

Abstract
A major conclusion from comparative genomics is that many sequences that do not code for proteins are conserved beyond neutral expectations, indicating that they evolve under the influence of purifying selection and are likely to have functional roles. Due to the degeneracy of the genetic code, synonymous sites within protein-coding genes have previously been seen as “silent” with respect to function and thereby invisible to selection. However, there are indications that synonymous sites of vertebrate genomes are also subject to selection and this is not necessarily because of potential codon bias. We used divergence in ancestral repeats as a neutral reference to estimate the constraint on 4-fold degenerate sites of avian genes in a whole-genome approach. In the pairwise comparison of chicken and zebra finch, constraint was estimated at 24–32%. Based on three-species alignments of chicken, turkey, and zebra finch, lineage-specific estimates of constraint were 43%, 29%, and 24%, respectively. The finding of significant constraint at 4-fold degenerate sites from data on interspecific divergence was replicated in an analysis of intraspecific diversity in the chicken genome. These observations corroborate recent data from mammalian genomes and call for a reappraisal of the use of synonymous substitution rates as neutral standards in molecular evolutionary analysis, for example, in the use of the well-known dN/dS ratio and in inferences on positive selection. We show by simulations that the rate of false positives in the detection of positively selected genes and sites increases several-fold at the levels of constraint at 4-fold degenerate sites found in this study.
Keywords: chicken, turkey, zebra finch, 4-fold degenerate sites, purifying selection, nearly neutral theory, comparative genomics
Introduction
Before detailed studies of genetic variation at the DNA and protein levels were possible, a common view held that most mutations in the genetic material have an effect on fitness (Dobzhansky 1970). As a consequence, they were thought to either relatively quickly reach fixation by positive selection or become removed from the population by negative selection. This view was challenged in the 1960s by the observation of significant within-species polymorphism (Hopkinson et al. 1963; Spencer et al. 1964; Lewontin and Hubby 1966), indicating that some of the variation in the genome might be more or less neutral with respect to fitness. Sparked in part by such data and armed with a mathematical approach based on diffusion equations to derive theoretical arguments, this soon led Kimura to develop the neutral theory of molecular evolution (Kimura 1968), a model positing that genetic drift of neutral alleles is an important driving force in evolution. In parallel, it became increasingly clear that a large fraction of the genome appears nonfunctional and is thereby potentially shielded from selection. DNA was found to consist of much else than genes, which were found to consist of exons and introns, and cracking the genetic code revealed that some positions within exons were “silent” with respect to which amino acid is encoded.
The historical perspective briefly sketched out above is of relevance for the development of our current view on genome composition and molecular evolution. To some extent, the shift in focus of the 1960s and 1970s, from natural selection being thought to have a prevailing role to acknowledging that neutral processes affect parts of the genome, is corroborated and substantiated by recent genomic data. This is particularly so, given Ohta's extension of Kimura's model to the nearly neutral theory of molecular evolution (Ohta 1973) and the many clear examples that slightly deleterious mutations will effectively behave as neutral at low effective population sizes (Wright and Andolfatto 2008; Ellegren 2009). However, in parallel, there has also been an opposite trend. One of the most important conclusions from comparative genomics is that many regions of the genome previously considered nonfunctional show evidence of sequence conservation beyond neutral expectations (reviewed by, e.g., Dermitzakis et al. 2005). This suggests that several other sequence categories than those directly coding for proteins are functional and subject to selection. The identification of the numerous ultra-conserved elements found in intergenic regions previously seen as genomic “deserts” in vertebrate genomes (Bejerano et al. 2004; Katzman et al. 2007) are an example of a transition in the perception of the genome and how it evolves.
Due to the degeneracy of the genetic code, synonymous substitutions are candidates to represent neutral changes. Accordingly, 4-fold degenerate sites have traditionally been seen as essentially free of selective constraint (Eyre-Walker and Keightley 1999; Nachman and Crowell 2000), at least in mammals where effective population sizes are often low and where mutations with a small effect on fitness should be expected to behave as neutral. The 4-fold degenerate sites have therefore been used as a neutral reference both in studies of constraint at nonsynonymous sites and in noncoding sequences. However, there is evidence that at least some silent sites are constrained also in mammals (Chamary et al. 2006). Importantly, two recent genome-wide studies have reported significant levels of constraint at 4-fold degenerate sites of mammalian genes. Eőry et al. (2010) obtained estimates of 22–27% in hominids and 11–12% in murids. Pollard et al. (2010) used alignments of 36 mammalian genomes to estimate a mean constraint in the mammalian phylogeny of 25%. In both these studies, divergence at ancestral repeats (ARs) was used as a neutral reference against which divergence at synonymous sites was contrasted. To widen the knowledge of selection at synonymous sites in vertebrates, we here estimate constraint at 4-fold degenerate sites of avian genes. We use data from three available bird genomes (chicken, turkey, and zebra finch) and find high levels of constraint: 24–43%. Selection on synonymous sites thus seems to be a ubiquitous feature of vertebrate genomes.
