

Abstract
Background:
The combination of the reversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) erlotinib with gemcitabine obtained FDA approval for treating patients with pancreatic cancer. However, duration of response is often limited and there is currently no reliable predictive marker.
Methods:
We determined the sensitivity of a panel of human pancreatic tumour cell lines to treatment with afatinib, erlotinib, monoclonal antibody (mAb) ICR62, and gemcitabine, using the Sulforhodamine B colorimetric assay. The effect of these agents on cell signalling and cell-cycle distribution was determined by western blot and flow cytometry, respectively.
Results:
At 200 nM, ICR62 had no effect on growth of these tumour cells with the exception of BxPC-3 cells. BxPC-3 cells were also sensitive to treatment with afatinib and erlotinib with respective IC50 values of 11 and 1200 nM. Compared with erlotinib, afatinib was also more effective in inhibiting the growth of the other human pancreatic tumour cell lines and in blocking the EGF-induced phosphorylation of tyrosine, EGFR, MAPK, and AKT. When tested in BxPC-3 xenografts, afatinib induced significant delay in tumour growth.
Conclusion:
The superiority of afatinib in this study encourages further investigation on the therapeutic potential of afatinib as a single agent or in combination with gemcitabine in pancreatic cancer.
Keywords: afatinib, erlotinib, ICR62, pancreatic cancer
Pancreatic cancer remains one of the most aggressive types of human cancer. Despite major advances in imaging technologies, surgery, chemotherapy and radiotherapy in the last few decades, survival rates for patients with pancreatic cancer are extremely poor with a mean 5-year survival rate of <10% (Klapman and Malafa, 2008). Worldwide, pancreatic cancer is responsible for >200 000 deaths each year (Parkin et al, 2005; Ferlay et al, 2010). In the United States alone, there were an estimated 43 140 new cases of pancreatic cancer and 36 800 deaths in 2010 (Jemal et al, 2010). One major contributing factor to mortality is the lack of specific marker(s) for the early detection of pancreatic cancer. Indeed, ∼80% of pancreatic cancer cases are diagnosed at an advanced stage of the disease and these patients are intrinsically resistant to treatment with radiation and chemotherapy (Li and Saif, 2009).
Since the early 1980s, aberrant expression and activation of epidermal growth factor receptor (EGFR) (ErbB1/HER1), which is the prototype member of the class-I growth factor receptor tyrosine kinase family (also called ErbB/HER family), has been reported in a wide range of human malignancies and in some cases it has been associated with poor prognosis (Modjtahedi and Dean, 1994; Nicholson et al, 2001; Normanno et al, 2006; Zhang et al, 2007). The ErbB/HER family consists of three additional members namely ErbB2 (neu/HER2), ErbB3 (HER3) and ErbB4 (HER4) (Normanno et al, 2006). The binding of a HER ligand to its respective receptor leads to conformational changes in the receptor's extracellular domain which allow the formation of homo- and/or heterodimers between different members of the ErbB family. Dimerisation results in auto- and transphosphorylation of specific tyrosine residues in the cytoplasmic region of the receptors, which in turn leads to the activation of numerous downstream mediator molecules. These downstream proteins mediate the activation of several signalling pathways the most significant of which are the PI3K/Akt and the RAS/RAF/MAPK pathways. These have an important role in the regulation of cellular processes such as cell proliferation, differentiation and survival (Olayioye et al, 2000; Yarden, 2001; Normanno et al, 2006). The association between aberrant activation of ErbB receptor family and a poor prognosis in a number of studies has led to the strategic development and approval of several ErbB targeted agents for the treatment of several human malignancies. These include anti-HER monoclonal antibodies (mAbs) like trastuzumab, cetuximab and panitunumab and small molecule HER tyrosine kinase inhibitors (TKIs) such as erlotinib, gefitinib and lapatinib (Zhang et al, 2007). Since the introduction of gemcitabine only the combination with erlotinib has been tested and approved for the treatment of patients with advanced pancreatic cancer (Kelley and Ko, 2008). This combination led to a marginal but statistically significant improvement in overall survival (6.24 months in combination vs 5.91 months with gemcitabine alone) and an increase in 1-year survival rate (23% with the combination vs 17% with gemcitabine alone; Moore et al, 2007). Despite the clinical benefit for a small fraction of treated patients, the majority of pancreatic cancer patients simply do not respond to treatment with erlotinib or acquire resistance following a few rounds of drug administration. Therefore, it is imperative not only to develop novel and more effective therapeutic agents for the treatment of pancreatic cancer, but also to identify those factors that are responsible for the poor response or development of resistance to the different types of therapies.
We reported previously the development of a large panel of anti-EGFR mAbs of which ICR62 was found to inhibit the binding of ligands to the EGFR and the subsequent ligand-induced EGFR tyrosine phosphorylation and activation of downstream cell signalling pathways (e.g., MAPK and PI3-K phosphorylation; Modjtahedi et al, 1993, 2003; Cunningham, 2006). In addition, mAb ICR62 has been shown to inhibit the growth of the EGFR overexpressing cell lines both in vitro and in vivo (Modjtahedi et al, 1993, 1996; Cunningham, 2006). Since small molecule TKIs and mAbs target two different domains of the receptor (extracellular and intracellular domains, respectively), a combination of these two strategies may be more effective than treatment with single agents. Indeed, in some studies, dual targeting of the EGFR with a combination of the two approaches (i.e., cetuximab with erlotinib or gefitinib) was shown to be superior to treatment with a single agent (Huang et al, 2004; Matar, 2004; Regales et al, 2009).
