

Abstract
Elevated MPO (myeloperoxidase) levels are associated with multiple human inflammatory pathologies. MPO catalyses the oxidation of Cl−, Br− and SCN− by H2O2 to generate the powerful oxidants hypochlorous acid (HOCl), hypobromous acid (HOBr) and hypothiocyanous acid (HOSCN) respectively. These species are antibacterial agents, but misplaced or excessive production is implicated in tissue damage at sites of inflammation. Unlike HOCl and HOBr, which react with multiple targets, HOSCN targets cysteine residues with considerable selectivity. In the light of this reactivity, we hypothesized that Sec (selenocysteine) residues should also be rapidly oxidized by HOSCN, as selenium atoms are better nucleophiles than sulfur. Such oxidation might inactivate critical Sec-containing cellular protective enzymes such as GPx (glutathione peroxidase) and TrxR (thioredoxin reductase). Stopped-flow kinetic studies indicate that seleno-compounds react rapidly with HOSCN with rate constants, k, in the range 2.8×103–5.8×106 M−1·s−1 (for selenomethionine and selenocystamine respectively). These values are ~6000-fold higher than the corresponding values for H2O2, and are also considerably larger than for the reaction of HOSCN with thiols (16-fold for cysteine and 80-fold for selenocystamine). Enzyme studies indicate that GPx and TrxR, but not glutathione reductase, are inactivated by HOSCN in a concentration-dependent manner; k for GPx has been determined as ~5×105 M−1·s−1. Decomposed HOSCN did not induce inactivation. These data indicate that selenocysteine residues are oxidized rapidly by HOSCN, with this resulting in the inhibition of the critical intracellular Sec-dependent protective enzymes GPx and TrxR.
Keywords: eosinophil peroxidase, glutathione peroxidase, hypothiocyanous acid (HOSCN), myeloperoxidase (MPO), selenium, thiocyanate, thioredoxin reductase
Abbreviations: DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); DTT, dithiothreitol; Fmoc, fluoren-9-ylmethoxycarbonyl; GPx, glutathione peroxidase; GR, glutathione reductase; LPO, lactoperoxidase; MetSeO, methionine selenoxide; MPO, myeloperoxidase; RBC, red blood cell; Sec, selenocysteine; SeMet, selenomethionine; t-BOOH, t-butyl hydroperoxide; TNB, 5-thio-2-nitrobenzoic acid; TrxR, thioredoxin reductase; UPLC, ultra-performance liquid chromatography
INTRODUCTION
Hypochlorous acid (HOCl), hypobromous acid (HOBr) and hypothiocyanous acid (HOSCN) are formed from the MPO (myeloperoxidase)-catalysed reaction of hydrogen peroxide (H2O2) with the halide ions Cl−, Br− and SCN− respectively (reviewed in [1,2]). These hypohalous acids are potent antibacterial agents and critical components of the human immune response against invading pathogens [2]. However, excessive or misplaced production leads to host tissue damage, with this postulated to contribute to a number of inflammatory diseases, including atherosclerosis, asthma, rheumatoid arthritis, cystic fibrosis and some cancers (reviewed in [1,3]). HOCl and HOBr are powerful oxidants that react rapidly with biological molecules, including thiol and amine groups on proteins, unsaturated lipids, antioxidants and DNA [4]. In contrast, HOSCN is less reactive than HOCl and HOBr and considerably more specific, with thiol residues being major targets [5–8]. Previous studies have shown that HOSCN induces a greater extent of apoptosis than HOCl and HOBr in macrophage-like cells [9], but not some types of endothelial cells [10], with this ascribed to specific targeting of critical thiol residues. Absolute rate constants have recently been reported for the reaction of HOSCN with low-molecular-mass thiols, and thiol-containing proteins, with values in the range 103–106 M−1·s−1 [5,6]. These values indicate that HOSCN may be a major initiator of thiol oxidation at sites of inflammation.
The thioredoxin and glutathione peroxidase systems are a large family of enzymes that play a critical role in the maintenance of thiols (both low-molecular-mass, such as GSH, and those on proteins) in their reduced form, and the removal of peroxides (via glutathione peroxidases and peroxiredoxins). These families are therefore critical to cellular protection against oxidative damage.
The thioredoxin system is composed of both thioredoxin and its corresponding NADPH-dependent reductase, TrxR (thioredoxin reductase). A major role of thioredoxins is maintenance of cysteine residues of intracellular proteins in a reduced state via cysteine thiol–disulfide exchange. Of particular importance is the reduction of members of the peroxiredoxin family that remove H2O2 [11]. TrxR has been implicated not only in protection against oxidative injury, but also in cell growth, transformation and the recycling of ascorbate [12]. Inhibition of the thioredoxin system results in increased oxidative stress and apoptosis [13]. GPxs (glutathione peroxidases) catalyse the reduction of hydroperoxides (H2O2, organic hydroperoxides or phospholipid hydroperoxides, depending on the isoform). This family of enzymes uses GSH as a source of reducing equivalents, with consequent oxidation to the disulfide, GSSG, which in turn is reduced by GR (glutathione reductase) at the expense of NADPH [12]. Both TrxR and GPx are seleno-enzymes, with critical Sec (selenocysteine) residues present in the active site [12,14].
Seleno-containing compounds react more rapidly with some oxidants than their sulfur analogues, owing to the low pKa of the Se–H bond (5.2 [15]) which results in this being predominantly ionized at physiological pH values (cf. a pKa of ~8.4 for free cysteine [16]), the protein environment of these residues and the greater nucleophilicity of the selenium atom [17]. It was therefore hypothesized that seleno-compounds would be more rapidly oxidized by hypohalous acids, and particularly HOSCN due to its specificity for thiols, than the corresponding sulfur species (cf. data in [5]) and that this might result in the inhibition of the activity of seleno-containing enzymes. The studies reported in the present paper support this hypothesis, with stopped-flow kinetic analyses indicating that seleno-compounds react very rapidly with HOSCN, and that this can result in inhibition of GPx and TrxR enzymatic activities.
EXPERIMENTAL
Materials
NADPH tetrasodium salt (~98%) was obtained from Roche; DTT (dithiothreitol) was from Astral Scientific; H2O2 (30%, v/v) was from Merck. LPO (lactoperoxidase) (from bovine milk) was from Calbiochem and its concentration was determined by absorbance measurement at 412 nm using ϵ=112000 M−1·cm−1. The diselenide forms of Gly-Sec-Gly and Glu-Sec-Gly were custom synthesized by Auspep. 3,3′-Diselenopropionic acid was synthesized and purified as described previously [18]. The diselenide form of selenocysteine methyl ester [-SeCH2CH(NH2)C(=O)OCH3]2 was kindly donated by Mr C. Storkey and Professor C. Schiesser (University of Melbourne, Melbourne, VIC, Australia). Isolated GPx was from bovine RBCs (red blood cells) (Sigma) and TrxR was from rat liver (Sigma). The isolated GPx used for the kinetic studies was purified by gel filtration to remove DTT present in the commercial preparations, under an atmosphere of N2 to prevent auto-oxidation of the Sec residues. Protein concentrations were determined by the BCA (bicinchoninic acid) assay using BSA standards (0–25 μg). All other chemicals were from Sigma–Aldrich/Fluka and used without further purification. All studies were performed in chelex-treated sodium phosphate buffer (pH 7.4, 100 mM) prepared using Milli Q water. RBC lysates were prepared from blood obtained from healthy human volunteers who gave informed consent. Blood was collected in EDTA tubes and lysed by diluting 5-fold in nanopure water. RBCs were obtained from healthy non-smoking male subjects, into tubes containing protease inhibitors (trypsin inhibitor, aprotinin, D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone) and EDTA. The tubes were immediately centrifuged at 2000 g at 4°C to separate the RBCs from plasma and the buffy coat, which were removed. The RBCs were washed four times by resuspending in 3 volumes of 0.9% NaCl, and spinning at 650 g for 10 min at 10°C. Following the final spin, the RBCs were resuspended at the original haematocrit in isotonic PBS (Amresco) (pH 7.5) and kept at 4°C until use (within 24 h).