Material and Methods
Sequence Data and Divergence Estimates
Protein-coding sequences for 1:1 orthologs of chicken and zebra finch and for 1:1:1 orthologous of chicken, turkey, and zebra finch were downloaded from BioMart (Ensembl 61) (http://biomart.org) and, in both cases, aligned using MAFFT 6.716b (Katoh et al. 2009). Pairwise chicken-zebra finch alignments of intronic sequences and three species EPO alignments of intronic sequences, including information about intron coordinates, were downloaded from Ensembl (Version 61). Due to alternative splicing, some introns are (partly) annotated as exonic sequences and intronic sites annotated as exons were removed. Repeats were masked using RepeatMasker 3.2.9 (Smit et al. 2010), and all alignments were then cleaned using Gblocks 0.91b (Castresana 2000) with a minimal block length of 30 bases and a maximum number of eight contiguous nonconserved positions. Chicken annotations (Ensembl 61) were used to identify intronic transposable elements for the chicken–zebra finch comparison, and we defined AR as elements present in orthologous positions of chicken and zebra finch introns. For the three-species comparison of chicken, turkey, and zebra finch, we defined AR as elements present in orthologous positions of introns of all three species, again using chicken repeat annotations.
We masked all CpG dinucleotides by excluding sites preceded by cytosine (C) or followed by guanine (G) (CpG-prone sites, see Keightley and Gaffney 2003); all divergence estimates reported herein are thus non-CpG divergences. Divergences were estimated using a general time-reversible model with a gamma distribution of variable rate among sites (REV model of baseml from the PAML package version 4.4b; Yang 2007). For two-species alignments, we estimated divergence in the different sequence categories (0-fold and 4-fold degenerate sites, ARs, and introns) for each gene separately and then obtained the genome-wide mean based on these estimates. For the three-species alignments where fewer genes were available, we concatenated all gene sequences and obtained a genome-wide divergence estimate from the concatenated data. However, in order to be able to test for a correlation between divergence and gene expression parameters in chicken, we also estimated divergence at 4-fold degenerate sites for each gene separately in the three-species alignments. To reduce the risk of incorrectly inferred orthology and to avoid saturation problems, genes with a total divergence d > 1.8 were excluded in the pairwise comparison, and genes exceeding a divergence of 0.9 in at least one branch were excluded from further analysis in the three-species comparison.
Pairwise estimates of dN and dS for each gene between chicken and zebra finch were taken from Nam et al. (2010).
Chicken polymorphisms derived from genome resequencing of pools of unrelated individuals were obtained from (Rubin et al. 2010). In the absence of available allele frequency estimates for these data, we used the density of single nucleotide polymorphisms (SNPs; number of SNPs per base pair) as a measure of polymorphism level.
Estimates of Selective Constraint
Selective constraint was estimated using an approach introduced by Kondrashov and Crow (1993) and extended by Eyre-Walker and Keightley (1999). For the pairwise alignments with divergence estimates obtained for individual genes, we used the formula:
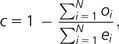 |
where N is the number of genes analyzed, oi is the observed divergence at tested sequence category for gene i (in most cases 4-fold degenerate sites) and ei is the expected divergence obtained from the divergence estimated for AR of gene i. For three-species alignments, we used the genome-wide divergence estimates obtained from concatenated sequences to directly estimate constraint as 1 − o/e. Note that we only included genes for which data on both divergence at 4-fold degenerate sites and at one or more intronic AR were available. This selection was applied to exclude the possibility of differences in rate and pattern of nucleotide substitution between regions with and without intronic AR affecting the estimates of constraint. Weighted estimates of constraint were obtained by a method similar to the approach of Halligan and Keightley (2006). For estimating the weighting factor, we divided the number of alignable non-CpG AR sites by the number of all non-CpG AR sites. Weighted constraints were estimated dividing the original constraint estimates by the weighting factor.
Simulations of Tests for Positive Selection
To evaluate the impact of constraint on tests for positive selection, we simulated sequences using a branch-site model of evolution using Evolver from the PAML package (Yang 2007). The data were generated using the tree shown in Supplementary figure 1 and with two types of selection schemes. In the first, we simulated two classes of sites, one evolving under w1 = 0.2 in background branches (black lineages in Supplementary figure 1) and w2 = 1.0 in foreground branch (gray lineage) and the other with a constant w = 0.2 in all the lineages. In the second scheme, the first class had w1 = 0.1 in background branches and w2 = 2.0 in the foreground branch, whereas the second class had a constant w = 0.1 in all the lineages. In both schemes, we allocated 20% of the sites to the first class and 80% to the second.