Unlike the reversible EGFR TKIs (gefitinib or erlotinib), afatinib (BIBW 2992) is an anilino-quinazoline derivative that was designed to covalently bind and irreversibly inhibit the ErbB family members EGFR, HER2 and HER4 (Solca, 2007; Minkovsky and Berezov, 2008; Perera et al, 2009; Nam et al, 2011). Afatinib was shown to have superior growth inhibitory efficacy compared with erlotinib in preclinical lung cancer models and in particular against lung cancer cells which display the EGFR (T790M) mutation and which are resistant to treatment with erlotinib or gefitinib (Li et al, 2008). In this study, we investigated the sensitivity of seven human pancreatic tumour cell lines to treatment with mAb ICR62, afatinib, erlotinib and gemcitabine used alone or in combinations. In addition, we investigated whether there was an association between the expression level of ErbB family members (EGFR, HER-2, HER-3 and HER-4) and response to treatment with the anti-HER agents as well as the effect of these agents on the cell-cycle distribution of pancreatic cancer cell lines.
Materials and methods
Tumour cell lines
A panel of seven human pancreatic cancer cell lines was used in this study including BxPC-3, PT-45, MiaPaCa-2, PANC-1, AsPc-1, Capan-1 and FA6. AsPc-1 and Capan-1 cell lines were kindly provided by Dr Charlotte Edling (Blizard Institute of Cell and Molecular Science, Barts and The London School of Medicine and Dentistry). Other pancreatic tumour cell lines (PANC-1, Mia PaCa2 and BxPC-3) were purchased from European Collection of Cell Cultures (ECACC, Porton Down, UK). The EGFR overexpressing (MDA-MB468), HER-2 overexpressing (SKBR3) and MCF-7 human breast tumour cell lines were used as controls in this study (Cunningham, 2006). All cell lines were cultured routinely at 37 °C in a humidified atmosphere (5% CO2). MiaPaCa-2 and PANC-1 were cultured in Dulbecco's Modified Eagle's Medium (Sigma-Aldrich, Gillingham, UK) supplemented with 10% Fetal Bovine Serum (FBS) (heat inactivated; PAA Laboratories, Yeovil, UK) and antibiotics penicillin (50 units per ml), streptomycin (0.05 mg ml−1) and neomycin (0.1 mg ml−1) (Sigma-Aldrich). BxPC-3, PT-45, AsPc-1, Capan-1 and FA6 were cultured in RPMI-1640 medium (Sigma-Aldrich) supplemented with 10% FBS, antibiotics penicillin, streptomycin and neomycin and 2 mM Glutamine (Sigma-Aldrich).
Antibodies and other reagents
mAB ICR62 (IgG2b) was raised against the external domain of the EGFR on the breast cancer cell line MDA-MB468 as described previously (Modjtahedi et al, 1993). The primary mouse antibodies used in this study, HM50.67A and HM43.16B, were raised against the external domain of the HER-2 and EGFR, respectively (Cunningham, 2006). Mouse mAbs MAB3481 (anti-HER-3) and MAB11311 (anti-HER-4) were purchased from R&D Systems (Abingdon, UK). Secondary FITC-conjugated rabbit anti-mouse mAb STAR9B was obtained from AbD Serotec (Kidlington, UK). Erlotinib was kindly provided by OSI Pharmaceuticals (Farmingdale, NY, USA). Gemcitabine was acquired from Healthcare at Home (Burton on Trent, UK) and the irreversible ErbB family blocker afatinib was developed by Boehringer Ingelheim (Vienna, Austria).
Cell surface expression of HER family members
The cell surface expression of all HER family members (EGFR, HER-2, HER-3 and HER-4) was assessed by flow cytometry as described previously (Cunningham, 2006). Briefly, ∼1 × 106 cells were suspended in 1 ml of growth medium supplemented with 2% FBS and incubated for 1 h by rotation at 4 °C, with medium alone or medium containing 10 μg ml−1 of mouse primary antibodies. Cancer cells were then washed three times by centrifugation at 1200 r.p.m. (264 g) for 5 min, resuspended in 2% FBS growth medium and then incubated for 1 h by rotation at 4 °C with FITC-conjugated rabbit anti-mouse IgG STAR9B (AbD Serotec) antibody (1 : 200 dilution). Following incubation with the secondary antibody, cells were washed three times by centrifugation and finally resuspended in 0.5 ml of FACS Flow buffer (Becton Dickinson Ltd, Oxford, UK). A minimum of 10 000 events were recorded by excitation with an argon laser at 488 nm using the FL-1 detector (525 nm) of a BD FACsCalibur flow cytometer (Becton Dickinson Ltd). Mean fluorescence Intensity values were calculated using the CellQuest Pro software (Becton Dickinson Ltd) and compared with those of negative controls.
Growth inhibition studies
The effect of the various agents on the growth of human cancer cell lines was investigated using the Sulforhodamine B (SRB; Sigma-Aldrich) colorimetric assay. Briefly, tumour cells were seeded at a density of 5 × 103 cells per well in 100 μl of growth medium (RPMI-1640 or DMEM according to the cell line) supplemented with 2% FBS in a 96-well plate. After 4 h incubation at 37 °C (in a humidified atmosphere, in 5% CO2), 100 μl aliquots of doubling dilutions of the agents were added to triplicate wells. Cells were incubated at 37 °C (3–5 days depending on the cell line) until the control cells (no treatment) became confluent. Tumour cells were then fixed with 10% trichloroacetic acid (Fisher Scientific, Loughborough, UK) for 1 h, washed five times with tap water, air dried and stained with 0.4% SRB in 1% acetic acid for 1 h. Following that, cells were washed with 1% acetic acid, air dried and the SRB stain was solubilised with 10 mM Tris-base (100 μl per well; Fisher Scientific). The absorbance of each well was measured at 565 nm using an Epoch plate reader (Biotek, Potton, UK). For the determination of the initial number of cells (before treatment), an extra plate was set up and processed similarly after 4 h incubation at 37 °C without the inhibitors. Growth as a percentage of control was determined from the formula:
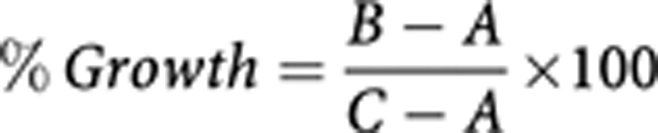 |
where: A=Absorbance at 565 nm (A565 nm) before treatment, B=A565 nm after treatment with inhibitors and C=A565 nm after incubation in medium alone. The 50% inhibitory concentration of cell growth (IC50) was calculated by non-linear least squares curve fitting (Four parameter analysis, log (inhibitor) vs response, variable slope) using Gen5 software (Biotek).