HOSCN preparation
HOSCN was prepared by LPO-catalysed reaction of H2O2 with SCN− as described previously [5,9,19]. Briefly, LPO (1.5–2 μM) was incubated with H2O2 (3.75 mM) and NaSCN (7.5 mM) in 10 mM potassium phosphate buffer (pH 6.6) for 15 min at 22°C. Catalase (1 mg·ml−1; from bovine liver) was added to remove unreacted H2O2 followed by filtration by centrifugation at 11300 g for 5 min using a 10 kDa molecular-mass cut-off filter to remove catalase and LPO. The concentration of HOSCN was determined by TNB (5-thio-2-nitrobenzoic acid) assay at 412 nm using ϵ=14150 M−1·cm−1 [20].
Determination of molar absorption coefficients
The diselenide forms of Sec, selenocystamine, 3-selenopropionic acid and selenocysteine methyl ester were reduced to the corresponding selenols using a 2–10-fold molar excess of NaBH4, with the conversion monitored spectroscopically to determine conditions under which complete conversion occurs. After preparation of the reduced form, concentrated stocks were diluted with N2-gassed 100 mM phosphate buffer (pH 7.4) to the desired concentration, and kept under a N2 atmosphere. Molar absorption coefficients were determined using a PerkinElmer Lambda 40 spectrometer relative to a 100 mM phosphate buffer baseline between 200 and 350 nm (at 1 nm interval) at 22°C (n≥11).
Kinetic measurements
Stopped-flow studies were carried out using an Applied Photophysics SX.18MV stopped-flow system as described previously [5,21,22]. Rate constants for the reactions of HOSCN with the selenol (reduced, RSeH) forms of Sec, selenocystamine, 3-selenopropionic acid, selenocysteine methyl ester, and the peptides Gly-Sec-Gly and Glu-Sec-Gly were determined by direct kinetics at the appropriate wavelengths after reduction of the diselenides with a 10-fold excess of NaBH4 under a N2 atmosphere. The reduced selenols were diluted with degassed phosphate buffer to the required concentration (9.9–86.4 μM), with the concentrations determined using their corresponding molar absorption coefficients. The selenols were then reacted with HOSCN (2.5 μM) and the kinetics were measured at 243 nm (Sec, selenocystamine, selenocysteine methyl ester, Gly-Sec-Gly and Glu-Sec-Gly) or 248 nm (3-selenopropionic acid) under a N2 atmosphere. HOSCN was kept as the limiting reagent with at least a 4-fold excess of the substrate, facilitating analysis. The temperature of the sample chamber was maintained at 22°C throughout the experiments, and each stopped-flow measurement was carried out ten times and averaged to improve the signal to noise ratio. The rate constant for the reaction of HOSCN (2.5 μM) with SeMet (selenomethionine) and selenomethylcysteine (10–100 μM) was determined by competition with TNB (10 μM) at 412 nm using kTNB 3.8×105 M−1·s−1 [5].
The reaction of HOSCN with GPx was investigated by competition kinetics using SeMet derivatized with Fmoc (fluoren-9-ylmethoxycarbonyl) (Fmoc–SeMet) in an adaptation of a previous method [22,23]. Oxidation of Fmoc–SeMet to the corresponding selenoxide [Fmoc–MetSeO (methionine selenoxide)] was monitored by UPLC (ultra-performance liquid chromatography) (Shimadzu Nexera system) with fluorescence detection (RF-20Axs detector; λex 265 nm; λem 310 nm). Parent and oxidized samples were separated on a Shim-pack XR-ODS (Shimadzu, 100 mm×4.6 mm, 2.2 μm) column at 40°C with a flow rate of 1.2 ml·min−1 using a gradient of buffer A [50 mM sodium acetate (pH 5.3) in 20% (v/v) methanol with 2.5% (w/v) tetrahydrofuran) and buffer B [50 mM sodium acetate (pH 5.3) in 80% (v/v) methanol with 2.5% (w/v) tetrahydrofuran]. The total run time was 12 min, with the gradient profile programmed as follows: 75% buffer B and 25% buffer A at 0 min, increasing to 87.5% buffer B over 5 min, increasing further to 100% buffer B over the next 0.5 min and washing with 100% buffer B for 2.5 min, before returning to 75% buffer B over 0.5 min and re-equilibrating for 3.5 min. Under these conditions, Fmoc–SeMet was eluted with a retention time of 3.05 min, and Fmoc–MetSeO at 1.65 min. Samples containing Fmoc–SeMet (5 μM) and increasing concentrations of GPx (0–3.9 μM tetramer, equivalent to a maximum reduced Sec concentration of 15.6 μM) were prepared under an atmosphere of N2 in a glove bag, before addition of deoxygenated HOSCN (~1 μM). The samples were allowed to react for at least 15 min before the GPx was removed by centrifugation at 11300 g for 10 min using a 10 kDa molecular-mass cut-off spin column (Pall Nanosep 10K Omega), and subsequent centrifugation at 11300 g for 5 min through a 0.2 μm filter (Pall Nanosep MF 0.2 μm). The samples were then placed in the UPLC autosampler at 10°C and analysed within 8 h. Fmoc–SeMet was employed for two reasons: to facilitate detection of low levels of these materials (by use of fluorescence spectroscopy with λex 265 nm and λem 310 nm), and to aid retention of the polar parent and product materials on the UPLC column employed. The corresponding non-derivatized species cannot be quantified accurately with UV–visible detection as they have little absorbance in this region. In order to assess the effect of Fmoc derivatization of SeMet on the reactivity of the side chain with HOSCN, control experiments in which SeMet (5–50 μM) and Fmoc–SeMet (5 μM) were competitive substrates for HOSCN (5 μM) were undertaken.
Direct kinetic data were analysed by single-wavelength analysis using Pro-Data viewer 4.0 (Applied Photophysics) and OriginPro 7.0 (OriginLab) software. All second-order rate constants reported were derived from at least four independent experiments employing four to ten different substrate concentrations and errors are specified as 95% confidence limits. Stock solutions of ebselen [2-phenyl-1,2-benzisoselenazol-3(2H)-one] were made up in methanol then diluted to the required concentrations with buffer (25–100 μM; ≤20% methanol) and reacted with oxidant (5 μM) with absorbance changes monitored at 322 nm.
TrxR assay
TrxR enzyme activity was determined spectrophotometrically from the TrxR-catalysed reduction of DTNB [5,5′-dithiobis-(2-nitrobenzoic acid)] with NADPH to form TNB, which has a strong absorbance at 412 nm (reaction 1) using a previously developed method [24]. The enzyme activity in samples containing TrxR (0.1 unit) with and without added oxidant (5–100 μM) was measured after 15 min at 22°C. Control incubations without added enzyme were also examined. TrxR activity was determined from the linear rate of DTNB reduction, measured as the absorbance change at 412 nm, over a period of 60 s with readings taken at 10 s intervals, and calculated from the difference between the control and the enzyme-containing samples.
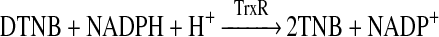 |
(1) |
The reversibility of enzyme inhibition was examined by incubating the oxidant-treated enzyme with DTT (0.5 mM) or NADPH (0.5 mM) for 15 min at 22°C before assay of enzyme activity. In the case of DTT, excess reagent (which interferes with the formation of TNB from DTNB) was removed by centrifugation at 11300 g for 2 min using a 10 kDa molecular-mass cut-off filter and subsequent washing with two aliquots of buffer before assay of the enzyme activity.
GPx and GR assay
GPx activity was measured by use of a coupled reaction with GR in the presence of GSH and NADPH as described previously [25] with minor modifications. Oxidized glutathione produced upon reduction of hydroperoxide by GPx (reaction 2) is reduced to its reduced state by GR using NADPH as a co-factor (reaction 3). The oxidation of NADPH to NADP+ results in a decrease in absorbance at 340 nm (A340), and therefore the rate of decrease in A340 is proportional to the GPx activity. The test mixture contained GSH (3 mM), NADPH (200 μM), EDTA (100 μM), desferrioxamine (50 μM), t-BOOH (t-butyl hydroperoxide) (200 μM) and GR (baker's yeast; 4 units) in phosphate buffer (100 mM, pH 7.4). For the isolated enzyme experiments, samples containing GPx (1.5 or 2.5 μM protein) were incubated for 120 min at 21°C in the absence or presence of added HOSCN (10–100 μM). The time course of NADPH oxidation was determined by diluting the incubation mixture 100-fold into the assay mixture, and following the reaction spectrophotometrically at 340 nm for 3.2 min at 0.2 min intervals. For the RBC experiments, samples containing intact RBCs (diluted 7.5-fold relative to their initial concentration in blood) were incubated for 120 min at 37°C in the absence or presence of added HOSCN (100–1000 μM). Following incubation, the RBCs were immediately lysed by the addition of an equal volume of water and incubation at 4°C for 5 min, followed by freezing on solid CO2 for 15 min. The subsequently thawed lysates were kept at 4°C until they were assayed for GPx activity (within 1 h), whereupon they were diluted 33.3-fold into the test mixture, with NADPH oxidation assayed as per the isolated GPx experiments. Results for both isolated GPx and RBC experiments were corrected for any apparent activity in the reaction mixture without added GPx or RBCs.