To mimic constraint acting on synonymous sites, we ignored a fraction (0.25, 0.35, or 0.45) of the synonymous substitutions simulated. This was done by comparing the simulated sequences with the ancestral sequences—provided by Evolver—generated during simulation. Each time a synonymous substitution was identified, it was replaced by the ancestral state with a probability equal to the constraint. In the case of two substitutions per codon, we considered all the mutational paths between the two codons, except those leading to a stop codon. For simplification, we considered only one substitution randomly in case of three substitutions per codon and did not consider multiple substitutions at the same site. The process was repeated for internal lineages in order to apply the same constraint throughout the phylogenetic tree. For each simulation scheme, we simulated 200 data sets of 1,000 codons with and without constraint acting on synonymous sites and applied the branch-site likelihood test of positive selection (Zhang et al. 2005). We repeated simulations with three different codon frequency spectra obtained for chicken–turkey–zebra finch orthologous alignments chosen to represent GC-poor, GC-average, and GC-rich genes.
Gene Expression and Codon Usage
Median-subtracted arcsinh expression data from 20 different chicken tissues were taken from Chan et al. (2009). Gene-wise expression breadth (τ) was estimated following Yanai et al. (2005):
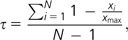 |
where N is the number of tissues, xi, the expression intensity for gene x in tissue i, and xmax, the maximum expression intensity of gene x across all tissues. For analyses of gene expression level, we used xmax. As a measure for codon bias, we calculated the Codon Adaptation Index (CAI; Sharp and Li 1987) obtained for chicken protein-coding sequences using CODONW (http://codonw.sourceforge.net).
Statistical Analyses
Statistical analyses were performed using R version 2.13.1 (R Development Core Team 2008). If not stated otherwise, nonparametric bootstrapping (1,000 iterations on concatenated sequences) was used to estimate confidence intervals for divergence and level of selective constraint.
Results
Estimates of Constraint at 4-Fold Degenerate Sites in the Chicken–Zebra Finch Comparison
We identified 13,245 chicken–zebra finch 1:1 orthologs and among these 3,772 genes had at least one intronic AR element. After alignment, filtering, and removal of CpG-prone sites, the data set could broadly be defined as composed of 2.97 million 0-fold degenerate sites, 0.34 million 4-fold degenerate sites, 7.94 million bp of intronic AR, and 58.19 million bp of non-AR intron sequence. Divergence in these three categories of sequence classes was lowest among 0-fold degenerate sites and highest in AR (fig. 1). Divergence at 4-fold degenerate sites and at 0-fold sites was significantly different from AR divergence (Mann–Whitney U test, P < 0.001 for both comparisons). If we use AR as a neutral reference to estimate constraint in the other sequence categories, 0-fold degenerate sites show an 86.7% (±0.6%) and 4-fold degenerate sites a 24.2% (±1.6%) constraint. Intronic divergence was similar to the AR divergence.
FIG. 1.—

Estimated sequence divergence of ARs, introns, 4-fold degenerated sites, and 0-fold sites in the chicken–zebra finch comparison estimated gene by gene. ***Denotes significantly lower divergence in comparison to ARs (P < 0.001).
Compared with the noncoding regions, the high degree of sequence conservation within the coding sequence and the ability to project DNA sequences onto alignments of protein sequences greatly reduce the frequency of gaps and the need for filtering of those regions difficult to align. However, filtering of regions that are difficult to align within a presumably neutral sequence may lead to the divergence being underestimated because the rapidly evolving sites can be excluded from the analysis. One way to handle this potential problem is to weight estimated constraint by the proportion of sequence removed in the neutral reference. Using a procedure similar to Halligan and Keightley (2006) (for details, see Material and Methods), we obtained a weighted estimate of constraint of 31.5% (±2.0%) for the 4-fold degenerate sites.
Previous work in several different organisms (Lercher et al. 2004; Webster et al. 2004), including birds (Axelsson et al. 2005), has demonstrated regional consistency in nucleotide substitution rates, suggested to reflect regional mutation rate variation. This can also be seen in our data with a significant correlation in divergence between the neighboring genes, both for divergence at 4-fold degenerate sites (Breusch–Godfrey serial correlation LM test, ut = 26.8599, degree of freedom [df] = 1, P < 0.001) and for divergence in ARs (ut = 5.9362, df = 1, P = 0.015). However, we found no evidence for the clustering of genes in the genome with similar levels of constraint at 4-fold degenerate sites (ut = 0.193, df = 1, P = 0.66) nor did the constraint at 0-fold sites correlate with the genomic location (ut = 0.0148, df = 1, P = 0.90). Thus, selection does not display regional variation over the long evolutionary time scale analyzed (whereas such effects are expected over short time scales, due to the background selection and selective sweeps).