Determination of combination index
The growth inhibitory effect of the agents under investigation was also assessed when used in combination. Interactions between the different agents were assessed, using the combination index (CI) as described by Chou and Talalay (1984). For each combination, the two drugs were mixed at their 4 × IC50 followed by eight doubling dilutions. Combination index <0.9 indicates a synergistic effect while CI between 0.90 and 1.10 denotes an additive effect. Combination index >1.1 indicates antagonistic effects. Data analysis was performed using the Calcusyn software (Biosoft, Cambridge, UK).
Cell-cycle distribution analysis
The effect of HER inhibitors and gemcitabine on the cell-cycle distribution of the cancer cell lines was investigated using flow cytometry. Briefly, ∼2.5 × 105 cells were seeded to 25 cm2 flasks containing 10 ml of 2% FBS growth medium and the inhibitors at different concentrations or control medium. Once the cells containing only medium were almost confluent, treated cells were harvested and pooled together with the supernatant. Cancer cells were washed three times with cold PBS by centrifugation at 1200 r.p.m. (264 g) for 5 min. The final cell pellet was resuspended in 200 μl of cold PBS, fixed by the addition of 70% ethanol overnight at 4 °C. Tumour cells were then collected by centrifugation (264 g for 5 min) washed once with PBS and incubated with PI/RNAse mix (500 μl per 106cells) (Becton Dickinson Ltd) for 35 min at room temperature. A minimum of 10 000 events were recorded by excitation with an argon laser at 488 nm using the FL-3 detector (620 nm) of a BD FACsCalibur flow cytometer (Becton Dickinson Ltd) and analysed using the CellQuest Pro software (Becton Dickinson Ltd).
Western blot analysis
Cancer cells were grown to near-confluency in 6-well culture plates containing 5 ml of 10% FBS RPMI growth medium. Cells were washed once with 5 ml of RPMI/0.5% FBS and incubated in 5 ml of RPMI/0.5% FBS containing no inhibitor, afatinib (400 nM), erlotinib (400 nM), ICR62 (400 nM) or gemcitabine (100 nM) for 24 h at 37 °C. Following incubation with the inhibitors, cells were stimulated with 20 nM of EGF (Austral Biologicals, San Ramon, CA, USA) for 15 min and washed once with 5 ml of PBS. Cancer cells were lysed using 400 μl of Lysis buffer (Invitrogen, Paisley, UK) containing a protease inhibitor cocktail (Sigma-Aldrich) and cell lysates were heated at 90 °C for 4 min. Protein samples (20 μg) were separated on 4–12% Bis-Tris gels (Invitrogen) using the XCell II Surelock Mini-Cell system (Invitrogen) and transferred onto polyvinylidene difluoride (PVDF) membranes using the XCell II Mini-Cell Blot Module kit (Invitrogen). The PVDF membranes were probed with antibodies using the SNAP i.d System (Millipore, Watford, UK). Mouse antibodies against phospho-Tyr-100 and β-actin were purchased from Cell Signalling (Hitchin, UK), while anti-EGFR antibody from Sigma-Aldrich. Rabbit antibodies against Akt, MAPK and phospho-MAPK (Thr202/Tyr204) were purchased from Cell Signalling while anti-phospho EGFR (Tyr1173) and anti-phospho Akt (S473) rabbit antibodies were obtained from Biosource (Paisley, UK). The specific signals were detected using the WesternBreeze chemiluminescence kit (alkaline phosphatase-conjugated secondary antibody) (Invitrogen).
In vivo xenograft experiments
Five- to six-week-old female athymic BomTac:NMRI-Foxn1nu mice were maintained under specific pathogen-free conditions. All experiments complied with the Declaration of Helsinki and European Policy Legislations (FELASA and GV-SOLAS) on the Care and Use of Laboratory Animals. After acclimatisation mice were inoculated subcutaneously with 1 × 106 BxPC-3 cells (in 100 μl matrigel) into the right flank of the animal. After tumours reached an average volume of 50–100 mm3, the mice were randomised and treated orally daily with 15 mg kg−1 afatinib or vehicle control on the basis of individual weights. Tumours were measured three times a week with calipers, and tumour volumes were calculated by the formula π/6 × length × (width)2. Afatinib was formulated in 1.8% HP-beta-CD (Hydroxypropyl-β-cyclodextrin, Sigma-Aldrich), 5% of a 10% acetic acid stock and aqueous Natrosol (0.5%), and administered by intragastral gavage. The administration volume was 10 ml kg−1 body weight.
Statistical analysis
Linear regression analysis was used for the assessment of the relationship between the HER family receptors expression and response to treatment with afatinib. The unpaired two-tailed Student's t-test was used for comparing mean values between two groups. Data are presented as mean±s.d. P<0.05 was considered statistically significant.
Results
Cell surface expression of HER family members
We determined the expression levels of HER family of receptors in seven different pancreatic cancer cell lines using flow cytometry. All pancreatic tumour cell lines were found to be positive for both EGFR and HER-2 (Table 1). Compared with the MFI value of 415 in the positive-control EGFR overexpressing cell line MDA-MB468, the MFI values for the EGFR in this panel of human pancreatic tumour cell lines were lower and ranged from 34.54 (FA6) to 181.82 (PT-45) (Table 1). Similarly, compared with the MFI value of 831 in control SKBR3 cells, the MFI values for HER-2 expression in pancreatic tumour cells were much lower and ranged from 8.65 (FA6) to 29.25 (MiaPaCa-2) (Table 1). Most of the human pancreatic tumour cell lines were found to be HER-4 negative while expressing extremely low or undetectable levels of HER-3 (Table 1).