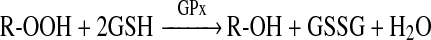 |
(2) |
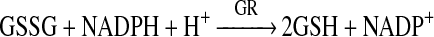 |
(3) |
GR activity was assessed using a similar coupled assay with NADPH, but with no GPx present and added GSSG (i.e. reaction 3). Thus 2.5 μM GR was incubated for 120 min at 21°C in the absence or presence of added HOSCN (25–200 μM). Following incubation, this was diluted 100-fold into a test mixture containing 1 mM GSSG and 150 μM NADPH in 59 mM phosphate buffer (pH 7.4). NADPH oxidation was subsequently monitored at 340 nm for 3.2 min at 0.2 min intervals.
RBC thiol concentrations
Low-molecular-mass thiols (predominantly GSH) were quantified in oxidant-treated and control RBCs after incubation for 2 h at 37°C and subsequent lysis (as per the GPx experiments above). Immediately following lysis, the RBC lysates were diluted 10-fold in water, and centrifuged to dryness through 10 kDa molecular-mass cut-off filters (13400 g, 20 min). The filtrate thiol concentrations were determined using ThioGlo 1 (Calbiochem) with GSH used to construct standard curves as described previously [26].
Statistics
Statistical analysis of the effect of HOSCN on enzyme activity compared with the control (enzyme only) used one-way ANOVA with Newman–Keuls or Dunnett's multiple comparison post-hoc test (GraphPad Prism), with P≤0.05 taken as statistically significant. Enzyme activity data are means±S.D. for n≥3 experiments each with duplicate samples unless stated otherwise.
RESULTS
Determination of the stability and molar absorption coefficients for selenols
Selenols (RSeH; analogous to thiols) are prone to rapid oxidation to the oxidized, diselenide forms (RSeSeR; analogous to disulfides), and are consequently commonly synthesized in the diselenide form. These materials therefore required reduction, and determination of their molar absorption coefficients, in order to allow rate constants to be obtained for the reactions of the selenols. Reduction by sodium borohydride (NaBH4) was examined using UV spectrophotometry and deoxygenated solutions, with anoxia maintained by continual gassing with N2 gas. Initial absorption spectra of the diselenides were recorded in 100 mM phosphate buffer (pH 7.4) at 22°C, and the reduction to the selenol was titrated with increasing amounts of added NaBH4. With a 10-fold excess of NaBH4, complete reduction to the reduced form was achieved with all of the compounds examined. The resulting solutions of the selenols were stable for considerable periods when maintained under anoxic conditions, but rapidly re-oxidized to the diselenide in the presence of O2. The selenide solutions were subsequently diluted with 100 mM phosphate buffer (pH 7.4) under a N2 atmosphere to the desired concentration (determined from the starting diselenide concentrations and assuming 100% conversion into the selenol), and the molar absorption coefficients were determined relative to a 100 mM phosphate buffer baseline; these values are collected in Table 1.
Table 1. Structures, molecular absorption coefficients and second-order rate constants for the reactions of HOSCN with selenols and selenoethers.
Reactions were carried out in 0.1 M phosphate buffer (pH 7.4) at 22°C for the reactions of HOSCN with selenols and selenoethers. Second-order rate constants are given with 95% confidence limits. Comparative rate constants for the corresponding thiols [k2(HOSCN+RSH)] are also included, where available [5], for comparison. ND, not determined.
| Substrate | Structure | λ (nm) | ϵ (M−1·cm−1) | k2(HOSCN) (M−1·s−1) | k2(HOSCN+RSH) (M−1·s−1) |
|---|---|---|---|---|---|
| Sec |  |
243 | 2650 | (1.24±0.04)×106* | (7.8±1.4)×104¶ |
| Sec methyl ester |  |
243 | 4760 | (3.7±0.2)×106* | (1.6±0.1)×105¶ |
| Selenocystamine |  |
243 | 7510 | (5.8±0.2)×106* | (7.1±0.7)×104¶ |
| 3-Selenopropionic acid |  |
248 | 3030 | (2.0±0.1)×106* | ND |
| Gly-Sec-Gly | 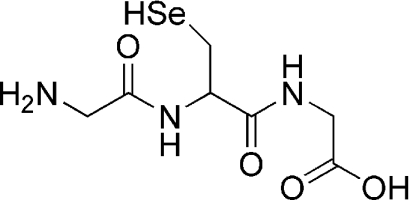 |
243 | 2650 | (1.65±0.03)×106* | ND |
| γ-Glu-Sec-Gly |  |
243 | 2650 | (1.7±0.2)×106* | (2.5±0.4)×104¶ |
| SeMet |  |
− | − | (2.8±0.2)×103† | <103¶ |
| Fmoc—SeMet |  |
− | − | (1.2±0.5)×104‡ | ND |
| Selenomethylcysteine |  |
− | − | <~500† | ND |
| Ebselen |  |
322 | − | ~30§ | ND |
| GPx1 | − | − | − | ~5×105‖ | ND |
*Determined by direct stopped-flow methods using analysis at the indicated wavelengths.
†Determined by competition kinetics relative to TNB (at 412 nm) using k(HOSCN+TNB)=3.8×105 M−1·s−1 at pH 7.4 [5].
‡Determined by competition kinetics relative to SeMet [k(HOSCN+SeMet)=2.8×103 M−1·s−1], by monitoring the formation of Fmoc–MetSeO from Fmoc–SeMet with HOSCN by UPLC.
§Determined by direct UV–visible spectroscopy at the indicated wavelength.
‖Determined by competition kinetics relative to the oxidation of Fmoc–SeMet to Fmoc–MetSeO as determined by UPLC analysis and k(HOSCN+Fmoc–SeMet)=1.2×104 M−1·s−1.
¶From [5].
Determination of rate constants for reaction of HOSCN with low-molecular-mass and peptide selenols
The rate constants for reaction of HOSCN with a range of low-molecular-mass and peptide selenols (Gly-Sec-Gly and γ-Glu-Sec-Gly, the seleno analogue of GSH) were determined by direct stopped-flow methods. Individual molar absorption coefficients for the peptides were not determined, owing to the low amount of material available, with the value for free Sec (see Table 1) used to determine the peptide concentrations. The selenols (9.9–86.4 μM; for structures, see Table 1) were allowed to react with HOSCN (2.5 μM) under a N2 atmosphere at 22°C, and the changes in absorbance were measured at their absorbance maxima (Table 1). Absorbance changes (averages of ten stopped-flow runs) were fitted satisfactorily to a single-exponential function (see, for example, Figure 1a) yielding an observed rate constant (kobs) for each compound, which was subsequently plotted against the substrate concentration (Figure 1b). The gradients of the resulting straight lines yielded the second-order rate constants given in Table 1.
Figure 1. Kinetic analysis of the reaction of HOSCN with Sec by direct stopped-flow studies.

(A) Kinetic trace obtained for the reaction of Sec (59.4 μM) with HOSCN (2.5 μM) at 22°C in 0.1 M phosphate buffer (pH 7.4). The kinetic data obtained at 243 nm are represented as data points (only 100 of 1000 data points are shown for clarity) with the exponential fit to the data shown by the continuous line. (B) Plot of the observed rate constants (kobs) against Sec concentrations, with kobs determined by fitting the absorbance changes at 243 nm to a single-exponential decay. Each data point represents a single determination of the observed rate constant, for which the error in the fit is typically <1%.