Lineage-Specific Estimates of Constraint in the Chicken–Turkey–Zebra Finch Comparison
Using three-species alignments of available avian genomes (chicken, turkey, and zebra finch) allows for lineage-specific estimates of divergence and also for polarizing substitutions onto lineages by parsimony principles. In addition, the evolutionary distance between the two galliforms, chicken and turkey, is considerably shorter than that between chicken and zebra finch, which should facilitate alignment and make divergence estimates more accurate. We identified 9,531 1:1:1 orthologs among the three species, and for 1,667 of these, there was at least one intronic AR present.
Table 1 reports the estimated divergences of AR and 4-fold degenerate sites in the chicken, turkey, and zebra finch lineages. Note that in the unrooted tree of the three species, the lineage from the split between chicken and turkey to zebra finch (“the zebra finch lineage”) includes the basal galliform branch, the short Galloanserae and Neoaves internal branches, the basal passeriform branch and the terminal branch leading to zebra finch (Supplementary figure 2). The trends were similar to those obtained for the chicken–zebra finch comparison with AR evolving more rapidly than 4-fold degenerate sites (table 1). The estimated constraint for 4-fold degenerate sites was 43.1% (±1.6%), 28.8% (±1.8%), and 24.2% (±1.4%) in the chicken, turkey, and zebra finch lineages, respectively. Weighting the estimated constraints to take regions difficult to align in AR into account increases the estimates to 49.9% (±1.9%), 33.4% (±2.1%), and 28.0% (±1.6%), respectively. These estimates are generally higher than what we obtained in the more distant chicken—zebra finch pairwise comparison.
Table 1.
Lineage-Specific Divergence (Mean ± Standard Deviation) of Different Sequences Categories Estimated from Concatenated Three-Species Alignments of Chicken, Turkey, and Zebra Finch
| 4-Fold Sites | AR | |
| Chicken | 0.028 (±0.001) | 0.049 (±0.001) |
| Turkey | 0.038 (±0.001) | 0.054 (±0.001) |
| Zebra finch | 0.302 (±0.005) | 0.399 (±0.004) |
Note.—The lineages are from an unrooted tree of the three species.
Chicken Polymorphisms
An analysis of within-species polymorphism essentially circumvents potential problems related to the varying confidence by which different sequence categories can be aligned in distant evolutionary comparisons. It also more directly pinpoints the selective constraints of sequence evolution currently in place. Based on genomic resequencing of pools of chicken population samples, Rubin et al. (2010) gathered genome-wide data on the location of 7 million of SNPs. We calculated the mean density of SNPs (i.e., the number of segregating sites divided by the length of sequence analyzed) in different sequence categories and found that 0-fold degenerate sites had the lowest incidence (0.0039 ± 0.0001), followed by 4-fold degenerate sites (0.0058 ± 0.0001) and AR (0.0080 ± 0.0002). The density of SNPs in AR was significantly higher than in the other categories (two-sided Mann–Whitney U test, P < 0.001 in all cases). The level of polymorphism at 4-fold degenerate sites in chicken was 28% lower than in AR.
Effect of Constraint at 4-Fold Degenerate Sites on dN/dS and Tests for Positive Selection
Constraint at 4-fold degenerate sites has implications for the interpretation of selection from the dN/dS ratio and for tests of positive selection. To illustrate this, we compared estimates of dN/dS and dN/dAR for the 3,772 genes in the chicken–zebra finch comparison (dAR being divergence at ARs). On average, dN/dS estimates (mean 0.133 ± 0.142) were about 20% higher than dN/dAR estimates (0.109 ± 0.157; two-tailed Mann–Whitney U test, P < 0.001). At the level of individual genes, dAR was higher than dS for 72% of the genes. As expected from the relatively low number of available sites per gene and the associated stochastic influence on divergence estimates, the difference between dAR and dS was largest for genes with high estimates of dAR (fig. 2).
FIG. 2.—

Gene-by-gene differences between divergence estimates of AR and synonymous sites. The dashed horizontal line marks where estimates of dS and dAR are equal. Values below the line are genes where divergence at AR is estimated higher than divergence at synonymous sites (and vice versa for values above the line). The red line denotes the lowess curve.
It is conceivable that selective constraint at 4-fold degenerate sites can affect inferences on positive selection. Specifically, if 4-fold degenerate sites are constrained, the proportion of nonsynonymous sites interpreted to evolve more rapidly than the presumed neutral reference should be elevated, potentially leading to an increased rate of false positives in the detection of positively selected sites and genes. To investigate this, we simulated sequence evolution under different levels of constraint at 4-fold degenerate sites and applied a standard maximum likelihood test of positive selection (PAML, branch-site model). The constraint levels (25%, 35%, and 45%) were chosen to reflect the range of estimates obtained in this study. For simulations without positive selection (simulations 1–3 in table 2), the proportion of genes detected as positively selected (i.e., false positives) increased from 3–9% without constraint to 14–33% with constraint. Moreover, the frequency of positively selected sites increased from 0.4–0.6% to 0.6–7.9%. Simulations letting a fraction of sites evolve under positive selection (see Material and Methods) found similar effects. For example, at a level of 35% constraint at 4-fold degenerate sites, the proportion of genes detected as positively selected increased from 47–54% to 76–90% and the frequency of positively selected sites increased from 4.2–7.4% to 8.1–8.8%.