Table 1. Expression (MFI) of HER family members (EGFR, HER-2, HER-3 and HER-4) in human pancreatic cancer cell lines.
|
Mean fluorescence intensity (MFI)
|
|||||
|---|---|---|---|---|---|
| Cell line | Control | EGFR | HER-2 | HER-3 | HER-4 |
| Capan-1 | 4.2±0.7 | 138.5±15.2 | 28.9±3.1 | 7.4±0.7 | 4.9±0.4 |
| PT-45 | 2.9±0.6 | 184.7±21.1 | 21±1.9 | 6.1±0.4 | 3.9±0.3 |
| PANC-1 | 3±0.2 | 119.7±7.6 | 13.6±0.6 | 3.4±0.1 | 4.1±0.2 |
| BxPC-3 | 8±1.2 | 98.0±12.7 | 29.4±3.3 | 15.5±1.5 | 9.8±0.5 |
| MiaPaCa-2 | 3.7±0.3 | 39.5±3 | 32.9±3.8 | 4±0.3 | 4.4±0.3 |
| AsPc-1 | 3.6±0.7 | 97.4±12.8 | 19±2 | 5.9±0.5 | 4.1±1.3 |
| FA6 | 5.9±0.4 | 40.4±8.2 | 14.5±1.9 | 9.8±1.4 | 5.5±0.5 |
| SKBR3 (breast) | 4.3±0.2 | N/A | 831.3±74.6 | 21.2±8.7 | N/A |
| MDA-MB468 (breast) | 3.8±0.3 | 415.3±42 | N/A | N/A | N/A |
| MCF-7 (breast) | 3.5±0.3 | N/A | N/A | N/A | 24.6±2.3 |
Abbreviation: EGFR=epidermal growth factor receptor.
Breast cancer cell lines SKBR3, MDA-MB468 and MCF-7 were used as positive controls for HER-2/HER-3, EGFR and HER-4 receptors, respectively.
Growth response of human pancreatic tumour cells to treatment with anti-EGFR mAb ICR62, erlotinib, afatinib and gemcitabine
Of the seven human pancreatic tumour cell lines examined, BxPC-3 cells were the most sensitive cell line to treatment with afatinib with an IC50 value of 11 nM. The growth of other human pancreatic tumour cells was also inhibited by afatinib with IC50 values ranging from 367 nM (AsPc-1) to 1.37 μM (FA6) (Table 2; Figure 1B). In addition, BxPC-3 cells were the most sensitive to treatment with erlotinib with an IC50 value of 1.26 μM followed by AsPc-1 with an IC50 value of 5.8 μM (Table 2; Figure 1C). The mAb ICR62 has previously been shown to completely inhibit the growth of EGFR overexpressing tumour cell lines HN5 and DiFi in vitro in the low nanomolar range. In these experiments, ICR62 did not have any effect on the growth of the human pancreatic tumour cell lines tested at 200 nM (Figure 1D; Cunningham, 2006). The only exception was BxPC-3 cells, which were growth inhibited by 13%, however, with no statistical significance (P=0.061, Figure 1D). Gemcitabine inhibited the growth of all human pancreatic tumour cell lines with IC50 values ranging from 5 nM (FA6) to 105 nM (Capan-1) (Figure 1A; Table 2). Morphology of BxPC-3 cells following the treatment with growth inhibitory concentrations of erlotinib, afatinib and gemcitabine is presented in Figure 2. Next, we investigated the interactions between the different agents when used in combination. Median effect analysis showed that a combination of afatinib or erlotinib with gemcitabine led to a synergistic effect (CI<0.9) in the majority of cell lines tested (Table 2). We found no enhancement of growth inhibition following treatment with a combination of ICR62 with afatinib or gemcitabine or erlotinib in these in vitro models (data not shown).
Table 2. IC50 values for erlotinib, afatinib and gemcitabine in pancreatic cancer cell lines assessed by the SRB colorimetric assay and combination index (CI) values of gemcitabine plus afatinib or erlotinib in pancreatic cancer cell lines.
|
IC50 value
|
|||||
|---|---|---|---|---|---|
| Cell line | Erlotinib | Afatinib | Gemcitabine | Afatinib+gemcitabine combination index (effect) | Erlotinib+gemcitabine combination index (effect) |
| BxPC-3 | 1.26 μM | 11 nM | 7.4 nM | 0.31 (Synergism) | 0.46 (Synergism) |
| AsPc-1 | 5.8 μM | 367 nM | 87.3 nM | 0.62 (Synergism) | 0.69 (Synergism) |
| FA6 | >20 μM | 1.37 μM | 5.23 nM | 0.85 (Slight synergism) | N/A |
| PANC-1 | >20 μM | 966 nM | 21.4 nM | 0.73 (Moderate synergism) | N/A |
| Capan-1 | >20 μM | 781 nM | 104.9 nM | 0.88 (Slight synergism) | 0.66 (Synergism) |
| MiaPaCa-2 | >20 μM | 709 nM | 6.4 nM | 1.25 (Moderate antagonism) | 1.30 (Moderate antagonism) |
| PT-45 | >20 μM | 1.02 μM | 14.3 nM | 1.40 (Moderate antagonism) | 0.54 (Synergism) |
Abbreviations: N/A=not available; IC50=50% inhibitory concentration; SRB=Sulforhodamine B.
Interpretation of the results was based on the proposed descriptions for presenting the degrees of antagonism or synergism by Calcusyn software
Figure 1.

Effect of doubling dilutions of gemcitabine (A), afatinib (B) or erlotinib (C) on the growth of human pancreatic cancer cells. Tumour cells were grown in growth medium (2% FBS) with the inhibitors or medium alone until control cells (only medium) were confluent. Cancer cell proliferation was calculated as a percentage of control cell growth, as described in the Materials and Methods. Each point represents the mean±s.d. (D) The effect of gemcitabine at 100 nM, ICR62 at 200 nM and erlotinib or afatinib at 1.5 μM in pancreatic cancer cell lines (as percentage of control growth) is shown (columns, mean of triplicate values; bars, ±s.d.).
Figure 2.