Determination of rate constants for the reaction of HOSCN with low-molecular-mass selenoethers
Kinetic data for the reaction of HOSCN with selenoethers, such as SeMet, was obtained from competition kinetic experiments using the rate constant determined previously for the reaction of HOSCN with TNB as a reference, as, unlike the selenols, the selenoethers do not have distinct UV–visible absorbance bands. The kinetics of the reaction of HOSCN (2.5 μM) with increasing concentrations of SeMet (10–100 μM) were investigated using TNB (10 μM) at 22°C over a period of 0.1–2 s. Absorbance changes arising from the reaction of TNB with HOSCN were monitored at 412 nm, as this allowed the decay of TNB to be measured without the confounding influence of other species. The changes in TNB absorbance obtained in the absence of any competing substrate (yieldmax), and in the presence of a range of SeMet concentrations (yieldquench) were quantified at fixed time points for each concentration after the cessation of absorbance changes detected in time-resolved experiments (Figure 2). In the absence of added SeMet, the extent of TNB loss was found to depend on the concentration of HOSCN, and the final absorbance values of the residual TNB were stable over the durations used in the experiment. These data indicate that secondary reactions and/or the formation of products do not interfere in a significant manner in these measurements. Control experiments in which the product of TNB oxidation, DTNB, was allowed to react with HOSCN, showed no significant absorbance changes over the durations employed, indicating that further oxidation of the disulfide by HOSCN is not a major confounding reaction.
Figure 2. Kinetic analysis of the reaction of HOSCN with SeMet determined by competition kinetics with TNB.

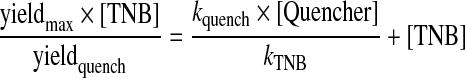 |
Under these conditions, the change in TNB absorbance was found to decrease with increasing concentrations of added SeMet, consistent with competition between TNB and SeMet for the HOSCN. Typical kinetic traces at 412 nm showing the change in absorbance due to TNB oxidation, and the decrease in the extent of absorbance loss with increasing concentrations of SeMet, are shown in Figure 2(a). These data yielded a linear competition plot, in which the gradient of the line is equal to kSeMet/kTNB and the intercept is given by the TNB concentration (Figure 2b). Using the rate constant obtained previously for TNB (kTNB 3.8×105 M−1·s−1 [5]), the second-order rate constant for reaction of HOSCN with SeMet was determined (Table 1). Analogous experiments, using competitive reaction with TNB, were carried out for selenomethylcysteine (i.e. SeMet with one fewer methylene group in the side chain; for the structure, see Table 1). However, under the conditions employed, no significant change in the absorbance due to TNB oxidation was detected with the concentrations (up to 2.5 mM) of selenomethylcysteine employed, indicating that this reaction is slow. These data yield an upper limit for the reaction of selenomethylcysteine with HOSCN of k<500 M−1·s−1.
The reaction of HOSCN with Fmoc–SeMet was also investigated by competition kinetics using an adaptation of a previous HPLC-based method [22,23], as the significantly lower solubility of Fmoc–SeMet compared with SeMet precluded the use of the stopped-flow method described above. Reaction of HOSCN (5 μM) with Fmoc–SeMet (5 μM) was carried out in competition with SeMet (0–50 μM). The extent of formation of the derivatized selenoxide (Fmoc–MetSeO) decreased as the concentration of SeMet was increased (yieldquench) and this was compared with the Fmoc–MetSeO yield in the absence of SeMet (yieldmax). The background yield of Fmoc–MetSeO in the absence of HOSCN was also determined (yieldblank) and a linear competition kinetics plot of [Fmoc–SeMet]{(yieldmax−yieldblank)/(yieldquench/yieldblank)} against [SeMet] (Figure 3a) was obtained with a gradient of kSeMet/kFmoc–SeMet, from which kFmoc–SeMet was calculated (Table 1).
Figure 3. Kinetic analysis of the reaction of HOSCN with Fmoc–SeMet and isolated purified GPx by competition kinetics.

(A) Linear analysis (see the text and [23] for derivation of equation) for competitive reaction between Fmoc–SeMet (5 μM) and SeMet (0–50 μM) with HOSCN. Results are means±S.E.M. (B) UPLC (with fluorescence detection) was used to monitor the formation of the selenoxide of Fmoc–SeMet (retention time, 1.65 min) following reaction of Fmoc–SeMet (5 μM) with HOSCN (1 μM) in 0.1 M phosphate buffer (pH 7.4) at 22°C. Increasing concentrations of GPx (up to 3.9 μM; tetramer concentrations for each trace shown) were included in the reaction mixture to assess the relative rate constants for GPx compared with Fmoc–SeMet. a.u., absorbance units. (C) Linear analysis (as above) for the competitive kinetic data for GPx with HOSCN. Individual data points for each replicate are shown. Y, yield.
Reaction of ebselen (25–100 μM, containing ≤20% methanol) with HOSCN (5 μM) was examined by monitoring absorbance changes at 322 nm arising from ebselen. Reaction of HOSCN with this compound was relatively slow, with reactions monitored over 2 h. Analysis of the resulting kinetic data gave an approximate rate constant for the reaction of HOSCN with this compound of ~30 M−1·s−1 (Table 1).
Determination of the rate constant for the reaction of HOSCN with GPx
Kinetic data for the reaction of HOSCN with purified GPx was obtained using the UPLC method described above for Fmoc–SeMet. Reaction of HOSCN with GPx was carried out in competition with Fmoc–SeMet (5 μM) using the rate constant determined above for oxidation of Fmoc–SeMet (Figure 3). The yields of Fmoc–MetSeO were determined in reaction mixtures that were not treated with oxidant, with 1 μM HOSCN, and with this concentration of oxidant and increasing concentrations of GPx (0–3.9 μM tetramer) under an atmosphere of N2 to prevent oxidation of the Sec residue. In the complete system, increasing concentrations of GPx decreased the conversion of parent Fmoc–SeMet into the selenoxide, Fmoc–MetSeO, consistent with competition between the selenoether and GPx for the oxidant (Figure 3b). The concentrations of Fmoc–MetSeO in the absence and presence of competing GPx were subsequently analysed as outlined above for Fmoc–SeMet, together with the value of k for Fmoc–SeMet (see Table 1) to give a second-order rate constant for reaction of HOSCN with the GPx tetramer of ~5×105 M−1·s−1. For individual Sec residues, k is ~105 M−1·s−1 (using the theoretical ratio of Sec residues per tetramer of 4). This value should be considered as an approximate value, as, despite extensive precautions to prevent oxidation of the GPx (which was isolated from DTT-containing samples to keep the Sec residues in a reduced state), we cannot exclude the possibility that some of the residues became oxidized during isolation, which would result in an underestimate of this rate constant.
Effect of HOSCN on the activity of TrxR
The above data indicate that the selenium atom of Sec residues can be rapidly oxidized by HOSCN. In the light of these data, experiments were carried out to determine whether the Sec residue present in TrxR was subject to oxidation by HOSCN and whether this resulted in enzyme inactivation. The assessment of enzyme activity was carried out using the NADPH-dependent reduction of DTNB to the corresponding thiolate anion (TNB) which is catalysed by TrxR, with the rate of reaction quantified by the rate of change of absorbance, arising from the formation of TNB, at 412 nm. Although we cannot exclude the possibility of some artefactual oxidation of the Sec residues in the isolated protein, the presentation of these data relative to the control (untreated) enzyme should minimize significant errors.
Incubation of purified TrxR (~0.1 unit, 1 μg of protein, ~60 nM) with 5–100 μM HOSCN for 15 min at 22°C and pH 7.4, before assessment of residual enzymatic activity, resulted in a dose-dependent decrease in the amount of DTNB reduced to TNB. This decrease was significant statistically at concentrations ≥25 μM oxidant (Figure 4). This decrease in enzyme activity was rapid, with longer incubation times not giving rise to any significant increase in the extent of inhibition. Experiments carried out with either 25 or 100 μM decomposed HOSCN (incubated at 37°C overnight before use) gave rise to significantly less enzyme inhibition than the active oxidant, indicating that HOSCN is likely to be the active agent rather than decomposition products (such as −OCN) derived from this species.
Figure 4. Inhibition of TrxR activity by HOSCN.

TrxR activity (assayed as the absorbance change at 412 nm over 1 min; data are normalized to the 0 μM HOSCN control) was quantified after reaction of the isolated enzyme with the indicated concentrations of HOSCN. TrxR (0.1 unit in 200 μl; ~60 nM) was incubated for 15 min with HOSCN before assay of residual activity. Significance relative to the 0 μM HOSCN control (as assessed by one-way ANOVA on the raw data) is indicated (**P<0.01; ***P<0.001).
The reversibility of this damage was investigated by incubation of the HOSCN-treated TrxR with or without added DTT (0.5 mM) or NADPH (0.5 mM), before assessment of residual activity. Neither of these agents was able to restore, in a significant manner, the activity of the oxidant-treated enzyme under these conditions (results not shown).