Table 2.
Simulation Results for the Proportion of Significant Likelihood Ratio Tests (LRT) for Positive Selected Genes and for the Number of Positively Selected Sites with Constraint (Denoted by “Constr.”) and without Constraint (Denoted by “No con.”)
| Simulation | GC-Content | Positive Selection Simulated | Proportions of Significant LRT |
Mean Number of Positively Evolving Sites |
||||||||||
| 25% Constraint |
35% Constraint |
45% Constraint |
25% Constraint |
35% Constraint |
45% Constraint |
|||||||||
| No Con. | Constr. | No Con. | Constr. | No Con. | Constr. | No Con. | Constr. | No Con. | Constr. | No Con. | Constr. | |||
| 1 | Low | No | 0.09 | 0.14 | 0.04 | 0.16 | 0.05 | 0.33 | 5.5 | 6 | 5 | 33 | 5 | 79.5 |
| 2 | Average | No | 0.04 | 0.14 | 0.05 | 0.17 | 0.06 | 0.3 | 6 | 6 | 5 | 25.5 | 4.5 | 74 |
| 3 | High | No | 0.08 | 0.15 | 0.09 | 0.22 | 0.03 | 0.32 | 6 | 6 | 6 | 13 | 6.5 | 77 |
| 4 | Low | Yes | 0.53 | 0.71 | 0.47 | 0.86 | 0.51 | 0.96 | 56.5 | 71 | 59.8 | 85.8 | 53.3 | 90.1 |
| 5 | Average | Yes | 0.49 | 0.73 | 0.49 | 0.76 | 0.45 | 0.94 | 74 | 77 | 74 | 81 | 74 | 86 |
| 6 | High | Yes | 0.53 | 0.74 | 0.54 | 0.9 | 0.52 | 0.96 | 39.5 | 70 | 42.5 | 88 | 50.5 | 96 |
Note.—Results were obtained from simulating 200 data sets of 1,000 codons with and without constraint acting on synonymous sites and applied the branch-site likelihood test of positive selection as implemented in PAML. LRT, Likelihood Ratio Tests.
As expected, the rate of false positives increased with increasing constraint. In simulations without positive selection, the proportion of genes identified as positively selected was on average 2.2 times higher at 25% constraint on 4-fold degenerate sites than when such sites were unconstrained. For levels of 35% and 45% constraint, this increased to 3.5 and 7.8 times higher, respectively. For identification of positive sites, the frequency was 1.0, 4.6, and 14.7 times higher at 25%, 35%, and 45% constraint, respectively. Under the more realistic scenario of some sites evolving under positive selection, the proportion of genes identified as positively selected was 1.4, 1.7, and 1.9 times higher at 25%, 35%, and 45% constraint, respectively. The corresponding numbers for positively selected sites were 1.4, 1.5, and 1.6 times the higher frequencies.
Divergence and 4-Fold Degenerate Sites and Gene Expression
In order to search for possible causative correlates of constraint at 4-fold degenerate sites, we considered variables related to gene expression: expression level, expression breadth, and codon usage. Microarray gene expression data is available from chicken so we focused on divergence in the chicken lineage using the three-species data set. However, divergence at 4-fold degenerate sites was not correlated to any of the variables tested (gene expression level: Pearson r = 0.0008, P = 0.98; breadth of gene expression (τ): r = −0.0262, P = 0.48; CAI: r = 0.0210, P = 0.57).
Discussion
The main conclusion from this study is that 4-fold degenerate sites of avian genes evolve under significant constraint, at least when constraint is estimated using divergence in ARs as a neutral reference. This conclusion is supported both by data on intraspecific polymorphism and interspecific divergence. In the relatively distant comparison of chicken and zebra finch, which are estimated to have diverged 90 mya (van Tuinen and Hedges 2001), we obtained constraint estimates of 24–32%. The split between chicken (super order Galloanserae) and zebra finch (Neoaves) lineages represents the most basal divergence within Neognathae (Supplementary figure 2), a group that contains >99% of all extant bird species. The deep split of the lineages investigated could suggest that the estimated constraint constitutes a representative average for birds. However, it should be noted that contemporary birds are classified in some 25 orders, most of which originated around or soon after the K/T boundary (i.e., there are probably only short internal branches, in particular within Neoaves; Hackett et al. 2008; Pacheco et al. 2011). The lineages that we sampled thus only constitute a minor part of evolution among modern birds.
