-alkylation (Scheme 6), which improves yields and simplifies purification. Second, there is no need to deprotect prior to dye synthesis, eliminating a synthetic step. Finally, the use of silyl ethers expands the range of functionality that is compatible with oxazine dye synthesis. Typically, anisoles have been used for protection of phenolic intermediates in dye synthesis,7,10a,14a and the requirement for HBr or BBr3 deprotection greatly limits functional group compatibililty. We anticipate that this method of dye synthesis will also find use in the preparation of rhodamine dyes bearing sensitive chemical functionality.
The AcOTFMB-protected sulfonated oxazine dyes 24 and 27 are readily unmasked by pig liver esterase (PLE) to afford the zwitterionic free sulfonate dyes 22 and 25, respectively (Figure 2). Similarly, treatment of 34 with PLE rapidly formed the highly polar bis-sulfonate (Figure S1). In contrast, TFMB-protected dyes are stable to esterase activity (Figure S1), and both TFMB and AcOTFMB dyes are stable to treatment with biological nucleophiles such as 5 mM glutathione and 1 mg/mL ovalbumin (Figure 2, Figure S1). These findings are consistent with our results with dansyl dyes,4 and suggest that the AcOTFMB group is generally suitable as a chemically-stable, esterase-labile protecting group for dyes containing either aryl, allyl, or alkyl sulfonates.
Figure 2.

AcOTFMB-oxazine dyes 24 and 27 were incubated in PBS (P), 5 mM glutathione (G), 1 mg/mL ovalbumin (O) or 1U/mL pig liver esterase (E) for 30 minutes, then separated by TLC (10% MeOH/CH2Cl2) and imaged. The respective free sulfonate dyes 22 and 25 are shown for reference.
We have described complex near-IR fluorophores that contain the esterase-labile sulfonate protecting group AcOTFMB. Moreover, intermediates such as 7, 21, and 32 are basic building blocks for the incorporation of esterase-labile sulfonates into oxazines and other dyes such as rhodamines, and intermediate 13 can allow the incorporation of the esterase-labile AcOTFMB sulfonate into an even wider variety of molecules. Further optimization of this approach and cell-based applications of these molecules are currently underway.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM087460). We thank Anjan Bhunia (formerly University of Massachusetts Medical School) for preliminary work on oxazine dye synthesis.
Footnotes
Supporting Information Available Experimental procedures and full spectroscopic data for all new compounds.
References
- 1.Weissleder R. Nat Biotechnol. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 2.(a) Johnson I. Histochem J. 1998;30:123–140. doi: 10.1023/a:1003287101868. [DOI] [PubMed] [Google Scholar]; (b) Cunningham CW, Mukhopadhyay A, Lushington GH, Blagg BSJ, Prisinzano TE, Krise JP. Molecular Pharm. 2010;7:1301–1310. doi: 10.1021/mp100089k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunting JR, Phan TV, Kamali E, Dowben RM. Biophys J. 1989;56:979–993. doi: 10.1016/S0006-3495(89)82743-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rusha L, Miller SC. Chem Commun. 2011;47:2038–2040. doi: 10.1039/c0cc04796a. [DOI] [PubMed] [Google Scholar]
- 5.Miller SC. J Org Chem. 2010;75:4632–4635. doi: 10.1021/jo1007338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Bioconjugate Chem. 1993;4:105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zilles A, Arden-Jacob J, Drexhage KH, Kemnitzer NU, Hamers-Schneider M. US 2006/0179585 A1. 2006; (b) Toutchkine A. WO 2009/152024 A1. 2009
- 8.Kolmanov K, Belov VN, Bierwagen J, Ringemann C, Muller V, Eggeling C, Hell SW. Chem Eur J. 2010;16:158–166. doi: 10.1002/chem.200902309. [DOI] [PubMed] [Google Scholar]
- 9.Niu SL, Ullrich G, Ziessel R, Kiss A, Renard PY, Romieu A. Org Lett. 2009;11:2049–2052. doi: 10.1021/ol900302n. [DOI] [PubMed] [Google Scholar]
- 10.(a) Hintersteiner M, Enz A, Frey P, Jaton AL, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold KH, Gremlich HU. Nature Biotech. 2005;23:577–583. doi: 10.1038/nbt1085. [DOI] [PubMed] [Google Scholar]; (b) Wakata A, Lee HM, Rommel P, Toutchkine A, Schmidt M, Lawrence DS. J Am Chem Soc. 2010;132:1578–1582. doi: 10.1021/ja907226n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eggeling C, Widengren J, Brand L, Schaffer J, Felekyan S, Seidel CAM. J Phys Chem A. 2006;110:2979–2995. doi: 10.1021/jp054581w. [DOI] [PubMed] [Google Scholar]
- 12.Theoclitou ME, Robinson LA. Tetrahedron Lett. 2002;43:3907–3910. [Google Scholar]
- 13.Kanitz A, Hartmann H. Eur J Org Chem. 1999:923–930. [Google Scholar]
- 14.(a) Herrmann R, Josel HP, Drexhage KH, Marx NJ. EP 757447. 1996; (b) Lieberwirth U, Arden-Jacob J, Drexhage KH, Herten DP, Muller R, Neumann M, Schultz A, Siebert S, Sagner G, Klingel S, Sauer M, Wolfrum J. Anal Chem. 1998;70:4771–4779. doi: 10.1021/ac980230k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.