Effect of HOSCN on GPx activity and RBC thiol levels
In the light of the above data, the effect of HOSCN on GPx activity was examined by pre-incubating the isolated purified enzyme with oxidant, before assessment of residual enzymatic activity as outlined in the Experimental section. GPx (1.5 μM protein) was incubated at room temperature (22°C) for 120 min in the absence (buffer added) or presence of added HOSCN (10–100 μM) in 100 mM phosphate buffer (pH 7.4), before assaying enzyme activity. Under these conditions, a significant loss of GPx activity was detected with oxidant concentrations ≥10 μM (results not shown). With a higher concentration of GPx (2.5 μM protein), significant inhibition was not detected until concentrations of 25 μM HOSCN were employed (Figure 5). The decrease in enzyme activity was determined to be time-dependent, with the extent of inhibition induced by 10 μM HOSCN and 1.5 μM protein, significantly different between reactions examined after 15 and 120 min. Experiments with 100 μM decomposed HOSCN gave higher levels of activity than the corresponding concentrations of freshly prepared oxidant, but this difference was not statistically significant.
Figure 5. Inhibition of GPx activity on the reaction of the isolated enzyme with HOSCN.

GPx (2.5 μM) was incubated for 120 min at room temperature with 25–100 μM HOSCN, with residual enzyme activity subsequently measured in a coupled system with excess GR, as detailed in the Experimental section. Data are presented as the mean±S.D. percentages of activity of the GPx control incubated in the absence of HOSCN for duplicate experiments. Statistical analysis, using raw data, was by one-way ANOVA with Dunnett's post-hoc test; significance relative to the 0 μM HOSCN control is indicated (*P<0.05; **P<0.01; ***P<0.001).
In order to confirm that the observed changes in consumption of NADPH do not arise from the reaction of residual HOSCN with the GR required for the coupled assay, experiments were carried out to examine the potential inhibition of GR in isolation. Incubation of isolated GR (2.5 μM protein) with 0–200 μM HOSCN was carried out for either 15 or 120 min, before assessment of enzyme activity. No loss of enzyme activity was detected at either time point with any of the HOSCN concentrations examined (results not shown), indicating that this enzyme is insensitive to HOSCN under the conditions employed; the observed inhibition in the rate of NADPH consumption in the coupled GPx–GR assay is therefore attributed to direct inhibition of GPx by HOSCN.
The ability of HOSCN to inhibit GPx in more complex systems was examined using intact human RBCs, a cell type with high levels of this enzyme [27]. Incubation of RBCs (diluted 7.5-fold in terms of protein concentration) with 100 μM HOSCN or higher concentrations, at 37°C for 120 min, resulted in a significant loss of GPx activity as assessed using the coupled GPx–GR assay (Figure 6). In analogous experiments, the levels of low-molecular-mass thiols (<10 kDa, predominantly GSH), which may act as a competing target for the oxidant, were assessed; a dose-dependent loss of thiols was detected across the same oxidant concentration range (Figure 7).
Figure 6. Inhibition of GPx activity in human RBCs on reaction with HOSCN.

RBCs (diluted to give a protein concentration 7.5-fold lower than in blood) were incubated for 120 min at 37°C with 100–1000 μM HOSCN, with residual enzyme activity subsequently measured following RBC lysis in a coupled system with excess GR, as detailed in the Experimental section. Data are normalized to the 0 μM HOSCN control, and are the means±S.D. for three experiments, each performed in duplicate. Statistical analysis, using raw data, was by one-way ANOVA with Dunnett's post-hoc test; significance relative to the 0 μM HOSCN control is indicated (*P<0.05; ***P<0.0001).
Figure 7. Oxidation of low-molecular-mass thiols (<10000 Da) in human RBCs on reaction with HOSCN.

RBCs were incubated as described in the legend to Figure 6, with residual low-molecular-mass thiol concentrations subsequently measured following RBC lysis and filtering through 10 kDa molecular mass cut-off filters using ThioGlo 1 (see the Experimental section). Data are the means±S.D. for three independent experiments. Statistical analysis, using raw data, was by one-way ANOVA with Dunnett's post-hoc test; significance relative to the 0 μM HOSCN control is indicated (***P<0.0001).
DISCUSSION
Previous studies have indicated that HOSCN, one of the major oxidants generated by MPO [1,28], reacts rapidly and with great selectivity with thiols (e.g. cysteine, GSH and related species) [5–7]. Rate constants, k, for reaction of HOSCN with such residues are in the range 7.3×103–1.6×105 M−1·s−1 for reaction with low-molecular-mass species at pH 7.4 [5], and (1–7)×104 M−1·s−1 for cysteine residues present on a number of proteins (e.g. creatine kinase, BSA, β-lactoglobulin and crystallins) [5]. It has been shown that these values vary considerably with the pH of the reaction mixture, with rate constants for reaction of HOSCN with TNB ranging from 3.8×105 to 7.7×106 M−1·s−1 for reactions at pH 7.4 and 6.0 respectively [5,6]. This increase in rate constant at lower pH values and a reported inverse correlation between thiol pKa value and rate constant [5] is consistent with reaction occurring most rapidly between the neutral form of HOSCN (rather than −OSCN) and the ionized form of the thiol (i.e. the thiolate anion, RS−) [5,6]. In the light of these data, it was hypothesized that reaction of HOSCN with selenols should be more rapid than with thiols, as selenols are predominantly ionized at neutral pH values (cf. pKa values of 5.2 and 8.4 for Sec and cysteine residues respectively [15,16]), and are more nucleophilic than sulfur centres.
The results of the present study support this hypothesis, with the rate constants determined for the selenols being between 15- and 82-fold higher than for the corresponding thiol species determined under identical conditions (Table 1). Quantitative comparison between the data for thioethers (such as methionine) and the corresponding selenoethers (e.g. SeMet) cannot be made, as it has proved impossible (to date) to obtain an accurate value for the rate constant for reaction of HOSCN with methionine, on account of the slow nature of this reaction; the values obtained for SeMet (2.8×103 M−1·s−1) and Fmoc–SeMet (1.2×104 M−1·s−1) are, however, clearly higher. No direct comparison is possible for the rate constants for the reaction of HOSCN with Sec rather than cysteine residues on proteins, due to differences in the structures of the proteins examined to date: the value determined here for reaction of HOSCN with GPx (~5×105 M−1·s−1) is higher than those reported previously for reaction with protein cysteine residues [5]; however, these values have been obtained for different proteins and hence cannot be compared directly. The data obtained in the present study for the two Sec-containing peptides are ~10-fold higher than that for the corresponding free amino acid (Sec; see Table 1), suggesting that the environment of the Sec residue modulates its reactivity.
There are a few previous reports on other rate constants for reaction of oxidants with selenol and selenoether species in the literature; these data are collected in Table 2. A value of 9.7×102 M−1·s−1 has been reported for the rate constant for reaction of H2O2 with selenocystamine (generated via in situ γ-radiolysis of the parent diselenide); values for other hydroperoxides were slightly smaller [29]. This value for reaction with selenocystamine is ~50-fold higher than for reaction of the same oxidant with cysteine, consistent with the observed acceleration of rates between the thiols and selenols with HOSCN observed here. This value for reaction of H2O2 with selenocystamine is, however, much lower, ~6000-fold, than that determined in the present study for the reaction of HOSCN with the same substrate (5.8×106 M−1·s−1; Table 1). These data therefore indicate that HOSCN is a much more effective oxidant of both thiols (cf. data in [5]) and selenol species than H2O2. Data have also been reported for reaction of peroxynitrite (ONOOH) with some seleno-compounds. The values reported for ONOOH and SeMet are very similar to those determined in the present study for HOSCN (both ~2–3×103 M−1·s−1; Table 1 and [30]), whereas the values for ebselen are very different (1.6×106 M−1·s−1 for ONOOH [31] and ~30 M−1·s−1 for HOSCN; Table 1); the reasons for this marked difference are unknown.
Table 2. Second-order rate constants for reaction of two-electron oxidants with selenium-containing compounds and enzymes.