When using three-species alignments of chicken, turkey, and zebra finch, we found constraint for 4-fold degenerate sites to be the highest in the chicken lineage (43%) followed by the turkey (29%) and the zebra finch lineages (24%). This rank order might be seen as surprising if one considers that selection against slightly deleterious mutations should be more efficient in large populations, that is, the level of constraint should correlate positively with the effective population size, Ne (Ohta 1973). Although passeriforms (zebra finch) are typically small and short-lived birds, galliforms (chicken and turkey) are large and long-lived, and it is clear that Ne of natural populations of passeriforms are typically larger than that of galliforms. As a consequence, selection should be more efficient and constraint higher in the former than in the latter. However, as mentioned above, the zebra finch lineage in the unrooted tree of chicken, turkey, and zebra finch includes several internal branches, including basal Galliformes, so the large population size of passeriforms is unlikely to be representative for the entire zebra finch lineage in the unrooted tree.
Moreover, just as functional elements within noncoding DNA turn over during evolution (Smith et al. 2004; Siepel et al. 2005; Pheasant and Mattick 2007), with the consequence of the amount of shared functional sequence decreasing with increasing genetic distance (Meader et al. 2010), it is conceivable that the functional importance of individual synonymous sites also changes. We may thus expect estimates of selective constraint to be lower for more distant comparisons. These caveats suggest that the lower estimate of constraint at 4-fold degenerate sites in the zebra finch than in the chicken and turkey lineages should not be taken too far. Yet, it could be noted that Eőry et al. (2010) found constraint at 4-fold degenerate sites to be lower in murids than in hominids, despite the much large effective population sizes of the former than of the latter.
Although comparative genomic studies are powerful in detecting purifying selection from sequence conservation in particular regions or at particular sites, they cannot reveal the underlying functional role of these sequences. However, several selective processes may explain why synonymous sites are constrained (Chamary et al. 2006), including selection for mRNA stability, translational efficiency, and splicing regulation (Rocha 2004; Chamary and Hurst 2005; Parmley and Hurst 2007; Drummond and Wilke 2008). Moreover, there are increasing number of examples where mutations at synonymous sites cause human disease, demonstrating the critical role of such sites (e.g., Brest et al. 2011). We failed to detect a significant correlation between the divergence at 4-fold degenerate sites and either breadth or level of gene expression. Moreover, we did not find a correlation between the divergence and codon usage. There is little evidence for codon usage bias in birds, and the codon adaptation index was not correlated with gene expression level (r = 0.0462, P = 0.21), as would have been expected under the selection for preferred codons in highly expressed genes (Hershberg and Petrov 2008). As selection for codon usage is typically weak (Duret 2002), it may very well be that the relatively low effective population sizes (Ne) of birds means that Nes for codon usage is in the neutral range.
Clearly, any inference of selective constraint in a particular sequence category is only relative to a presumed neutral reference. Do ARs represent the “ideal” neutral reference? A possible confounding factor is the homogenizing effects on sequence evolution of events of nonallelic gene conversion. Such events are known to occur among transposable elements although their incidence is highest among young repeat elements with high sequence similarity (Aleshin and Zhi 2010). It may thus not be an issue for ARs present in distantly related genomes, like those of chicken and zebra finch. Another factor would be positive or negative selection at individual elements that have attained functional roles, for example, because of exonization (Schwartz et al. 2009; Shen et al. 2011). There is increasing awareness of the importance of Alu elements as gene regulators during human evolution, however, for birds there has been no such documentation. Indeed, avian genomes are low in repeat numbers (Hillier et al. 2004), and there is no prominent occurrence of short interspersed elements (SINE) (like mammalian Alu elements). A third aspect relates to the fact that transposable elements tend to be hypermethylated as a host defense mechanism against transcription and further spread of repeat elements (Yoder et al. 1997). This will clearly affect the substitution rate at CpG sites due to the strong tendency for cytosines at CpG sites to be methylated and replaced by thymine upon spontaneous deamination (e.g., Holliday and Grigg 1993). Divergence at AR-CpG sites should thus be higher than at many other CpG sites even though they evolve neutrally. However, methylation status does not appear to affect the substitution rate at non-CpG sites (Mugal and Ellegren 2011), which is the rate we studied herein. Overall, conclusions reached by recent studies of mammalian genomes suggest that AR currently represent the most appropriate neutral reference for molecular evolutionary analyses (Thomas et al. 2003; Lunter et al. 2006; Eőry et al. 2010; Pollard et al. 2010).