(A) Morphology of BxPC-3 cells following growth inhibitory concentrations of erlotinib, afatinib and gemcitabine (μM) compared with treatment with medium alone (original magnification × 20). (B) Effect of afatinib, erlotinib and ICR62 on EGF-induced phosphorylation of tyrosine, EGFR, MAPK and Akt in BxPC-3 cells. BxPC-3 cells were cultured to near-confluency in growth medium containing 10% FBS, then treated in 0.1% FBS medium containing 400 nM of TKI, mAb ICR62 (400 nM) or gemcitabine (100 nM) for 24 h at 37 °C. Following incubation with the inhibitors, cells were stimulated with 20 nM of EGF for 15 min. Then, treated cells were lysed, protein samples were separated by SDS–PAGE, transferred onto PVDF membranes, an probed with antibodies specific for the molecule of interest.
Correlation of HER family member expression and sensitivity to ErbB inhibitors
Linear regression analysis was carried out in order to determine whether sensitivity to treatment with ErbB inhibitors was dependent on the expression level of the HER receptors. There was no correlation between the expression of EGFR (R=0.010, P=0.982) or HER-2 (R=0.458, P=0.301) and the IC50 values for afatinib nor any association between EGFR expression levels and response to treatment with erlotinib (R=0.153, P=0.743) or other members of HER family.
Cell-cycle distribution analyses
We examined the changes in cell-cycle distribution of the pancreatic cancer cells of various origins after treatment with afatinib and gemcitabine. Treatment with gemcitabine increased the proportion of pancreatic tumour cells in the subG1 phase (apoptotic/dead cells) of the cell cycle and this was accompanied by a significant increase in the percentage of cells in S phase consistent with inhibition of DNA synthesis (Table 3). For example, treatment of BxPC-3 cells with gemcitabine increased the proportion of cells in the subG1 phase from 9% to 36% and in the S phase from 22% to 46%, respectively (Table 3). In contrast, treatment of human pancreatic cells with afatinib increased the proportion of cells in subG1 phase of the cell cycle, accompanied by a decrease in the percentage of cells in G0/G1 phase of the cell cycle in the majority of cell lines investigated. Erlotinib had a similar effect to afatinib on the cell-cycle distribution of the two cell lines examined (BxPC-3 and AsPc-1) (Table 3).
Table 3. Effect of afatinib, erlotinib and gemcitabine on the cell-cycle distribution of pancreatic cancer cell lines.
| Cell line/treatment | Sub-G1 | G0/G1 | S | G2/M |
|---|---|---|---|---|
| BxPc-3 | ||||
| Control | 8.7 | 57.3 | 21.9 | 12.2 |
| Afatinib | 21.8 | 50.1 | 16.5 | 9.9 |
| Gemcitabine | 35.5 | 11 | 46.3 | 5.6 |
| Erlotinib | 13.3 | 51.6 | 20.7 | 13.5 |
| AsPC-1 | ||||
| Control | 1 | 45.9 | 32 | 19.6 |
| Afatinib | 13.3 | 43.5 | 30.9 | 13.6 |
| Gemcitabine | 3.3 | 15.7 | 54.5 | 24.9 |
| Erlotinib | 8 | 44.8 | 31.7 | 14.5 |
| Capan-1 | ||||
| Control | 3.6 | 49.2 | 27.7 | 19.4 |
| Afatinib | 9.5 | 49.3 | 28.9 | 10.8 |
| Gemcitabine | 23 | 26.3 | 31.9 | 18.1 |
| PT-45 | ||||
| Control | 2.8 | 71.4 | 14.3 | 10.8 |
| Afatinib | 9 | 67.9 | 15.4 | 6.8 |
| Gemcitabine | 23.7 | 33.6 | 36.9 | 5.4 |
| MiaPaCa-2 | ||||
| Control | 6.5 | 74.6 | 9.1 | 8.7 |
| Afatinib | 15.1 | 45.9 | 19.6 | 17.6 |
| Gemcitabine | 9.1 | 34 | 33.4 | 20 |
| PANC-1 | ||||
| Control | 6.7 | 51.6 | 11.7 | 29.4 |
| Afatinib | 42.9 | 28 | 10.5 | 17.9 |
| Gemcitabine | 20.1 | 38.8 | 30.1 | 9.4 |
| FA6 | ||||
| Control | 15.2 | 50.5 | 22 | 11.5 |
| Afatinib | 27.1 | 37.9 | 23.7 | 8.2 |
| Gemcitabine | 26.1 | 40.4 | 25.2 | 9 |
Each population is expressed as a percentage of gated cells.
Effect of anti-EGFR mAb ICR62, erlotinib, afatinib and gemcitabine treatment on EGF-induced tyrosine phosphorylation of the proteins in BxPC-3 cells
In contrast to gemcitabine, all three inhibitors blocked the EGF-induced tyrosine phosphorylation of EGFR and the phosphorylation of the tyrosine residue at position 1173 of the EGFR in BxPC3 cells. However, of the three inhibitors, afatinib was the only agent that successfully blocked the EGF-induced phosphorylation of both MAPK (ERK 1/2) and Akt (Figure 2B).
Xenograft experiments in mice
In vivo, afatinib showed potent anti-tumour activity in the BxPC-3 human pancreatic xenograft model. Daily administration of 15 mg kg−1 afatinib as a single agent to mice carrying established tumours did not induce long-lasting tumour regressions but significantly delayed tumour growth with a T/C value of 18% and a tumour growth inhibition (TGI) value of 89% as assessed at the end of the experiment (Figure 3).
Figure 3.

Effect of afatinib on the growth of BxPC-3 xenografts. Mice carrying established tumours (50–100 mm3) were treated orally, once daily with 15 mg kg−1 afatinib. Graph (A) shows tumour growth kinetics and insert (B) reports individual tumour volume on the last day of the experiment (day 24).