Data for radical-mediated oxidation reactions are given in [18,45,46]. Values were determined at room temperature (20–25°C) and pH 7.0–7.4 unless stated otherwise. PC-OOH, phosphatidylcholine hydroperoxide.
| Substrate | Oxidant | k2 (M−1·s−1) | Reference |
|---|---|---|---|
| Sec | HOSCN | 1.24×106 | The present study |
| Selenocysteine methyl ester | HOSCN | 3.7×106 | The present study |
| Selenocystamine | H2O2 | 9.7×102 | [29] |
| t-BOOH | 1.2×102 | [29] | |
| Cumene-OOH | 2.5×102 | [29] | |
| HOSCN | 5.8×106 | The present study | |
| 3-Selenopropionic acid | HOSCN | 2.0×106 | The present study |
| Gly-Sec-Gly | HOSCN | 1.65×106 | The present study |
| γ-Glu-Sec-Gly | HOSCN | 1.7×106 | The present study |
| SeMet | ONOOH | 2.4×103 | [30] |
| HOSCN | 2.8×103 | The present study | |
| Fmoc—SeMet | HOSCN | 1.2×104 | The present study |
| Selenomethylcysteine | HOSCN | <500 | The present study |
| Ebselen | H2O2 | 4.8 | [47] |
| ONOOH | 2×106 (25°C, pH≥8) | [48] | |
| HOSCN | ~30 | The present study | |
| Ebselen–GSH conjugate (selenosulfide) | H2O2 | <0.17 | [47] |
| Ebselen selenol | H2O2 | 47 | [47] |
| >350 | [49] | ||
| 2.05×102* | [50] | ||
| Ebselen diselenide | H2O2 | 5.3 | [47] |
| 2-(Methylseleno)benzanilide | ONOOH | 2.7×103 | [51] |
| GPx1 (per tetramer) | H2O2 | 4.1×107† | [52] |
| t-BOOH | 4.2×106† | [52] | |
| ONOOH | 8.0×106 | [53] | |
| ONOOH | 1.8×105 (37°C, pH 7.1) | [31] | |
| HOSCN | 5×105 | The present study | |
| Oxidized GPx1 (per tetramer) | ONOOH | 7.4×105 | [53] |
| Extracellular GPx | H2O2 | 4.0×107† | [52] |
| t-BOOH | 2.3×106† | [52] | |
| PC-OOH | 3.4×105† | [52] | |
| Phospholipid hydroperoxide GPx4 | PC-OOH | 1.5×107† | [52] |
| Selenoprotein P | PC-OOH | 8.6×104† | [52] |
*Measured under catalytic conditions in the presence of dehydrolipoate as a cofactor with the tetrameric enzyme; these values are therefore not strictly comparable with direct rate constant data.
†Measured under catalytic conditions in the presence of GSH as a cofactor with the tetrameric enzyme; these values are therefore not strictly comparable with direct rate constant data.
The suggestion that the environment of the selenol centre plays a key role in determining the magnitude of the rate constant for reaction is supported by the observed variation in the experimental data for the range of selenol species studied. Of the selenols examined, the lowest rate constant is for Sec itself. Higher rate constants have been determined for both the analogue without the amine function (3-selenopropionic acid), which is ~16-fold more reactive, and that without the carboxyl function (selenocystamine) that has an even higher rate constant (by a further factor of ~3). The methyl ester derivative of Sec also has a much higher rate constant than that of Sec consistent with either the presence of a positively charged amine function or absence of a negatively charged carboxy group, having a marked attenuating effect on the reactivity of the selenium centre. This conclusion is supported by the data for the selenoethers, where k for selenomethylcysteine could not be measured, whereas a value was determined for SeMet, with the increased length of the side chain (i.e. distance of the selenium atom from the carboxy group) appearing to increase the rate of reaction. Whether the variation in rate constants for the selenols reflects the influence of these nearby functional groups on the pKa of the selenol function or other factors (e.g. steric constraints) remains to be determined as pKa values for most of these selenols have not been determined as far as we can ascertain. Previous studies on the reaction of HOSCN with thiols have shown that there is an inverse correlation between the thiol pKa and the rate of HOSCN-mediated oxidation [5], and similar data have been reported for chloramine reactions [32], so it is possible that the enhanced rate of reactivity, and higher molar absorption coefficients, of selenocystamine and selenocysteine methyl ester may arise from the presence of the protonated amine functions in these compounds modulating the pKa of the selenol function. A structural dependence on the rate constants for reaction is also clear for the commercial drug ebselen, a GPx mimetic drug [33] that has been employed in clinical trials for the treatment of stroke [34,35], which reacts much more slowly than with SeMet (~90-fold). This may reflect a mixture of steric interactions and the lower availability of the selenium atom lone pairs due to conjugation with both the aromatic ring and amide functions in ebselen.
The experiments with both isolated TrxR and GPx and intact RBCs indicate that HOSCN is capable of diminishing the activity of these enzymes. Loss of activity may arise from both reversible and irreversible alteration of the active-site Sec residue, but as the assessment of enzyme activity involves the use of reducing species (which may repair reversible damage), the observed loss of enzyme activity is believed to arise from irreversible modification of the enzyme structure. In contrast with TrxR and GPx, GR appears to be insensitive to HOSCN, probably due to the activity of this enzyme being dependent on a disulfide moiety that is relatively unreactive with HOSCN [5].
Whereas the present study has provided evidence as to the rapidity of the reactions of HOSCN with Sec and related species, the products of these reactions remain to be defined. For the corresponding thiol reactions, evidence has been presented [8,36] for the formation of short-lived adduct species (RS-SCN; reaction 4) that subsequently undergo reaction with another thiol to give a disulfide and regenerate SCN− (reaction 5) [36,37], or hydrolysis to give a sulfenic acid (RS-OH; reaction 6) (reviewed in [37]). Disulfide formation appears to be the more favourable process, with sulfenic acid generation only occurring to a significant extent when the former cannot occur (e.g. due to steric constraints on proteins or absence of suitable thiols). The sulfenic acids can undergo subsequent reaction with another thiol (to give a disulfide) or further oxidation to give sulfinic (RSO2H) or sulfonic (RSO3H) acids. Disulfide formation can be viewed as the initial stages of a repair process for the oxidized target, owing to the presence of multiple disulfide reductases, whereas formation of the higher oxyacids is typically irreversible, with the only (current) exception being the reduction of sulfinic acid forms of some peroxiredoxins by mitochondrial sulfiredoxins [38].
 |
(4) |
 |
(5) |
 |
(6) |
We propose that reaction of HOSCN with Sec residues (and selenols in general) occurs via a similar scheme, involving initial formation of an RSe-SCN species (reaction 7). For low-molecular-mass species, subsequent reaction may occur with another parent selenol molecule to give a diselenide (reaction 8), or hydrolysis to give a RSe-O− species (or its neutral form RSe-OH) (reaction 9). Both RSe-SCN and RSe-O− species may react (by analogy to thiol chemistry and the enzymatic cycle of GPx [39]) with a thiol molecule (R′SH) to give a mixed RSe-SR′ species (reaction 10). The RSe-SR′ may in turn be reduced by a further thiol (cf. the enzymatic cycle of GPx [39]) to regenerate the reduced selenol (RSe-H) and a disulfide (reaction 11). The intermediate species (e.g. RSe-SCN and RSe-O−) may also undergo further oxidation to products such as RSeO2H and RSeO3H (reaction 12) [40]. The rate constants obtained in the present study are presumed to be those for the first of these reactions (reaction 7). By analogy with the behaviour of HOCl and HOBr, we would expect that reaction of HOSCN with the selenoethers (e.g. SeMet) would give rise to the corresponding selenoxide (reaction 13, and similar species for ebselen).
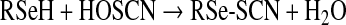 |
(7) |
 |
(8) |
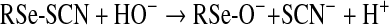 |
(9) |
 |
(10) |
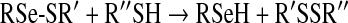 |
(11) |
 |
(12) |
 |
(13) |
The inactivation of GPx and TrxR is presumed to occur via a (currently unknown) sequence of reactions resulting in reaction 12. The very different time courses of reaction 7 (i.e. the rate constant data) and the enzyme inactivation indicates that at least some of the reactions subsequent to reaction 7 are slow, with reaction 12 possibly being the slow (and rate-determining) step corresponding to enzyme inactivation. The generation of species such as RSe-SCN, RSe-O−, RSe-SR′ (and possibly RSe-NHR′ in the case of GPx [39]) may result in a transient decrease in enzyme activity, but the presence of reductants in the enzyme assay mixtures may repair these species, and hence a decreased overall loss of activity. The generation of these species may account for the incomplete inhibition of GPx activity induced by HOSCN. In contrast, reaction of TrxR with HOSCN does not appear to be readily reversed under the conditions employed in the present study. Thus, unlike GPx, treatment of TrxR with high concentrations of HOSCN resulted in complete inhibition of enzyme activity (cf. Figures 4 and 5). One possible interpretation of these data is that rapid reaction occurs with TrxR via reaction 7 to give a RSe-SCN adduct, and that this species, in the absence of reducing species, rapidly converts into irreversible oxidation products such as RSeO2H, RSeO3H or dehydroalanine (via loss of the seleno group from these species; cf. data in [40]). The non-reversibility of the TrxR inactivation supports the proposition that the Sec residue is the site of oxidation, given the known ease of repair of oxidation at cysteine residues [40].