The rationale for studying the strength of selection at nonsynonymous sites by taking the dN/dS ratio, rather than just dN, is that scaling dN by dS will take variation in nonsynonymous divergence due to variation in the underlying mutation rate (supposedly manifested in dS) into account (Hurst 2002). However, a consequence of constrained evolution at 4-fold degenerate sites is that the dN/dS metric will not be a proper measure of the rate of protein evolution. For example, the common inference of neutral evolution when dN/dS = 1 will not be valid. This will clearly need further investigation, not least because the degree of constraint at synonymous sites may very well vary among genes and so also the effect on dN/dS. Related to this, it will be important to address the correlation in constraint between dN and dS. These two parameters are obviously correlated due to mutation rate variation (Wolf et al. 2009); however, the question is if purifying selection adds to the correlation. Moreover, variation in the level of constraint at 4-fold degenerate sites due to variation in Ne or life history will have implications to attempts to evaluate the effects of the same parameters on the rate of protein evolution via dN/dS (cf. Wright and Andolfatto 2008; Ellegren 2009).
One important consequence of constraint at synonymous sites is that it may impede on the identification of positively selected genes and sites. Based on simulations of sequence evolution and inferences of positive selection using PAML, we found a significant increase in the frequency of false positives when constraint at synonymous sites was introduced. For example, at 35% constraint and at a GC-content close to the genomic average, the frequency of falsely identified positively selected genes, as well as falsely identified positively selected sites, increased by a factor of 3–5 under a scenario of no positive selection. Under the same constraint and GC-content and with positive selection introduced, the incidence of false positives increased by a factor of 1.1–1.5. We therefore foresee the need for improved maximum likelihood protocols for detection of positive selection that take into account deviations from neutrality in the sequence category used as a neutral reference.
Divergence at synonymous sites is often used as a measure of genetic distance between species. Another consequence of the observation that 4-fold degenerate sites evolve under constraint is that this divergence will not represent an unbiased distance metric, and this is particularly so if the level of constraint varies among lineages. For example, in a previous study, we estimated genome-wide mean dS in the chicken–zebra finch comparison at 0.42, an estimate that would be recognized as similar to that typically seen among eutherian orders. If we assume that the level of constraint at 4-fold degenerate sites is 30%, then a more unbiased (neutrality-based) distance would be 0.42/0.7 = 0.60.
Supplementary Material
Supplementary figures 1 and 2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
Financial support was obtained from the Swedish Research Council, the European Research Council, and the Knut and Alice Wallenberg Foundation. Useful comments from two anonymous reviewers were appreciated.
References
- Aleshin A, Zhi D. Recombination-associated sequence homogenization of neighboring Alu elements: signature of nonallelic gene conversion. Mol Biol Evol. 2010;27:2300–2311. doi: 10.1093/molbev/msq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Webster MT, Smith NGC, Burt DW, Ellegren H. Comparison of the chicken and turkey genomes reveals a higher rate of nucleotide divergence on micro chromosomes than macro chromosomes. Genome Res. 2005;15:120–125. doi: 10.1101/gr.3021305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- Brest P, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Chamary JV, Hurst L. Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 2005;6:R75. doi: 10.1186/gb-2005-6-9-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 2006;7:98–108. doi: 10.1038/nrg1770. [DOI] [PubMed] [Google Scholar]
- Chan ET, et al. Conservation of core gene expression in vertebrate tissues. J Biol. 2009;8:33. doi: 10.1186/jbiol130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Reymond A, Antonarakis SE. Conserved non-genic sequences—an unexpected feature of mammalian genomes. Nat Rev Genet. 2005;6:151–157. doi: 10.1038/nrg1527. [DOI] [PubMed] [Google Scholar]
- Dobzhansky T. Genetics of the evolutionary process. New York: Columbia University Press; 1970. [Google Scholar]
- Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L. Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev. 2002;12:640–649. doi: 10.1016/s0959-437x(02)00353-2. [DOI] [PubMed] [Google Scholar]
- Ellegren H. A selection model of molecular evolution incorporating the effective population size. Evolution. 2009;63:301–305. doi: 10.1111/j.1558-5646.2008.00560.x. [DOI] [PubMed] [Google Scholar]
- Eőry L, Halligan DL, Keightley PD. Distributions of selectively constrained sites and deleterious mutation rates in the hominid and murid genomes. Mol Biol Evol. 2010;27:177–192. doi: 10.1093/molbev/msp219. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. doi: 10.1038/16915. [DOI] [PubMed] [Google Scholar]