Discussion
Currently, among the several ErbB-targeting inhibitors developed, only the reversible EGFR TKI erlotinib has been approved in combination with gemcitabine for the treatment of patients with pancreatic cancer (Mendelsohn and Baselga, 2006; Ciardiello and Tortora, 2008; Modjtahedi and Essapen, 2009). While erlotinib improves survival, the duration of response is often limited and the majority of patients acquire resistance following a few rounds of treatment. Unlike first-generation reversible EGFR TKIs such as erlotinib and gefitinib, afatinib covalently binds and irreversibly inhibits the tyrosine kinase activity of all ErbB family members and is therefore expected to block both partners in the ErbB receptor dimer. This should result in a more effective signalling blockade and greater anti-tumour efficacy when compared with agents targeting EGFR alone. In this study, for the first time, we investigated the growth response of a panel of human pancreatic tumour cells to treatment with afatinib, ICR62, erlotinib and gemcitabine when used alone or in combination. We also investigated whether there was any association between the expression of HER family members and response to treatment with ErbB inhibitors.
We reported previously that, at concentrations >3.2 nM, mAb ICR62 inhibits completely the growth of the EGFR overexpressing cell lines HN5 and DiFi in vitro (Cunningham, 2006). In the present experiments, at a maximum concentration of 200 nM, mAb ICR62 had no significant effect on the growth of any of the human pancreatic tumour cell lines investigated (Figure 1D). This may be due to the lower expression of EGFR in these pancreatic cell lines compared with the EGFR expression in HN5 or DiFi cells (Modjtahedi et al, 1993; Cunningham, 2006). Another possible explanation for the lack of response to treatment with mAb ICR62 could be the presence of autocrine loops. Previous studies have shown that EGFR ligands may compete with mAbs for binding to the receptor and reduce their anti-proliferative activity (Castillo, 2004). In another study, Buchsbaum and colleagues investigated the effect of anti-EGFR mAb erbitux on the proliferation of BxPC-3 and MiaPaCa-2 cells. They found that while exposure to erbitux for 96 h resulted in a 17% reduction in proliferation of BxPC-3 cells, it did not have any inhibitory effect on MiaPaCa-2 cells(Buchsbaum et al, 2002). Of all cell lines examined in our study, mAb ICR62 only inhibited the growth of BxPC-3 cells by 13%, but this was not statistically significant (P=0.061, Figure 1D) and blocked the EGF-induced tyrosine phosphorylation of EGFR in these cells (Figure 2B).
Unlike mAb ICR62 which targets the extracellular ligand binding domain of the EGFR, afatinib is an irreversible inhibitor of the ErbB family (Solca, 2007; Berezov, 2008; Minkovsky and Berezov, 2008; Nam et al, 2011). Afatinib was found to inhibit the growth of all seven human pancreatic cancer cell lines with IC50 values ranging from 11 nM (BxPC3) to 1.37 μM (FA6) (Figure 1B; Table 2). However, the majority of cell lines were found to be insensitive to treatment with erlotinib with only BxPC-3 and AsPc-1 cell lines responding to treatment with this agent. The IC50 values were 1.26 and 5.8 μM for BxPC-3 and AsPc-1 cells, respectively (Table 2). In agreement with our findings in a previous study, Buck et al (2006) investigated the sensitivity of three pancreatic tumour cell lines, BxPC-3, MiaPaCa-2 and PANC-1 to treatment with erlotinib and of these only BxPC-3 was marginally sensitive to treatment with erlotinib with an IC50 value of 1.3 μM. In another study, erlotinib was found to inhibit the growth of BxPC-3 and Capan-1 cells with IC50 values of 7.6 and >20 μM, respectively (Cai et al, 2010). This comparative data analysis suggests that afatinib inhibits more potently the proliferation of pancreatic cancer cells in vitro. In addition, of all agents investigated, only afatinib inhibited the EGF-induced phosphorylation of MAPK (ERK 1/2) and Akt in BxPc-3 cells. Several studies have evaluated the anti-tumour activity of lapatinib, which is a reversible dual EGFR/HER-2 TKI. In one study, Komoto et al (2010) found that lapatinib, at the maximum concentration of 30 μM, inhibited the growth of Capan-1, Capan-2, PANC-1 and MiaPaCa-2 by <8%. Recently, in a recent phase II study 125 patients with metastatic pancreatic cancer were treated with 1500 mg of Lapatinib per day and 1g m−2 per week of gemcitabine for 3 weeks until the disease progression. However, no therapeutic benefit was reported for such combination (Safran et al, 2011).
In this study, we found no correlation between the expression level of EGFR (R=0.010, P=0.982) or HER-2 (R=0.458, P=0.301) and response to treatment with afatinib. Similarly, Komoto et al (2010) found that there was no statistically significant association between EGFR or HER-2 expression and the in vivo anti-tumour effect of the dual EGFR/HER-2 TKI lapatinib. These findings suggest that ErbB receptor expression levels are not of major relevance and may not be used as predictive biomarkers to identify responding models in vitro. Other factors such as the mutational status of ErbB family receptors or downstream molecules like K-RAS may be implicated in the sensitivity to ErbB inhibition. For example, in glioblastoma and non-small-cell lung carcinoma (NSCLC) patients the presence of EGFRvIII or T790M EGFR was found to be associated with resistance to treatment with gefitinib and erlotinib, respectively (Learn et al, 2004; Kobayashi et al, 2005; Pao et al, 2005). In addition, K-Ras mutations, which lead to the activation of the K-RAS protein in an EGFR-independent manner, have been associated with resistance to treatment with EGFR targeting mAbs in NSCLC and metastatic colorectal cancer patients (Linardou et al, 2008). While EGFR gene mutation is a rare phenomenon in pancreatic cancer, the presence of K-Ras mutations is a very common event and occurs in 75–95% pancreatic cancer patients (Hruban et al, 1993; Berrozpe et al, 1994; Laghi et al, 2002; Immervoll et al, 2006). It is noteworthy that of all the cell lines investigated in this study, BxPC-3 is the only one which carries a wild-type K-Ras gene which could explain its greater sensitivity to afatinib (IC50=11 nM) and erlotinib (IC50=1.26 μM) treatment compared with the rest of the tested cell lines (Table 2; Sipos et al, 2003). Further studies aimed at replacing wild-type K-Ras with mutant K-Ras in isogenic models will be necessary to substantiate this hypothesis. The BxPC-3 model could be suitable for such activities as in vitro observations can be verified in the in vivo setting. In this model, afatinib as a single agent demonstrated significant anti-tumour activity achieving an 89% TGI at the end of the experiment (day 24) with a T/C value of 18% (Figure 3). For comparison, gemcitabine when tested as a single agent, in the same model (different experiment) at a dose of 150 mg kg−1 twice weekly for 4 weeks (day 28), also induced tumour growth delay with a T/C value of 23%, thus comparing in activity with afatinib (data not shown).