As HOSCN reacts rapidly with thiols, in addition to seleno centres (Table 1), it would be expected that competition would occur, in cells, between selenoproteins and protein thiols/GSH for available oxidant. Although reaction with some of the Sec-containing species examined in the present study is more rapid than with GSH or protein thiols, the concentration of the cysteine-containing species within cells is likely to be significantly higher (GSH concentrations in RBCs are ~2 mM [41] and GPx protein is ~2 μM, calculated from data in [42]). The data obtained with intact RBCs, which contain competing protein thiols and GSH, indicate that loss of GPx activity occurs over the same oxidant concentration range as GSH depletion. Whereas the above rate constant and abundance data would suggest that GSH is the predominant target, the decrease in GPx activity observed in these intact cells is unlikely to be due to GSH depletion, as excess GSH is present in the enzyme activity assay buffer. Conversely, whether the observed GSH depletion arises solely from direct oxidation by HOSCN (a process likely to occur to at least some extent due to the combination of high abundance of GSH and high rate constant for this reaction [5]), or also from the enzymatic activity of GPx, which uses this material as a co-factor, is unclear.
Overall, the results of the present study indicate that Sec and SeMet residues, and other seleno species, are targets for HOSCN, with the rate constants for reaction being between 15- and 82-fold higher than for the corresponding thiols. Evidence has also been presented for rapid reaction of this oxidant with Sec-containing proteins, with these reactions resulting in irreversible inhibition of the major intracellular protective enzymes TrxR and GPx. Damage to these enzymes may play a major role in altering the redox balance within cells as TrxR is critical to the maintenance of thioredoxin in its reduced form, with this in turn being a key co-factor for the peroxiredoxin family of enzymes, which play a critical role in removing peroxides (H2O2 [11] and also amino acid/protein hydroperoxides [43]) from cells. Inhibition of GPx would be expected to affect cellular redox status, as this enzyme also plays a key role in H2O2 removal. Thus exposure of cells to HOSCN may result in both direct GSH consumption and also inhibition of two of the major protective systems against H2O2-mediated damage. The high reactivity of seleno-compounds with these inflammatory oxidants also suggests that novel seleno drugs [44] may have therapeutic potential in the amelioration of tissue damage and certain human pathologies.
AUTHOR CONTRIBUTION
Ojia Skaff performed most of the research, analysed data and contributed to preparation of the paper. David Pattison performed some of the research, carried out data analysis, assisted with experimental design, and contributed to preparation of the paper. Philip Morgan performed some of the research, analysed data and contributed to preparation of the paper. Rushad Bachana carried out some of the research. Vimal Jain and K. Indira Priyadarsini contributed novel reagents and participated in experimental discussions. Michael J. Davies directed the research, designed the experimental plan and wrote the paper.
ACKNOWLEDGEMENTS
We thank Mr Corin Storkey and Professor Carl Schiesser (University of Melbourne) for the gift of the diselenide of selenocysteine methyl ester, Professor Peter Lay and Dr Aviva Levina (University of Sydney) for access to the rapid stopped-flow apparatus and Associate Professor Clare Hawkins for helpful discussions.
FUNDING
This work was supported by grants from the Australian Research Council under the ARC Centres of Excellence [grant number CE0561607] and Discovery [grant number DP0988311] programmes, the National Health and Medical Research Council [grant number 570829] and the National Heart Foundation [Grants in Aid numbers G09S4313 and G08S3769]. R.B. was supported by a University of Sydney Medical School summer scholarship.
References
- 1.Davies M. J., Hawkins C. L., Pattison D. I., Rees M. D. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid. Redox Signaling. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 2.Winterbourn C. C., Hampton M. B., Livesey J. H., Kettle A. J. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J. Biol. Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 3.van der Veen B. S., de Winther M. P., Heeringa P. Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signaling. 2009;11:2899–2937. doi: 10.1089/ars.2009.2538. [DOI] [PubMed] [Google Scholar]
- 4.Pattison D. I., Davies M. J. Reactions of myeloperoxidase-derived oxidants with biological substrates: gaining insight into human inflammatory diseases. Curr. Med. Chem. 2006;13:3271–3290. doi: 10.2174/092986706778773095. [DOI] [PubMed] [Google Scholar]
- 5.Skaff O., Pattison D. I., Davies M. J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: absolute rate constants and assessment of biological relevance. Biochem. J. 2009;422:111–117. doi: 10.1042/BJ20090276. [DOI] [PubMed] [Google Scholar]
- 6.Nagy P., Jameson G. N., Winterbourn C. C. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem. Res. Toxicol. 2009;22:1833–1840. doi: 10.1021/tx900249d. [DOI] [PubMed] [Google Scholar]
- 7.Aune T. M., Thomas E. L. Oxidation of protein sulfhydryls by products of peroxidase-catalyzed oxidation of thiocyanate ion. Biochemistry. 1978;17:1005–1010. doi: 10.1021/bi00599a010. [DOI] [PubMed] [Google Scholar]
- 8.Hawkins C. L., Pattison D. I., Stanley N. R., Davies M. J. Tryptophan residues are targets in hypothiocyanous acid-mediated protein oxidation. Biochem. J. 2008;414:271–280. doi: 10.1042/BJ20070941. [DOI] [PubMed] [Google Scholar]
- 9.Lloyd M. M., van Reyk D. M., Davies M. J., Hawkins C. L. Hypothiocyanous acid is a more potent inducer of apoptosis and protein thiol depletion in murine macrophage cells than hypochlorous acid or hypobromous acid. Biochem. J. 2008;414:271–280. doi: 10.1042/BJ20080468. [DOI] [PubMed] [Google Scholar]
- 10.Bozonet S. M., Scott-Thomas A. P., Nagy P., Vissers M. C. Hypothiocyanous acid is a potent inhibitor of apoptosis and caspase 3 activation in endothelial cells. Free Radical Biol. Med. 2010;49:1054–1063. doi: 10.1016/j.freeradbiomed.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Low F. M., Hampton M. B., Winterbourn C. C. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxid. Redox Signaling. 2008;10:1621–1630. doi: 10.1089/ars.2008.2081. [DOI] [PubMed] [Google Scholar]
- 12.Lu J., Holmgren A. Selenoproteins. J. Biol. Chem. 2009;284:723–727. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- 13.Marzano C., Gandin V., Folda A., Scutari G., Bindoli A., Rigobello M. P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radical Biol. Med. 2007;42:872–881. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 14.Zhong L., Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 15.Stadtman T. C. Selenocysteine. Annu. Rev. Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 16.Kortemme T., Creighton T. E. Ionisation of cysteine residues at the termini of model α-helical peptides: relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 17.Gromer S., Johansson L., Bauer H., Arscott L. D., Rauch S., Ballou D. P., Williams C. H., Jr, Schirmer R. H., Arner E. S. Active sites of thioredoxin reductases: why selenoproteins? Proc. Natl. Acad. Sci. U.S.A. 2003;100:12618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunwar A., Mishra B., Barik A., Kumbhare L. B., Pandey R., Jain V. K., Priyadarsini K. I. 3,3′-diselenodipropionic acid, an efficient peroxyl radical scavenger and a GPx mimic, protects erythrocytes (RBCs) from AAPH-induced hemolysis. Chem. Res. Toxicol. 2007;20:1482–1487. doi: 10.1021/tx700137a. [DOI] [PubMed] [Google Scholar]