- Hackett SJ, et al. A phylogenomic study of birds reveals their evolutionary history. Science. 2008;320:1763–1768. doi: 10.1126/science.1157704. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD. Ubiquitous selective constraints in the Drosophila genome revealed by a genome-wide interspecies comparison. Genome Res. 2006;16:875–884. doi: 10.1101/gr.5022906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- Hillier LW, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Holliday R, Grigg GW. DNA methylation and mutation. Mutat Res. 1993;285:61–67. doi: 10.1016/0027-5107(93)90052-h. [DOI] [PubMed] [Google Scholar]
- Hopkinson DA, Spencer N, Harris H. Red cell acid phosphatase variants: a new human polymorphism. Nature. 1963;199:969–971. doi: 10.1038/199969a0. [DOI] [PubMed] [Google Scholar]
- Hurst LD. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18:486–487. doi: 10.1016/s0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with mafft. In: Posada D, editor. Bioinformatics for DNA sequence analysis. New York: Humana Press; 2009. pp. 39–64. [DOI] [PubMed] [Google Scholar]
- Katzman S, et al. Human genome ultraconserved elements are ultraselected. Science. 2007;317:915. doi: 10.1126/science.1142430. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Gaffney DJ. Functional constraints and frequency of deleterious mutations in noncoding DNA of rodents. Proc Natl Acad Sci U S A. 2003;100:13402–13406. doi: 10.1073/pnas.2233252100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS, Crow JF. A molecular approach to estimating the human deleterious mutation rate. Hum Mutat. 1993;2:229–234. doi: 10.1002/humu.1380020312. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Chamary JV, Hurst LD. Genomic regionality in rates of evolution is not explained by clustering of genes of comparable expression profile. Genome Res. 2004;14:1002–1013. doi: 10.1101/gr.1597404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC, Hubby JL. A molecular approach to the study of genic heterozygosity in natural populations. II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudoobscura. Genetics. 1966;54:595–609. doi: 10.1093/genetics/54.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Ponting CP, Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol. 2006;2:e5. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meader S, Ponting CP, Lunter G. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res. 2010;20:1335–1343. doi: 10.1101/gr.108795.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugal CF, Ellegren H. Substitution rate variation at human CpG sites correlates with non-CpG divergence, methylation level and GC content. Genome Biol. 2011;12:R58. doi: 10.1186/gb-2011-12-6-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K, et al. Molecular evolution of genes in avian genomes. Genome Biol. 2010;11:R68. doi: 10.1186/gb-2010-11-6-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Pacheco MA, et al. Evolution of modern birds revealed by mitogenomics: timing the radiation and origin of major orders. Mol Biol Evol. 2011;28:1927–1942. doi: 10.1093/molbev/msr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmley JL, Hurst LD. Exonic splicing regulatory elements skew synonymous codon usage near intron-exon boundaries in mammals. Mol Biol Evol. 2007;24:1600–1603. doi: 10.1093/molbev/msm104. [DOI] [PubMed] [Google Scholar]
- Pheasant M, Mattick JS. Raising the estimate of functional human sequences. Genome Res. 2007;17:1245–1253. doi: 10.1101/gr.6406307. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2008. [Google Scholar]
- Rocha EPC. Codon usage bias from tRNA's point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004;14:2279–2286. doi: 10.1101/gr.2896904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin C-J, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- Schwartz S, et al. Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput Biol. 2009;5:e1000300. doi: 10.1371/journal.pcbi.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, et al. Widespread establishment and regulatory impact of Alu exons in human genes. Proc Natl Acad Sci U S A. 2011;108:2837–2842. doi: 10.1073/pnas.1012834108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker open-3.0. 2010. [cited 15 Mar 2011]. Available from: http://www.repeatmasker.org/ [Google Scholar]
- Smith NGC, Brandström M, Ellegren H. Evidence for turnover of functional noncoding DNA in mammalian genome evolution. Genomics. 2004;84:806–813. doi: 10.1016/j.ygeno.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Spencer N, Hopkinson DA, Harris H. Phosphoglucomutase polymorphism in man. Nature. 1964;204:742–745. doi: 10.1038/204742a0. [DOI] [PubMed] [Google Scholar]
- Thomas JW, et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424:788–793. doi: 10.1038/nature01858. [DOI] [PubMed] [Google Scholar]
- van Tuinen M, Hedges SB. Calibration of avian molecular clocks. Mol Biol Evol. 2001;18:206–213. doi: 10.1093/oxfordjournals.molbev.a003794. [DOI] [PubMed] [Google Scholar]
- Webster MT, Smith NGC, Lercher MJ, Ellegren H. Gene expression, synteny, and local similarity in human noncoding mutation rates. Mol Biol Evol. 2004;21:1820–1830. doi: 10.1093/molbev/msh181. [DOI] [PubMed] [Google Scholar]
- Wolf JBW, Künstner A, Nam K, Jakobsson M, Ellegren H. Nonlinear dynamics of nonsynonymous (dN) and synonymous (dS) substitution rates affects inference of selection. Genome Biol Evol. 2009;1:308–319. doi: 10.1093/gbe/evp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Andolfatto P. The impact of natural selection on the genome: emerging patterns in Drosophila and Arabidopsis. Annu Rev Ecol Evol Syst. 2008;39:193–213. [Google Scholar]
- Yanai I, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]