The combination potential of these agents was assessed in vitro. While the addition of ICR62 to either afatinib or gemcitabine had no additional growth inhibitory effect compared with treatment with a single agent, a synergistic effect was observed in five of the seven human pancreatic tumour cell lines when afatinib was used in combination with gemcitabine (Table 2), thus generating a rationale for assessing such combination in vivo. Similarly, the combination of erlotinib with gemcitabine led to a synergistic effect in the majority of cell lines tested (Table 2). The enhanced growth inhibitory effect of the combination of ErbB inhibitors and gemcitabine may be attributed to the different mode of action of these agents. Cell-cycle distribution analyses demonstrated that gemcitabine altered the cell cycle of the treated cells, resulting in an increased subG1 cell population as well as an increase in cells in S phase consistent with previous findings (Ng et al, 2000). Although treatment of human pancreatic cells with afatinib or erlotinib increased the population of cells in subG1 phase of the cell cycle, a reduction in the percentage of cells in G0/G1 and G2/M phases of the cell cycle was observed in the majority of the cell lines (Table 3). Intriguingly, the in vitro combination of afatinib with gemcitabine was also capable of producing moderate antagonistic effects in two of the human pancreatic tumour cell lines used in this study with CI values of 1.25 and 1.4 in MiaPaCa-2 and PT-45, respectively (Table 2). In addition, the combination of erlotinib and gemcitabine produced antagonistic effects in MiaPaCa-2 (Table 2). Antagonism has also been reported when afatinib was combined with another cytotoxic drug (5-FU) in some colorectal cancer cells (Khelwatty et al, 2011). The different outcome of the same drug combination in different cell lines indicates the complex nature of drug/drug interactions and illustrates the elaborate biology behind these agents, which simultaneously force cells to perform G1- and S-phase arrests. Further investigations are required to understand the subtle differences induced by the combination treatments in these different pancreatic cancer models.
In conclusion, our results show that afatinib is effective at inhibiting the growth of human pancreatic tumour cells and that it potentiates the effect of gemcitabine in the majority of cell lines examined. Taken together, our findings encourage future investigation on the therapeutic potential of afatinib, as a single agent or in combination with gemcitabine in pancreatic cancer.
Acknowledgments
This work is supported by Kingston University London. We are grateful to OSI Pharmaceuticals for providing OSI-774 for use in our study. AGD receives support from Ralph Bates Pancreatic Cancer Research fund and the Cancer Vaccine Institute. UG and FS are employed by Boehringer Ingelheim RCV GmbH & Co KG, Austria.
References
- Berezov NMA (2008) BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr Opin Investig Drugs 9(12): 11. [PubMed] [Google Scholar]
- Berrozpe G, Schaeffer J, Peinado MA, Real FX, Perucho M (1994) Comparative analysis of mutations in the p53 and K-ras genes in pancreatic cancer. Int J Cancer 58(2): 185–191 [DOI] [PubMed] [Google Scholar]
- Buchsbaum DJ, Bonner JA, Grizzle WE, Stackhouse MA, Carpenter M, Hicklin DJ, Bohlen P, Raisch KP (2002) Treatment of pancreatic cancer xenografts with erbitux (IMC-C225) anti-EGFR antibody, gemcitabine, and radiation. Int J Radiat Oncol Biol Phys 54(4): 1180–1193 [DOI] [PubMed] [Google Scholar]
- Buck E, Eyzaguirre A, Brown E, Petti F, McCormack S, Haley JD, Iwata KK, Gibson NW, Griffin G (2006) Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors. Mol Cancer Ther 5(11): 2676–2684 [DOI] [PubMed] [Google Scholar]
- Cai X, Zhai H-X, Wang J, Forrester J, Qu H, Yin L, Lai C-J, Bao R, Qian C (2010) Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem 53(5): 2000–2009 [DOI] [PubMed] [Google Scholar]
- Castillo L (2004) Pharmacological background of EGFR targeting. Ann Oncol 15(7): 1007–1012 [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55 [DOI] [PubMed] [Google Scholar]
- Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358(11): 1160–1174 [DOI] [PubMed] [Google Scholar]
- Cunningham MP (2006) Responses of human colorectal tumor cells to treatment with the anti-epidermal growth factor receptor monoclonal antibody ICR62 used alone and in combination with the EGFR tyrosine kinase inhibitor gefitinib. Cancer Res 66(15): 7708–7715 [DOI] [PubMed] [Google Scholar]
- Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127(12): 2893–2917 [DOI] [PubMed] [Google Scholar]
- Hruban RH, van Mansfeld AD, Offerhaus GJ, van Weering DH, Allison DC, Goodman SN, Kensler TW, Bose KK, Cameron JL, Bos JL (1993) K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 143(2): 545–554 [PMC free article] [PubMed] [Google Scholar]
- Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM (2004) Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR). Cancer Res 64(15): 5355–5362 [DOI] [PubMed] [Google Scholar]
- Immervoll H, Hoem D, Kugarajh K, Steine SJ, Molven A (2006) Molecular analysis of the EGFR-RAS-RAF pathway in pancreatic ductal adenocarcinomas: lack of mutations in the BRAF and EGFR genes. Virchows Arch 448(6): 788–796 [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60(5): 277–300 [DOI] [PubMed] [Google Scholar]
- Kelley RK, Ko AH (2008) Erlotinib in the treatment of advanced pancreatic cancer. Biologics 2(1): 83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelwatty SA, Essapen S, Seddon AM, Modjtahedi H (2011) Growth response of human colorectal tumour cell lines to treatment with afatinib (BIBW2992), an irreversible erbB family blocker, and its association with expression of HER family members. Int J Oncol 39: 483–491 [DOI] [PubMed] [Google Scholar]