- 19.Thomas E. L. Products of the lactoperoxidase-catalysed oxidation of thiocyanate and halides. In: Pruitt K. M., Tenovuo J. O., editors. The Lactoperoxidase System: Chemistry and Biological Significance. New York: Marcel Dekker; 1985. pp. 31–53. [Google Scholar]
- 20.Eyer P., Worek F., Kiderlen D., Sinko G., Stuglin A., Simeon-Rudolf V., Reiner E. Molar absorption coefficients for the reduced Ellman reagent: reassessment. Anal. Biochem. 2003;312:224–227. doi: 10.1016/s0003-2697(02)00506-7. [DOI] [PubMed] [Google Scholar]
- 21.Pattison D. I., Davies M. J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 22.Skaff O., Pattison D. I., Davies M. J. The vinyl ether linkages of plasmalogens are favored targets for myeloperoxidase-derived oxidants: a kinetic study. Biochemistry. 2008;47:8237–8245. doi: 10.1021/bi800786q. [DOI] [PubMed] [Google Scholar]
- 23.Pattison D. I., Davies M. J. A kinetic analysis of the reactions of hypobromous acid with protein components: implications for cellular damage and the use of 3-bromotyrosine as a marker of oxidative stress. Biochemistry. 2004;43:4799–4809. doi: 10.1021/bi035946a. [DOI] [PubMed] [Google Scholar]
- 24.Arnér E. S. J., Zhong L., Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- 25.Flohé L., Günzler W. A. Assays of glutathione peroxidase. Methods Enzymol. 1984;105:114–120. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins C. L., Morgan P. E., Davies M. J. Quantification of protein modification by oxidants. Free Radical Biol. Med. 2009;46:965–988. doi: 10.1016/j.freeradbiomed.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Cohen G., Hochstein P. Glutathione peroxidase: the primary agent for the elimination of hydrogen peroxide in erythrocytes. Biochemistry. 1963;2:1420–1428. doi: 10.1021/bi00906a038. [DOI] [PubMed] [Google Scholar]
- 28.van Dalen C. J., Whitehouse M. W., Winterbourn C. C., Kettle A. J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prutz W. A. Glutathione peroxidase-like activity of simple selenium compounds: peroxides and the heterocyclic N-oxide resazurin acting as O-atom donors. Z. Naturforsch. C. J. Biosci. 1995;50:209–219. [PubMed] [Google Scholar]
- 30.Padmaja S., Squadrito G. L., Lemercier J. N., Cueto R., Pryor W. A. Rapid oxidation of DL-selenomethionine by peroxynitrite. Free Radical Biol. Med. 1996;21:317–322. doi: 10.1016/0891-5849(96)00132-3. [DOI] [PubMed] [Google Scholar]
- 31.Padmaja S., Squadrito G. L., Pryor W. A. Inactivation of glutathione peroxidase by peroxynitrite. Arch. Biochem. Biophys. 1998;349:1–6. doi: 10.1006/abbi.1997.0407. [DOI] [PubMed] [Google Scholar]
- 32.Peskin A. V., Winterbourn C. C. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radical Biol. Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 33.Sies H. Ebselen, a selenoorganic compound as glutathione peroxidase mimic. Free Radical Biol. Med. 1993;14:313–323. doi: 10.1016/0891-5849(93)90028-s. [DOI] [PubMed] [Google Scholar]
- 34.Ogawa A., Yoshimoto T., Kikuchi H., Sano K., Saito I., Yamaguchi T., Yasuhara H. Ebselen in acute middle cerebral artery occlusion: a placebo-controlled, double-blind clinical trial. Cerebrovasc. Dis. 1999;9:112–118. doi: 10.1159/000015908. [DOI] [PubMed] [Google Scholar]
- 35.Parnham M., Sies H. Ebselen: prospective therapy for cerebral ischaemia. Expert Opin. Invest. Drugs. 2000;9:607–619. doi: 10.1517/13543784.9.3.607. [DOI] [PubMed] [Google Scholar]
- 36.Aune T. M., Thomas E. L., Morrison M. Lactoperoxidase-catalyzed incorporation of thiocyanate ion into a protein substrate. Biochemistry. 1977;16:4611–4615. doi: 10.1021/bi00640a013. [DOI] [PubMed] [Google Scholar]
- 37.Hawkins C. L. The role of hypothiocyanous acid (HOSCN) in biological systems. Free Radical Res. 2009;43:1147–1158. doi: 10.3109/10715760903214462. [DOI] [PubMed] [Google Scholar]
- 38.Chang T. S., Jeong W., Woo H. A., Lee S. M., Park S., Rhee S. G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 39.Flohé L., Toppo S., Cozza G., Ursini F. A comparison of thiol peroxidase mechanisms. Antioxid. Redox Signaling. 2011;15:763–780. doi: 10.1089/ars.2010.3397. [DOI] [PubMed] [Google Scholar]
- 40.Cho C. S., Lee S., Lee G. T., Woo H. A., Choi E. J., Rhee S. G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signaling. 2010;12:1235–1246. doi: 10.1089/ars.2009.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lentner C., editor. Geigy Scientific Tables: Physical Chemistry, Composition of Blood, Hematology, Somatometric Data. Basle: Ciba-Geigy; 1984. [Google Scholar]
- 42.Takahashi K., Cohen H. J. Selenium-dependent glutathione peroxidase protein and activity: immunological investigations on cellular and plasma enzymes. Blood. 1986;68:640–645. [PubMed] [Google Scholar]
- 43.Peskin A. V., Cox A. G., Nagy P., Morgan P. E., Hampton M. B., Davies M. J., Winterbourn C. C. Removal of amino acid, peptide and protein hydroperoxides by reaction with peroxiredoxins 2 and 3. Biochem. J. 2010;432:313–321. doi: 10.1042/BJ20101156. [DOI] [PubMed] [Google Scholar]
- 44.Storkey C., Davies M. J., White J. M., Schiesser C. Synthesis and antioxidant capacity of 5-selenopyranose derivatives. Chem. Commun. 2011;47:9693–9695. doi: 10.1039/c1cc13652f. [DOI] [PubMed] [Google Scholar]
- 45.Kumar B. S., Kunwar A., Ahmad A., Kumbhare L. B., Jain V. K., Priyadarsini K. I. In vitro radioprotection studies of organoselenium compounds: differences between mono- and diselenides. Radiat. Environ. Biophys. 2009;48:379–384. doi: 10.1007/s00411-009-0240-1. [DOI] [PubMed] [Google Scholar]
- 46.Mishra B., Kumbhare L. B., Jain V. K., Priyadarsini K. I. Pulse radiolysis studies on reactions of hydroxyl radicals with selenocystine derivatives. J. Phys. Chem. B. 2008;112:4441–4446. doi: 10.1021/jp709880b. [DOI] [PubMed] [Google Scholar]
- 47.Morgenstern R., Cotgreave I. A., Engman L. Determination of the relative contributions of the diselenide and selenol forms of ebselen in the mechanism of its glutathione peroxidase-like activity. Chem. Biol. Interact. 1992;84:77–84. doi: 10.1016/0009-2797(92)90122-2. [DOI] [PubMed] [Google Scholar]
- 48.Masumoto H., Kissner R., Koppenol W. H., Sies H. Kinetic study of the reaction of ebselen with peroxynitrite. FEBS Lett. 1996;398:179–182. doi: 10.1016/s0014-5793(96)01237-9. [DOI] [PubMed] [Google Scholar]
- 49.Zhao R., Holmgren A. A novel antioxidant mechanism of ebselen involving ebselen diselenide, a substrate of mammalian thioredoxin and thioredoxin reductase. J. Biol. Chem. 2002;277:39456–39462. doi: 10.1074/jbc.M206452200. [DOI] [PubMed] [Google Scholar]
- 50.Haenen G. R., De Rooij B. M., Vermeulen N. P., Bast A. Mechanism of the reaction of ebselen with endogenous thiols: dihydrolipoate is a better cofactor than glutathione in the peroxidase activity of ebselen. Mol. Pharmacol. 1990;37:412–422. [PubMed] [Google Scholar]
- 51.Masumoto H., Sies H. The reaction of 2-(methylseleno)benzanilide with peroxynitrite. Chem. Res. Toxicol. 1996;9:1057–1062. doi: 10.1021/tx9600560. [DOI] [PubMed] [Google Scholar]
- 52.Takebe G., Yarimizu J., Saito Y., Hayashi T., Nakamura H., Yodoi J., Nagasawa S., Takahashi K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 2002;277:41254–41258. doi: 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- 53.Briviba K., Kissner R., Koppenol W. H., Sies H. Kinetic study of the reaction of glutathione peroxidase with peroxynitrite. Chem. Res. Toxicol. 1998;11:1398–1401. doi: 10.1021/tx980086y. [DOI] [PubMed] [Google Scholar]