- Klapman J, Malafa MP (2008) Early detection of pancreatic cancer: why, who, and how to screen. Cancer Control 15(4): 280–287 [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong K-K, Tenen DG, Halmos B (2005) An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res 65(16): 7096–7101 [DOI] [PubMed] [Google Scholar]
- Komoto M, Nakata B, Nishii T, Kawajiri H, Shinto O, Amano R, Yamada N, Yashiro M, Hirakawa K (2010) In vitro and in vivo evidence that a combination of lapatinib plus S-1 is a promising treatment for pancreatic cancer. Cancer Sci 101(2): 468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laghi L, Orbetegli O, Bianchi P, Zerbi A, Di Carlo V, Boland CR, Malesci A (2002) Common occurrence of multiple K-RAS mutations in pancreatic cancers with associated precursor lesions and in biliary cancers. Oncogene 21(27): 4301–4306 [DOI] [PubMed] [Google Scholar]
- Learn CA, Hartzell TL, Wikstrand CJ, Archer GE, Rich JN, Friedman AH, Friedman HS, Bigner DD, Sampson JH (2004) Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res 10(9): 3216–3224 [DOI] [PubMed] [Google Scholar]
- Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK (2008) BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 27(34): 4702–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Saif MW (2009) Advancements in the management of pancreatic cancer. JOP 10(2): 109–117 [PubMed] [Google Scholar]
- Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S (2008) Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol 9(10): 962–972 [DOI] [PubMed] [Google Scholar]
- Matar P (2004) Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin Cancer Res 10(19): 6487–6501 [DOI] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J (2006) Epidermal growth factor receptor targeting in cancer. Semin Oncol 33(4): 369–385 [DOI] [PubMed] [Google Scholar]
- Minkovsky N, Berezov A (2008) BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr Opin Investig Drugs 9(12): 1336–1346 [PubMed] [Google Scholar]
- Modjtahedi H, Dean C (1994) The receptor for EGF and its ligands: expression, prognostic value and target for therapy in cancer. Int J Oncol 4: 277–296 [DOI] [PubMed] [Google Scholar]
- Modjtahedi H, Essapen S (2009) Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anticancer Drugs 20(10): 851–855 [DOI] [PubMed] [Google Scholar]
- Modjtahedi H, Hickish T, Nicolson M, Moore J, Styles J, Eccles S, Jackson E, Salter J, Sloane J, Spencer L, Priest K, Smith I, Dean C, Gore M (1996) Phase I trial and tumour localisation of the anti-EGFR monoclonal antibody ICR62 in head and neck or lung cancer. Br J Cancer 73(2): 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modjtahedi H, Moscatello DK, Box G, Green M, Shotton C, Lamb DJ, Reynolds LJ, Wong AJ, Dean C, Thomas H, Eccles S (2003) Targeting of cells expressing wild-type EGFR and type-III mutant EGFR (EGFRvIII) by anti-EGFR MAb ICR62: a two-pronged attack for tumour therapy. Int J Cancer 105(2): 273–280 [DOI] [PubMed] [Google Scholar]
- Modjtahedi H, Styles JM, Dean CJ (1993) The human EGF receptor as a target for cancer therapy: six new rat mAbs against the receptor on the breast carcinoma MDA-MB 468. Br J Cancer 67(2): 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25(15): 1960–1966 [DOI] [PubMed] [Google Scholar]
- Nam H-J, Kim H-P, Yoon Y-K, Hur H-S, Song S-H, Kim M-S, Lee G-S, Han S-W, Im S-A, Kim T-Y (2011) Antitumor activity of HM781-36B, an irreversible Pan-HER inhibitor, alone or in combination with cytotoxic chemotherapeutic agents in gastric cancer. Cancer Lett 302(2): 155–165 [DOI] [PubMed] [Google Scholar]
- Ng SSW, Tsao M-S, Chow S, Hedley DW (2000) Inhibition of phosphatidylinositide 3-kinase enhances gemcitabine-induced apoptosis in human pancreatic cancer cells. Cancer Res 60(19): 5451–5455 [PubMed] [Google Scholar]
- Nicholson RI, Gee JM, Harper ME (2001) EGFR and cancer prognosis. Eur J Cancer 37(Suppl 4): S9–S15 [DOI] [PubMed] [Google Scholar]
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366(1): 2–16 [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19(13): 3159–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2(3): e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global Cancer Statistics, 2002. CA Cancer J Clin 55(2): 74–108 [DOI] [PubMed] [Google Scholar]
- Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L, Borgman CL, Zaghlul S, Brandstetter KA, Kubo S, Takahashi M, Chirieac LR, Padera RF, Bronson RT, Shapiro GI, Greulich H, Meyerson M, Guertler U, Chesa PG, Solca F, Wistuba II, Wong KK (2009) HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci 106(2): 474–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, Koutcher JA, Spassova M, Ouerfelli O, Mellinghoff IK, Zakowski MF, Politi KA, Pao W (2009) Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 119(10): 3000–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran H, Miner T, Bahary N, Whiting S, Lopez CD, Sun W, Charpentier K, Shipley J, Anderson E, McNulty B, Schumacher A, Clark A, Vakharia J, Kennedy T, Sio T (2011) Lapatinib and gemcitabine for metastatic pancreatic cancer: a phase II study. Am J Clin Oncol 34(1): 50–52 [DOI] [PubMed] [Google Scholar]
- Sipos B, Möser S, Kalthoff H, Török V, Löhr M, Klöppel G (2003) A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform. Virchows Arch 442(5): 444–452 [DOI] [PubMed] [Google Scholar]
- Solca F (2007) Pharmacology and molecular mechanisms of BIBW2992 a potent irreversible dual EGFR/HER-2 kinase inhibitor of cancer therapy. Target Oncol 2: s15 [Google Scholar]
- Yarden Y (2001) The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer 37(Supplement 4): 3–8 [DOI] [PubMed] [Google Scholar]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI (2007) ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Investig 117(8): 2051–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]